

Summary
The angiