

Abstract
A convergent route has been developed to synthesize an antifungal tricyclic o-hydroxy-p-quinone methide diterpenoid and analogs. The Li/naphthalene mediated reductive alkylation was employed for coupling β-cyclocitral and the corresponding benzyl chloride while the BBr3–mediated one-pot bis-demethylation and intramolecular Friedel Crafts alkylation was used to assemble the tricyclic molecular skeleton. The structure-activity relationship of the diterpenoid was assessed based on anti-proliferation assays of the natural product and analogs against strains of pathogenic yeasts and filamentous fungi.
INTRODUCTION
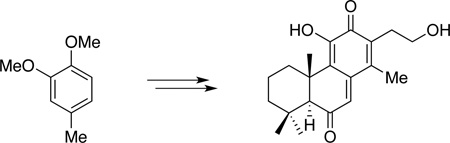
Quinone methides are typically reactive electrophilic species that are formed as transient intermediates in chemical transformations.1 However, in the presence of stabilizing structural elements, such as extended conjugation, relatively stable quinone methides may be formed. Indeed, a number of o-hydroxy-p-quinone methide diterpenoids and triterpenoids, such as fuerstion (1), taxodione (2) and its dimer (4), pristimerin (5), celastrol (6), tingenone (7), many of which show interesting biological activities, have been isolated from Nature (Figure 1).2 As part of our efforts in investigating natural products with useful pharmacological properties,3 we became interested in 3, an unnamed quinone methide diterpenoid isolated from the root bark of Bobgunnia madagascariensis.4 In particular, we were intrigued by its reported potent anti-fungal activity, which even rivals that of the clinical drugs amphotericin B and fluconazole. Our interest was partially fueled by the increasing demand for new antifungal drugs.5 The increasing incidence of invasive fungal infections associated with substantial mortality and high financial burden, especially among immunocompromised patient populations, rendered it increasingly important to develop new classes of drugs to combat fungal infection.
Figure 1.

Some o-hydroxy-p-quinone methide natural products
RESULTS AND DISCUSSION
Whereas a number of approaches have been reported for synthesis of taxodione and related compounds,6 many employing linear synthetic routes starting from natural precursors, no total synthesis had been reported for 3 since its isolation in 2000.5a To fully explore the biomedical potential of 3, we envisioned a convergent synthetic approach that not only would enable access of the natural product, but also allow preparation of analogs for structure-activity relationship (SAR) and mode-of-action studies. In this regard, we envisioned that the o-hydroxy-p-quinone methide moiety of 3 could be prepared by oxidation of catechol 8 (Scheme 1). An intramolecular Friedel-Crafts alkylation would be relied upon to build the tricyclic core of 8. The corresponding precursor 9 would be assembled by coupling of benzyl chloride 10 and commercially available β-cyclocitral. Herein we describe the results of our study, which led to the first synthesis of (±)–3 and elucidation of its preliminary SAR.
Scheme 1.

Synthetic design
Our synthesis started from the known bromobenzene 11, available in 4 steps from commercial 3,4-dimethyoxytoluene (Scheme 2).7 Stille coupling of 11 and tributylvinyltin was envisioned for introducing the ethylene group as a precursor to the 2-hydroxylethyl functionality of 3. After extensive screening of reaction conditions, the coupling was achieved using Pd2(dba)3 and P(t-Bu)3 to give 12 in 98% yield.8 The reduction of ester 12 with DIBAL-H yielded benzyl alcohol 13, which was converted to benzyl chloride 14 upon treatment with SOCl2. Whereas complex mixtures were obtained when Li/naphthalene was used for coupling benzyl chloride 14 and β-cyclocitral,6c no desired product (i.e. 15) could be isolated from our attempts of coupling through the corresponding Grignard and organolithium reagents of 14.
Scheme 2.

Synthesis of 14
We speculated that the extended conjugation of 14 might be responsible for the difficulty of the coupling. Thus, we explored the possibility of furnishing the 2-hydroxylethyl group prior to the coupling reactions. However, attempts to introduce the 2-hydroxylethyl group of 17 through hydroboration/oxidation of 12 was only partially successful as the undesired regio-isomer (i.e. 16) was formed as the major product under all the conditions except when BH2I·Me2S was used,9 which gave the desired regio-isomeric product 17 somewhat preferentially (2:1) (Scheme 3).
Scheme 3.

Hydroboration/oxidation of 12
In order to improve the efficiency of the synthesis, we turned to the allyl-substituted benzyl chloride 19, which was expected to be compatible with the coupling reaction (Scheme 4). The previously employed Stille reaction conditions proved to be also effective for coupling of 11 and allyltributyltin to give 18. DIBAL-H reduction followed by chlorination with SOCl2 converted 18 into benzyl chloride 19. As we had expected, the coupling of benzyl chloride 19 and β-cyclocitral went smoothly to give 20 (79% yield), which was converted to α, β-unsaturated ketone 21 through oxidation with IBX. After oxidative cleavage of the alkene of 21 through OsO4-catalyzed dihydroxylation and subsequent oxidative cleavage of the vicinal diol with NaIO4, aldehyde 22 was obtained in 85% yield. Selective reduction of the aldehyde in the presence of α, β-unsaturated ketone was achieved with NaBH(OAc)3 to give primary alcohol 23.
Scheme 4.

Synthesis of 23
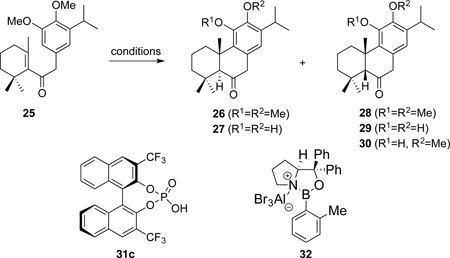
The generation of the tricyclic molecular skeleton of 3 by the intramolecular Friedel-Crafts alkylation reaction is challenging because it requires simultaneous dearomatization and the formation of an all-carbon quaternary center. Thus, to evaluate the feasibility and to elucidate the nuance of this transformation, we used 25 as a model substrate to test various conditions for the cyclization. This substrate (i.e. 25) was synthesized through Li/naphthalene mediated coupling of the known benzyl chloride 24 and β-cyclocitral followed by oxidation with IBX (Scheme 5).6j
Scheme 5.

Synthesis of 25
A number of achiral and chiral Brønsted/Lewis acids were evaluated for the intramolecular Friedel-Crafts alkylation of 25. The combination of formic acid and phosphoric acid was reported to be suitable for cyclizing 25 into 26 in 41% yield at 115 °C (not shown).6f Interestingly, extended reaction under such conditions led to formation of the undesired cis-isomeric 28 as the major product (Table 1, entry 1), suggesting its thermodynamic nature. The BINOL-based chiral phosphoric acid 31c was also effective for the cyclization (entry 2), but no enantioselectivity was observed under the harsh conditions necessary for the reaction to proceed. A number of Lewis acids were also tested (see Supporting Information for a complete list). Treatment of 25 with BBr3 led to one-pot bis-demethylation and the intramolecular Friedel-Crafts alkylation to give 27 (entry 3),10 which could be oxidized with Ag2O to give taxodione (i.e. 2, not shown).6a On the other hand, the starting material was recovered when 25 was treated with BF3·Et2O (entry 4). Surprisingly, the one-pot demethylation and Friedel-Crafts cyclization of 25 with TMSI led to diastereoselective formation of the cis-isomeric product 30, with only one of the methoxyl groups hydrolyzed (entry 5). In order to impose enantioselection during the cyclization, the oxazaborolidine-based Lewis acid 32 was also used.11 However, the cyclization proceeded with low yield and low enantioselectivity (entry 6). A series of chiral Brønsted and Lewis acidic conditions were also tested (see Supporting Information). However, no enantioselection was observed for the cyclization.
Table 1.
The intramolecular Friedel-Crafts alkylation of 25
 | |||||
|---|---|---|---|---|---|
| entry | conditions | T (°C) | time | product | % yield (ee) |
| 1 | HCO2H, H3PO4 | 115 | 12 h | 26+28 | 18 + 38 |
| 2 | HCO2H, 31c | 110 | 96 h | 26+28 | 43 + 14 |
| 3 | BBr3, CH2Cl2 | −78 to 0 | 30 min | 27 | 45 |
| 4 | BF3·Et2O, CH2Cl2 | −78 to rt | 2h | 27 | 0 |
| 5 | TMSI, toluene | 70 | 4h | 30 | 54 |
| 6 | 32, BBr3, CH2Cl2 | 0 | 20 min | 27 | 20 (< 20) |
The application of the BBr3-mediated one-pot reaction conditions allowed the completion of the synthesis of 3. Thus, treatment of 23 with BBr3 led to one-pot bis-demethylation and cyclization to form a tricyclic catechol, which was subjected to oxidation with Ag2O to give 3 (Scheme 6). Comparison of the 1H, 13C NMR and MS spectra with those reported for the natural product confirmed the identity of synthetic 3. Selective mono-etherificaiton of 3 with dimethoxymethane gave analog 33.
Scheme 6.

Synthesis of 3 and 33
To evaluate the effect of the 2-hydroxyethyl group of 3 over the antifungal activity, we also prepared analog 37 from the known carboxylic acid 347 (Scheme 7). Thus, reduction of 34 with borane dimethyl sulfide complex followed by chlorination with SOCl2 provided benzyl chloride 35. The coupling of 35 and β-cyclocitral went smoothly with Li/naphthalene to give the corresponding allyl alcohol, which was oxidized with IBX to give enone 36. Compound 37 was obtained by BBr3-mediated one-pot bis-demethylation and intramolecular Friedel-Crafts alkylation followed by oxidation of the resulting catechol with Ag2O.
Scheme 7.

Synthesis of 37
With the compounds synthesized above in hand, we evaluated their anti-fungal activity against strains of pathogenic yeasts (Candida spp. and Cryptococcu spp.) and the mold Aspergillus fumigatus. Candida, Cryptococcus, and Aspergillus species are the major fungal pathogens of global significance (Table 2).12 The strains were chosen because of existing data on their susceptibility to fluconazole or itraconazole,13,14 two azole drugs that are commonly used in clinic to treat infections caused by yeast and mold respectively. Consistent with the original report, the synthetic o-hydroxy-p-quinone methide 3 indeed was found to be potently cytotoxic towards all the yeast strains tested, with MIC100 values ranging from 0.4 to 6.4 mg/L. Interestingly, the values of MIC100 were found to be similar to those of MFC (minimal fungicidal concentration). By contrast, MFCs are often much higher than MICs for fungistatic drugs such as fluconazole (Table 2). This finding indicates the fungicidal rather than fungistatic nature of these compounds. Compounds 3 and 37 are particularly effective against all the yeast strains, including the Candida glabrata, Candida krusei, and Candida parapsilosis strains that are resistant to the commonly used antifungal fluconazole (Table 2).14 The lack of any apparent correlation between the fungal susceptibility against these new compounds and that against azole drugs suggests that these compounds might differ in their mode of action against fungi from the known drug fluconazole. Taxodione (2) showed potent activity as well. However, the methoxymethyl ether of 3 (i.e. 33) significantly diminished the growth inhibition activity. None of these compounds were found to be effective against the mold A. fumigatus. It appears that the cytotoxicity of these compounds is correlated with the fungal growth mode (yeast versus mold) rather than their evolutionary relatedness, given that Candida (yeast) and Aspergillus (mold) spp. are closely related and their common ancestors diverged from that of Cryptococcus (yeast) spp. about one billion years ago. The cause of the differential cytotoxicity of these compounds against yeasts and the filamentous fungal stains is currently unknown.
Table 2.
Taxodione (2), 3, and analogs exhibit fungicidal effect (mg/L)*
| Yeast Strains | MIC100 | MFC* | MIC100 | MFC | MIC100 | MFC | MIC100 | MFC | MIC90 | MFC |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 3 | 37 | 37 | 2 | 2 | 33 | 33 | Azole | Azole | |
| Cryptococcus gattii R265 | 0.4 | 0.4 | 0.8 | 0.8 | 0.8 | 0.8 | 6.4 | 12.8 | 4 | - |
| Cryptococcus neoformans H99α | 0.4 | 0.8 | 0.8 | 0.8 | 1.6 | 1.6 | 6.4 | 12.8 | 1 | 8 |
| Candida albicans SC5314 | 6.4 | 12.8 | 3.2 | 6.4 | 6.4 | >12.8 | >12.8 | >100 | 0.2 | 64 |
| Candida glabrata PAT2ISO3 | 1.6 | 6.4 | 1.6 | 3.2 | 3.2 | >6.4 | 6.4 | >12.8 | 8 | >64 |
| Candida krusei DUMC132.91 | 1.6 | 1.6 | 1.6 | 3.2 | 3.2 | 6.4 | 12.8 | >12.8 | 64 | 64 |
| Candida parapsilosis MMRL1594 | 3.2 | 6.4 | 1.6 | 3.2 | 6.4 | 12.8 | 12.8 | >100 | >64 | >64 |
| Aspergillus fumigatus Af293 | 25.6 | 25.6 | 17.1 | 17.1 | >100 | >100 | 100 | >100 | 0.4 | 12.8 |
| Aspergillus fumigatus CEA10 | 25.6 | 25.6 | 17.1 | 17.1 | >100 | >100 | 100 | >100 | 0.5 | - |
MFC is defined as the lowest drug concentration at which at least 99% of cells were killed compared to the original inocula. Suspensions from the microdilution assays after 24 h (for Candida species and Aspergillus fumigatus strains) or 48 h (for strains of Cryptococcus species) of incubation were plated on drug-free medium for obtaining the values of colony forming units (CFU) to determine the minimal fungicidal concentration (MFC). Successive 2× serial dilutions of the compounds were used. The data on the susceptibility of these strains against azole drugs (itraconazole for the mold A. fumigatus and fluconazole for all yeast species) were obtained from previous studies.13,14,15
CONCLUSION
In summary, we completed the first total synthesis of the antifungal tricyclic o-hydroxy-p-quinone methide diterpenoid 3. The Stille reaction was used to introduce the allyl group as a masked 2-hydroxyethyl side chain whereas the Li/naphthalene mediated reductive alkylation was employed for coupling benzyl chloride 19 and β-cyclocitral. We developed a BBr3-mediated one-pot bis-demethylation/intramolecular Friedel-Crafts alkylation to assemble the tricyclic molecular skeleton and completed the synthesis of 3. Taxodione 2 and analogs of 3 were also synthesized. Compounds 3, 37, and 2 showed potent cytotoxicity against various strains of pathogenic yeasts tested, but etherification of the 2-hydroxyethyl led to significantly attenuated activity. Surprisingly, these compounds were found to be ineffective against the fungal strain Aspergillus fumigatus Af293. The cause of this discrepancy of activity against the yeasts versus filamentous fungi is currently unknown.
EXPERIMENTAL SECTION
General Information
All moisture sensitive reactions were carried out in flame-dried flasks under nitrogen atmosphere. Dichloromethane and diethyl ether were dried using an activated molecular sieve solvent purification system. Tetrahydrofuran was freshly distilled over sodium and benzophenone. All other commercial reagents were used as received. Reactions were monitored by TLC performed on pre-coated glass-backed TLC plates, Silica Gel 60 F254 (EMD 250 µm thickness). Spots were visualized with UV or through staining with an ethanolic solution of phosphomolybdic acid. Flash column chromatography was performed using 60 Å silica gel (230–400 mesh) as the stationary phase. 1H NMR chemical shifts are reported as δ values in ppm relative to CDCl3 (7.26 ppm) or acetone-d6 (2.05 ppm), coupling constants (J) are reported in Hertz (Hz), and multiplicity follows normal convention. CDCl3 (77.0 ppm) or acetone-d6 (29.84 ppm) served as the internal standard for 13C NMR spectra. Infrared spectra (IR) resonance frequencies are given as wavenumbers in cm−1. HRMS spectra were recorded using a tandem TOF spectrometer.
Methyl 4,5-dimethoxy-2-methyl-3-vinylbenzoate (12)
To a suspension of ester 11 (1.57 g, 5.45 mmol), Pd2dba3 (113 mg, 0.11mmol), tributylvinyltin (1.75 mL, 6.0 mmol) in toluene (11 mL) was added P(t-Bu)3 (0.22 mL, 0.22 mmol, 1 M in toluene) under nitrogen. The reaction was stirred at 60 °C overnight. When the reaction was complete based on TLC, the solution was treated with KF (4 g), Et2O (30 mL), and activated carbon (4 g). The mixture was stirred for 5 min before it was filtered through a short pad of silica gel (washed with ethyl acetate). After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide 12 (1.26 g, 98 %) as yellow oil. IR (film, cm−1) 2999, 2955, 2842, 1723, 1590, 1478, 1427, 1324, 1199, 1167, 1110, 983; 1H NMR (500 MHz, CDCl3) δ 7.32 (s, 1H), 6.71 (dd, J = 17.9, 11.6 Hz, 1H), 5.61 (dd, J = 11.6, 2.0 Hz, 1H), 5.52 (dd, J = 17.9, 2.0 Hz, 1H), 3.90 (s, 3H), 3.89 (s, 3H), 3.79 (s, 3H), 2.47 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.4, 150.0, 149.7, 133.8, 131.2, 131.1, 126.3, 121.1, 112.9, 60.3, 55.9, 52.0, 17.6; HRMS: molecular ion not observed.
(4,5-Dimethoxy-2-methyl-3-vinylphenyl)methanol (13)
To a stirred solution of ester 12 (1.24 g, 5.25 mmol) in THF (30 mL) was added DIBAL-H (1 M in hexanes, 15.75 mL, 15.75 mmol) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 2 h before it was quenched with ethyl acetate (3 mL) and saturated aqueous Rochelle salt (30 mL). The mixture was stirred for another 1 h at room temperature. The organic layer was separated and aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/1) to provide 13 (992 mg, 91 %) as colorless oil. IR (film, cm−1) 2934, 1587, 1475, 1318, 1235, 1116, 974, 921, 944; 1H NMR (500 MHz, CDCl3) δ 6.92 (s, 1H), 6.74 (dd, J = 17.9, 11.7 Hz, 1H), 5.58 (dd, J = 11.7, 2.1 Hz, 1H), 5.54 (dd, J = 17.9, 2.1 Hz, 1H), 4.67 (s, 2H), 3.86 (s, 3H), 3.75 (s, 3H), 2.25 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.4, 146.3, 134.6, 132.9, 131.5, 126.6, 120.5, 111.2, 63.9, 60.2, 55.9, 15.1; HRMS (ESI): calculated for C12H16O3 [M+Li+] 215.1259, found 215.1266.
1-(Chloromethyl)-4,5-dimethoxy-2-methyl-3-vinylbenzene (14)
To a solution of alcohol 13 (832 mg, 4.0 mmol) in CH2Cl2 (20 mL) was added SOCl2 (0.35 mL, 4.8 mmol) dropwise at 0 °C under nitrogen. The reaction was stirred at room temperature for 30 min before it was carefully quenched with saturated aqueous NaHCO3 and taken into CH2Cl2. The organic layer was separated, washed with water, brine, and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/20) to provide 14 (832 mg, 92 %) as yellow oil. IR (film, cm−1) 3005, 2970, 2934, 2837, 1590, 1478, 1318, 1238, 1116, 1045, 980, 927, 853; 1H NMR (500 MHz, CDCl3) δ 6.84 (s, 1H), 6.73 (dd, J = 17.9, 11.7 Hz, 1H), 5.60 (dd, J = 11.7, 2.0 Hz, 1H), 5.54 (dd, J = 17.9, 2.1 Hz, 1H), 4.62 (s, 2H), 3.88 (s, 3H), 3.76 (s, 3H), 2.34 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.5, 147.4, 133.4, 131.3, 131.2, 128.2, 120.7, 113.0, 60.2, 55.9, 45.8, 15.4; HRMS: molecular ion not observed.
Ethyl 3-allyl-4,5-dimethoxy-2-methylbenzoate (18)
To a suspension of ester 11 (2.88 g, 10 mmol), Pd2dba3 (275 mg, 0.30 mmol), and allyltributyltin (3.72 mL, 12 mmol) in toluene (15 mL) was added P(t-Bu)3 (1 M in toluene, 0.60 mL, 0.60 mmol) under nitrogen. The reaction was stirred at 60 °C for overnight. When the reaction was complete (monitored by TLC, ethyl acetate/petroleum ether = 1:10 for 2~3 times), KF (5 g) was added followed by Et2O (30 mL) and activated carbon (5 g). The mixture was stirred for 5 min, and then it was filtered through a short pad of silica (washed with ethyl acetate). After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide 18 (2.59 g, 98%) as yellow oil. IR (film, cm−1) 2949, 2842, 1717, 1596, 1484, 1330, 1211, 1099, 1045, 989, 1H NMR (500 MHz, CDCl3) δ 7.29 (s, 1H), 5.95-5.89 (m, 1H), 5.00 (dd, J = 10.2, 1.8 Hz, 1H), 4.86 (dd, J = 17.1, 1.8 Hz, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.49 (dt, J = 5.6, 1.8 Hz, 2H), 2.41 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.6, 150.2, 150.0, 136.0, 133.3, 132.1, 126.1, 115.1, 112.2, 60.9, 55.7, 51.9, 30.8, 16.1; HRMS (ESI): calculated for C14H18O4 [M+H+] 251.1283, found 251.1274.
3-Allyl-1-(chloromethyl)-4,5-dimethoxy-2-methylbenzene (19)
To a solution of ester 18 (2.50 g, 10 mmol) in THF (50 mL) was added DIBAL-H (1 M in hexanes, 30 mL, 30 mmol) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 4 h before it was treated with ethyl acetate (5 mL) and saturated aqueous Rochelle salt (50 mL). The mixture was stirred for another 1 h at room temperature. The organic layer was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/1) to provide allyl alcohol S1 (2.01 g, 90 %) as colorless oil. IR (film, cm−1) 3414, 2937, 1640, 1599, 1487, 1309, 1232, 1111, 1042, 906, 850; 1H NMR (500 MHz, CDCl3) δ 6.87 (s, 1H), 5.92 (ddt, J = 17.1, 10.1, 5.8 Hz, 1H), 4.99 (dd, J = 10.2, 1.8 Hz, 1H), 4.90 (dd, J = 17.1, 1.9 Hz, 1H), 4.64 (d, J = 4.5 Hz, 2H), 3.84 (s, 3H), 3.78 (s, 3H), 3.47 (dt, J = 5.8, 1.8 Hz, 2H), 2.18 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.4 146.7, 136.5, 134.4, 132.6, 127.5, 115.0, 110.5, 63.9, 60.9, 55.7, 30.9, 13.9; HRMS (ESI): calculated for C13H18O3 [M+Li+] 229.1416, found 229.1406.
To a solution of S1 (1.85 g, 8.32 mmol) in CH2Cl2 (40 mL) was added SOCl2 (0.73 mL, 9.99 mmol) dropwise at 0 °C under nitrogen. The reaction was maintained at room temperature for 30 min before it was carefully quenched with saturated aqueous NaHCO3. The mixture was taken into CH2Cl2. The organic layer was separated, washed with water, brine, and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/10) to provide 19 (1.93 g, 97 %) as white amorphous solid. IR (film, cm−1) 2940, 1602, 1484, 1330, 1226, 1108, 1045, 986, 912; 1H NMR (500 MHz, CDCl3) δ 6.79 (s, 1H), 5.92 (ddt, J = 17.1, 10.2, 5.8 Hz, 1H), 5.01 (dd, J = 10.2, 1.8 Hz, 1H), 4.89 (dd, J = 17.1, 1.8 Hz, 1H), 4.60 (s, 2H), 3.86 (s, 3H), 3.80 (s, 3H), 3.48 (dt, J = 5.8, 1.8 Hz, 2H), 2.26 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.5, 147.7, 136.2, 133.1, 131.0, 129.1, 115.1, 112.2, 60.9, 55.7, 45.9, 31.0, 14.3; HRMS (ESI): calculated for C13H17ClO2 [M+H+] 241.0995, found 241.1003.
2-(3-Allyl-4,5-dimethoxy-2-methylphenyl)-1-(2,6,6-trimethylcyclohex-1-en-1-yl)ethanol (20)
A mixture of naphthalene (3.61 g, 28.13 mmol) and lithium (196 mg, 28.13 mmol) in THF (30 mL) was stirred at room temperature under nitrogen for 1.5 h. The mixture was then treated with a solution of β-cyclocitral (1.26 g, 8.25 mmol) and benzyl chloride 19 (1.80 g, 7.50 mmol) in THF (10 mL) dropwise via syringe at 0 °C. After stirring at room temperature for 2 h, the mixture was diluted with ether and treated with saturated aqueous NH4Cl. The organic phase was separated. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/4) to provide 20 (2.13 g, 79%) as colorless liquid. IR (film, cm−1) 3500, 2931, 1593, 1484, 1291, 1229, 1111, 1048, 989, 906; 1H NMR (500 MHz, CDCl3) δ 6.70 (s, 1H), 5.94 (dd, J = 17.1, 10.1 Hz, 1H), 5.01 (dq, J = 10.1, 1.7 Hz, 1H), 4.93 (dq, J = 17.1, 1.8 Hz, 1H), 4.45 (dd, J = 10.0, 4.3 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.49 (ddd, J = 5.8, 3.9, 1.9 Hz, 2H), 3.19 (dd, J = 14.2, 10.0 Hz, 1H), 2.88 (dd, J = 14.2, 4.3 Hz, 1H), 2.24 (s, 3H), 2.03-1.99 (m, 2H); 2.02 (s, 3H), 1.65-1.54 (m, 2H), 1.46-1.44 (m, 2H), 1.11 (s, 3H), 0.93 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.3, 146.0, 139.3, 136.7, 133.3, 132.4, 132.0, 128.3, 114.9, 113.0, 71.2, 60.9, 55.8, 40.5, 40.0, 34.8, 34.2, 31.2, 28.5, 28.3, 21.4, 19.3, 15.2; HRMS (ESI): calculated for C23H34O3 [M+Na+] 381.2406, found 381.2388.
2-(3-Allyl-4,5-dimethoxy-2-methylphenyl)-1-(2,6,6-trimethylcyclohex-1-en-1-yl)ethanone (21)
A solution of alcohol 20 (910 mg, 2.54 mmol) and IBX (1.07 g, 3.81 mmol) in DMSO (5 mL) was stirred at room temperature for 3 h. The reaction was quenched with H2O (5 mL) at 0 °C. The mixture was filtered and the aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/1) to provide 21 (780 mg, 86%) as yellow oil. IR (film, cm−1) 2937, 1691, 1596, 1487, 1285, 1226, 1113, 1048, 995; 1H NMR (500 MHz, CDCl3) δ 6.56 (s, 1H), 5.93 (ddt, J = 17.1, 10.2, 5.7 Hz, 1H), 4.99 (dq, J = 10.1, 1.7 Hz, 1H), 4.91 (dq, J = 17.1, 1.9 Hz, 1H), 3.87 (s, 2H), 3.82 (s, 3H), 3.78 (s, 3H), 3.48 (dt, J = 5.7, 1.8 Hz, 2H), 2.11 (s, 3H), 1.99 (t, J = 6.5 Hz, 2H), 1.72-1.67 (m, 2H), 1.63 (s, 3H), 1.48-1.46 (m, 2H), 1.11 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 207.8, 150.3, 146.4, 143.1, 136.6, 132.4, 129.5, 129.1, 128.4, 114.9, 112.9, 60.8, 55.8, 50.8, 39.0, 33.4, 31.3, 31.2, 28.8, 21.2, 18.9, 15.4; HRMS (ESI): calculated for C23H32O3 [M+H+] 357.2430, found 357.2423.
2-(2,3-Dimethoxy-6-methyl-5-(2-oxo-2-(2,6,6-trimethylcyclohex-1-en-1-yl)ethyl)phenyl)acetaldehyde (22)
To a solution of alkene 21 (530 mg, 1.49 mmol) in acetone/H2O (4/1, 15 mL) was added NMO (262 mg, 2.23 mmol) and OsO4 (2.5 wt% solution in t-BuOH, 758 mg, 7.45% mmol). After being stirred at room temperature overnight, the mixture was concentrated in vacuo. The residue was taken into EtOAc, washed with H2O, brine, and dried over Na2SO4. After concentration in vacuo, the reaction crude was dissolved in CH3CN/H2O (1/1, 16 mL) and treated with NaIO4 (478 mg, 2.24 mmol) at 0 °C. The mixture was allowed to room temperature and stirred for 1 h before it was filtered through a short pad of silica gel (eluted with EtOAc). The filtrate was washed with H2O, brine, and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide 22 (446 mg, 84% for 2 steps) as white amorphous solid. IR (film, cm−1) 2937, 1720, 1697, 1484, 1279, 1229, 1108, 1066; 1H NMR (500 MHz, CDCl3) δ 9.68 (s, 1H), 6.62 (s, 1H), 3.89 (s, 2H), 3.84 (s, 3H), 3.81 (d, J = 2.0 Hz, 2H), 3.78 (s, 3H), 2.07 (s, 3H), 2.00 (t, J = 6.5 Hz, 2H), 1.72-1.66 (m, 2H), 1.64 (s, 3H), 1.49-1.46 (m, 2H), 1.12 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 207.6, 199.7, 150.2, 146.6, 143.0, 129.7, 129.2, 128.8, 125.7, 114.2, 60.5, 55.8, 50.8, 42.7, 38.9, 33.5, 31.3, 28.8, 21.2, 18.8, 16.1; HRMS (ESI): calculated for C22H30O4 [M+Li+] 365.2304, found 365.2312.
2-(3-(2-Hydroxyethyl)-4,5-dimethoxy-2-methylphenyl)-1-(2,6,6-trimethylcyclohex-1-en-1-yl)ethanone (23)
To a solution of aldehyde 22 (423 mg, 1.18 mmol) in benzene (10 mL) was added NaBH(OAc)3 (376 mg, 1.77 mmol) and acetic acid (0.70 mL). After being stirred at room temperature under nitrogen for 6 h, the reaction was quenched with saturated aqueous NaHCO3 at 0 °C. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/1) to provide 23 (370 mg, 87%) as white amorphous solid. IR (film, cm−1) 3446, 2934, 1694, 1596, 1492, 1300, 1229, 1116, 1048; 1H NMR (500 MHz, CDCl3) δ 6.55 (s, 1H), 3.88 (s, 2H), 3.83 (s, 3H), 3.82 (s, 3H), 3.77 (t, J = 6.7 Hz, 2H), 3.00 (t, J = 6.8 Hz, 2H), 2.16 (s, 3H), 2.00 (t, J = 6.4 Hz, 2H), 1.72-1.67 (m, 2H), 1.64 (s, 3H), 1.49-1.46 (m, 2H), 1.12 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 207.8, 150.1, 146.5, 143.0, 131.1, 129.6, 128.8, 128.8, 113.1, 62.7, 60.5, 55.7, 50.9, 38.9, 33.5, 31.3, 30.7, 28.8, 21.2, 18.8, 15.7; HRMS (ESI): calculated for C22H32O4 [M+Li+] 367.2461, found 367.2469.
2-(3-Isopropyl-4,5-dimethoxyphenyl)-1-(2,6,6-trimethylcyclohex-1-en-1-yl)ethanone (25)
A mixture of naphthalene (4.92 g, 38.35 mmol) and lithium (266 mg, 38.35 mmol) in THF (40 mL) was stirred at room temperature for 1.5 h before it was treated with a solution of β-cyclocitral (1.28 g, 8.44 mmol) and benzyl chloride 24 (1.75 g, 7.67 mmol) in THF (10 mL) dropwise at 0 °C. The resulting mixture was stirred at room temperature for 2 h, diluted with ether and then treated with saturated aqueous NH4Cl. The organic phase was separated and the aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide allyl alcohol S2 (2.26 g, 85%) as light yellow liquid. 1H NMR (500 MHz, CDCl3) δ 6.70 (d, J = 1.9 Hz, 1H), 6.66 (d, J = 1.9 Hz, 1H), 4.42 (dd, J = 10.1, 3.6 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.34 (dt, J = 13.9, 6.9 Hz, 1H), 3.06 (dd, J = 13.9, 10.1 Hz, 1H), 2.82 (dd, J = 13.9, 3.5 Hz, 1H), 2.05-1.94 (m, 2H), 1.96 (s, 3H), 1.62-1.53 (m, 2H), 1.46-1.42 (m, 2H), 1.23 (s, 3H), 1.22 (s, 3H), 1.10 (s, 3H), 0.97 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 152.5, 144.9, 142.4, 138.9, 135.5, 131.8, 118.9, 110.7, 72.3, 60.9, 55.7, 43.4, 39.9, 34.8, 34.1, 28.6, 28.1, 26.8, 23.6, 23.5, 21.4, 19.3.
To a solution of S2 (370 mg, 1.07 mmol) in DMSO (4 mL) was added IBX (449 mg, 1.60 mmol) at room temperature. The resulting mixture was stirred at room temperature for 3 h before it was treated with H2O (5 mL) at 0 °C. The mixture was filtered and the aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration, the residue was purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide 25 (285 mg, 77%) as yellow amorphous solid. 1H NMR (500 MHz, CDCl3) δ 6.66 (d, J = 2.0 Hz, 1H), 6.64 (d, J = 2.0 Hz, 1H), 3.85 (s, 3H), 3.80 (s, 3H), 3.79 (s, 2H), 3.33 (dt, J = 13.9, 6.9 Hz, 1H), 1.99-1.96 (m, 2H), 1.69-1.67 (m, 2H), 1.56 (s, 3H), 1.47-1.44 (m, 2H), 1.20 (s, 3H), 1.19 (s, 3H), 1.08 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 152.3, 145.1, 143.1, 142.0, 129.7, 129.6, 119.7, 111.3, 60.9, 55.7, 52.2, 38.9, 33.4, 31.2, 28.8, 26.7, 23.5, 21.2, 18.8.
o-Hydroxy-p-quinone methide diterpenoid 3
To a solution of alcohol 23 (301 mg, 0.84 mmol) in dichloromethane (8 mL) was added a freshly solution of BBr3 (1 M in dichloromethane, 5.04 mL, 5.04 mmol) dropwise at −78 °C under nitrogen. The reaction was stirred for 30 min, allowed to 0 °C and stirred for another 2 h. The reaction was carefully quenched with saturated aqueous NaHCO3. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration in vacuum, the residue was taken into CHCl3 (10 mL) and treated with Ag2O (223 mg, 0.90 mmol). The mixture was stirred at 50 °C for 30 min and filtered through a cotton plug. The filtrate was concentrated and purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/1) to provide 3 (103 mg, 37% for 2 steps) as orange amorphous solid. IR (film, cm−1) 3325, 2925, 1670, 1610, 1377, 1223, 1185, 1146, 1045; 1H NMR (500 MHz, acetone-d6) δ 8.32 (s, 1H), 6.50 (s, 1H), 3.75 (t, J = 5.7 Hz, 1H), 3.63-3.59 (m, 2H), 3.05-3.02 (m, 1H), 2.85-2.81 (m, 2H), 2.65 (s, 1H), 2.34 (s, 3H), 1.76-1.70 (m, 2H), 1.59-1.54 (m, 1H), 1.40-1.36 (m, 1H), 1.29-1.26 (m, 1H), 1.27 (s, 3H), 1.24 (s, 3H), 1.10 (s, 3H); 1H NMR (500 MHz, acetone-d6+D2O) δ 6.52 (s, 1H), 3.59 (t, J = 7.1 Hz, 2H), 3.03 (d, J = 11.8 Hz, 1H), 2.83 (t, J = 7.2 Hz, 2H), 2.66 (s, 1H), 2.33 (s, 3H), 1.73-1.66 (m, 2H), 1.56-1.53 (m, 1H), 1.37 (d, J = 11.7 Hz, 1H), 1.26 (s, 3H), 1.23 (s, 3H), 1.10 (s, 3H); 13C NMR (125 MHz, acetone-d6) δ 201.1, 182.3, 146.4, 145.1, 142.0, 134.8, 131.3, 127.0, 62.3, 61.2, 43.0, 42.9, 37.7, 33.4, 33.3, 31.0, 22.2, 21.6, 19.3, 15.9; HRMS (ESI): calculated for C20H26O4 [M-H+] 329.1753, found 329.1738.
(4bS,8aS)-4-Hydroxy-2-(2-(methoxymethoxy)ethyl)-1,4b,8,8-tetramethyl-4b,5,6,7,8,8a-hexahydrophenanthrene-3,9-dione (33)
To a solution of alcohol 3 (9.9 mg, 0.03 mmol) in dichloromethane (2 mL) was added dimethyoxymethane (0.027 mL, 0.3 mmol) and P2O5 (43 mg, 0.15 mmol). The solution was stirred at room temperature for 2 h before it was diluted with dichloromethane, washed with saturated aqueous NaHCO3, and then dried over Na2SO4. After concentration, the residue was purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide 33 (7.4 mg, 66%) as yellow liquid. IR (film, cm−1) 3322, 2934, 1670, 1611, 1380, 1152, 1116, 1025; 1H NMR (500 MHz, CDCl3) δ 7.53 (s, 1H), 6.51 (s, 1H), 4.60 (s, 2H), 3.62 (t, J = 6.9 Hz, 2H), 3.33 (s, 3H), 2.98-2.95 (m, 1H), 2.93-2.88 (m, 2H), 2.59 (s, 1H), 2.29 (s, 3H), 1.74-1.70 (m, 2H), 1.62-1.59 (m, 2H), 1.44-1.40 (m, 1H), 1.28 (s, 3H), 1.26 (s, 3H), 1.13 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 201.2, 181.5, 146.1, 143.7, 141.1, 133.5, 131.1, 126.6, 96.3, 65.9, 62.2, 55.2, 42.6, 42.4, 37.1, 33.2, 32.7, 27.2, 21.9, 21.7, 18.6, 15.8; HRMS (ESI): calculated for C22H30O5 [M+Li+] 381.2248, found 381.2262.
(4bS,8aR)-4-Hydroxy-2-isopropyl-3-methoxy-4b,8,8-trimethyl-4b,5,6,7,8,8a-hexahydro phenanthren-9(10H)-one (30)
A mixture of hexamethyldisilizane (0.25 mL, 1.2 mmol) and iodine (305 mg, 1.2 mmol) was heated at 65 °C for 20 min under nitrogen. After cooling down to room temperature, a solution of ketone 25 (69 mg, 0.2 mmol) in toluene (1 mL) was added. The reaction mixture was stirred at 70 °C for 4 h before it was concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether, 1/5) to provide 30 (35.9 mg, 54 %) as yellow amorphous solid. IR (film, cm−1) 3414, 2955, 2931, 1694, 1448, 1418, 1300, 1229, 1057, 1033, 998; 1H NMR (500 MHz, CDCl3) δ 6.67 (s, 1H), 5.77 (s, 1H), 3.80 (s, 3H), 3.65 (d, J = 22.9, 1.1 Hz, 1H), 3.47 (d, J = 22.9 Hz, 1H), 3.27 (dt, J = 13.9, 6.9 Hz, 1H), 3.03-2.99 (m, 1H), 1.98 (s, 1H), 1.61-1.15 (m, 5H), 1.26 (d, J = 6.9 Hz, 3H), 1.24 (d, J = 6.9 Hz, 3H), 1.24 (s, 3H), 0.94 (s, 3H), 0.30 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 213.1, 145.8, 144.9, 133.7, 130.4, 126.4, 122.3, 68.5, 61.9, 44.0, 42.2, 40.1, 37.4, 34.4, 32.4, 31.9, 27.2, 22.8, 22.5, 22.4, 20.2; HRMS (ESI): calculated for C21H30O3 [M+H+] 331.2268, found 331.2264.
1-(Chloromethyl)-4,5-dimethoxy-2-methylbenzene (35)
To a solution of carboxylic acid 34 (1.0 g, 5.10 mmol) in THF (9 mL) was added a solution of borane dimethylsulfide complex (2 M in THF, 4.08 mL, 8.15 mmol) at 0 °C. The solution was allowed to room temperature and stirred overnight. The reaction was carefully quenched with H2O and the resulting mixture was diluted with EtOAc. The organic phase was separated, washed with H2O, and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/1) to provide the benzyl alcohol (836 mg, 90 %) as white amorphous solid.
The benzylic alcohol (1.82 g, 10 mmol) was taken into CH2Cl2 (20 mL) and treated with SOCl2 (0.87 mL, 12 mmol) dropwise at 0 °C under nitrogen. The mixture was stirred at room temperature for 30 min before it was carefully quenched with saturated aqueous NaHCO3. The organic phase was separated and the aqueous phase was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide 35 (1.90 g, 95 %) as yellow oil. IR (film, cm−1) 2963, 2934, 1611, 1519, 1466, 1338, 12821226, 1105, 998; 1H NMR (500 MHz, CDCl3) δ 6.84 (s, 1H), 6.71 (s, 1H), 4.60 (s, 2H), 3.89 (s, 6H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.1, 147.0, 129.8, 127.3, 113.7, 113.0, 56.0, 55.9, 45.2, 18.4; HRMS (ESI): calculated for C10H13O2Cl [M+H+] 201.0682, found 201.0692.
2-(4,5-Dimethoxy-2-methylphenyl)-1-(2,6,6-trimethylcyclohex-1-en-1-yl)ethanone (36)
A mixture of naphathalene (5.77 g, 45 mmol) and lithium (313 mg, 45 mmol) in THF (50 mL) was stirred at room temperature under nitrogen for 1.5 h. To this mixture was added a solution of β-cyclocitral (1.51 g, 9.9 mmol) and benzyl chloride 35 (1.80 g, 9 mmol) in THF (10 mL) via syringe at 0 °C. The mixture was then stirred at room temperature for 2 h before it was diluted with ether and treated with saturated aqueous NH4Cl. The organic phase was separated and the aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration, the residue was purified by flash column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/5) to provide allyl alcohol S3 (2.17 g,76%) as light yellow amorphous solid. IR (film, cm−1) 3538, 2934, 1608, 1513, 1466, 1270, 1226, 1102, 998; 1H NMR (500 MHz, CDCl3) δ 6.74 (s, 1H), 6.69 (s, 1H), 4.44 (dd, J = 10.2, 4.1 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.14 (dd, J = 14.2, 10.2 Hz, 1H), 2.79 (dd, J = 14.2, 4.1 Hz, 1H), 2.33 (s, 3H), 2.03-1.94 (m, 2H), 2.00 (s, 3H), 1.62-1.53 (m, 2H), 1.45-1.42 (m, 2H), 1.10 (s, 3H), 0.94 (s, 3H); 13C NMR (125 MHz, CDCl3) 147.3, 147.0, 139.3, 131.9, 129.6, 128.8, 113.8, 113.7, 71.4, 56.1, 55.9, 40.0, 39.5, 34.8, 34.2, 28.5, 28.3, 21.4, 19.6, 19.3; HRMS (ESI): calculated for C20H30O3 [M+Li+] 325.2355, found 325.2339.
A solution of S3 (960 mg, 3.03 mmol) in DMSO (6 mL) was treated with IBX (1.27 g, 4.55 mmol) and stirred at room temperature for 2 h. The reaction mixture was treated with H2O (6 mL) at 0 °C and filtered. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/3) to provide 36 (701 mg, 73%) as light yellow amorphous solid. IR (film, cm−1) 2931, 1700, 1516, 1460, 1270, 1229, 1108, 1039, 998; 1H NMR (500 MHz, CDCl3) δ 6.71 (s, 1H), 6.61 (s, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.82 (s, 2H), 2.23 (s, 3H), 1.99 (t, J = 6.4 Hz, 2H), 1.71-1.66 (m, 2H), 1.62 (s, 3H), 1.48-1.46 (m, 2H), 1.11 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 208.0, 147.8, 146.8, 143.0, 129.5, 129.4, 124.5, 113.7, 113.6, 56.0, 55.8, 49.7, 38.9, 33.4, 31.2, 28.8, 21.1, 19.6, 18.8; HRMS (ESI): calculated for C20H28O3 [M+H+] 317.2117, found 317.2126.
(4bS,8aS)-4-Hydroxy-1,4b,8,8-tetramethyl-4b,5,6,7,8,8a-hexahydrophenanthrene-3,9-dione (37)
To a solution of ketone 36 (95 mg, 0.30 mmol) in CH2Cl2 (3 mL) was added a freshly prepared solution of BBr3 (1 M in CH2Cl2, 1.80 mL, 1.80 mmol) dropwise at −78 °C under nitrogen. The reaction was stirred for 30 min, warmed to 0 °C, and further stirred for 3 h. The reaction was carefully quenched with saturated aqueous NaHCO3. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration in vacuo, the residue was taken into CHCl3 (10 mL) and treated with Ag2O (104 mg, 0.45 mmol). The mixture was stirred at 50 °C for 30 min and filtered through a plug of cotton. The filtrate was concentrated and purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/4) to provide 37 (35.9 mg, 42% for 2 steps) as orange amorphous solid. IR (film, cm−1); 3432, 2925, 1673, 1626, 1413, 1380, 1356, 1223, 1146, 773; 1H NMR; δ 7.42 (s, 1H), 6.43 (s, 1H), 6.42 (d, J = 1.2 Hz 1H), 2.97-2.94 (m, 1H), 2.59 (s, 1H), 2.26 (d, J = 1.3 Hz, 3H), 1.77-1.66 (m, 2H), 1.62-1.57 (m, 1H), 1.43-1.39 (m, 1H), 1.27 (s, 3H), 1.26 (s, 3H), 1.24-1.16 (m, 1H), 1.12 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 201.1, 181.6, 150.3, 144.5, 140.9, 131.3, 126.9, 126.4, 62.4, 42.9, 42.4, 37.1, 33.2, 32.8, 21.9, 21.7, 19.8, 18.6; HRMS (ESI): calculated for C18H22O3 [M-H+] 285.1496, found 285.1501.
Taxodione (2)
To a solution of ketone 25 (103 mg, 0.30 mmol) in CH2Cl2 (3 mL) was added a freshly prepared solution of BBr3 (1 M in CH2Cl2, 1.80 mL, 1.80 mmol) at −78 °C under nitrogen. The reaction was stirred for 30 min, warmed to 0 °C, and stirred for another 30 min. The reaction was carefully quenched with saturated aqueous NaHCO3. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. After concentration in vacuo, the residue was taken into CHCl3 (10 mL) and treated with Ag2O (104 mg, 0.45 mmol). The mixture was stirred at 50 °C for 30 min before it was filtered through a plug of cotton. The filtrate was concentrated and purified by column chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/4) to provide 2 (36.8 mg, 39% for 2 steps) as orange amorphous solid.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mr. Gaines Taylor (NSF REU student) of University of Maryland, Baltimore County for preparing some of the starting materials. Financial support was provided by the Robert A. Welch Foundation (A-1700 to JY), the National Science Foundation (CHE-1150606 to JY, CHE-1062840 to GT), the Norman Hackerman Advanced Research Program (grant 01957 to XL), and National Institute of Allergy and Infectious Diseases (AI097599 to XL).
Footnotes
SUPPORTING INFORMATION
1H and 13C NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCE
- 1.For a review, see: Van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367–5405.
- 2.For a review, see: Zhou Q. Natural Diterpene and Triterpene Quinone Methides: Structures, Synthesis and Biological Potentials. In: Rokita SE, editor. Quinone Methides. Wiley: Hoboken, NJ; 2009.
- 3.a) Fang L, Yang J, Yang F. Org. Lett. 2010;12:3124–3127. doi: 10.1021/ol1011423. [DOI] [PubMed] [Google Scholar]; b) Huang J, Yang J. Synlett. 2012;23:737–740. [Google Scholar]; c) Huang J, Yang JR, Zhang J, Yang J. J. Am. Chem. Soc. 2012;134:8806–8809. doi: 10.1021/ja303529z. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Huang J, Yang JR, Zhang J, Yang J. Org. Biomol. Chem. 2013;11:3212–3222. doi: 10.1039/c3ob40120k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Schaller F, Rahalison L, Islam N, Potterat O, Hstettmann K, Stoeckli-Ecans H, Mavi S. Helv. Chem. Acta. 2000;83:407–413. [Google Scholar]; b) Schaller F, Wolfender J-L, Hostettmann K, Mavi S. Helv. Chem. Acta. 2001;84:222–229. [Google Scholar]
- 5.For a review: Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH. Nat. Reviews Drug Discovery. 2010;9:719–727. doi: 10.1038/nrd3074.
- 6.For some examples: Mori K, Matsui M. Tetrahedron. 1970;26:3467–3473. doi: 10.1016/s0040-4020(01)92926-6. Mastumoto T, Tachibana Y, Uchida J, Fukui K. Bull. Chem. Soc. Jpn. 1971;44:2766–2770. Mastumoto T, Usui S, Morimoto TA. Bull. Chem. Soc. Jpn. 1977;50:1575–1579. Snitman DL, Himmelsbach RJ, Haltiwanger RC, Watt DS. Tetrahedron Lett. 1979:2477–2480. Johnson WS, Shenvi AB, Boots SG. Tetrahedron. 1982;38:1397–1404. Stevens RV, Bisacchi GS. J. Org. Chem. 1982;47:2396–2399. Burnell RH, Jean M, Poirier D. Can. J. Chem. 1987;65:775–781. Harring SR, Livinghouse T. Tetrahedron Lett. 1989;30:1499–1502. Engler TA, Sampath U, Naganathan S, Velde DV, Takusagawa F, Yohames D. J. Org. Chem. 1989;54:5712–5727. Harring S, Livinghouse T. J. Chem. Soc., Chem. Commun. 1992:502–503. Sanchez AJ, Konopelski JP. J. Org. Chem. 1994;59:5445–5452. Harring SR, Livinghouse T. Tetrahedron. 1994;50:9229–9254. Tada M, Kurabe J, Yoshida T, Ohkanda T, Matsumoto Y. Chem. Pharm. Bull. 2010;58:818–824. doi: 10.1248/cpb.58.818. and references cited therein.
- 7.Kim JY, Kim YH, Nam HT, Kim BT, Heo J-N. Org. Lett. 2008;10:3543–3546. doi: 10.1021/ol801291k. [DOI] [PubMed] [Google Scholar]
- 8.Littke AF, Schwarz L, Fu GC. J. Am. Chem. Soc. 2002;124:6343–6348. doi: 10.1021/ja020012f. [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran PV, Madhi S, O' Donnell MJ. J. Fluorine Chem. 2006;127:1252–1255. [Google Scholar]
- 10.a) Kim K, Kim I. Org. Lett. 2010;12:5314–5317. doi: 10.1021/ol102322g. [DOI] [PubMed] [Google Scholar]; b) Selvakumar J, Makriyannis A, Ramanathan CR. Org. Biomol. Chem. 2010;8:4056–4058. doi: 10.1039/c0ob00269k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For a review: Corey EJ. Angew. Chem. Int. Ed. 2009;48:2100–2117. doi: 10.1002/anie.200805374.
- 12.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. Sci. Transl. Med. 2012;4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 13.Ngamskulrungroj P, Price J, Sorrell T, Perfect JR, Meyer W. PLoS One. 2011;6:e16076. doi: 10.1371/journal.pone.0016076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhai B, Zhou H, Yang L, Zhang J, Jung K, Giam CZ, Xiang X, Lin X. J. Antimicrob. Chemother. 2010;65:931–938. doi: 10.1093/jac/dkq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, Ramage G, Denning DW, Bowyer P. J. Antimicor. Chemother. 2013;68:1487–1496. doi: 10.1093/jac/dkt075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.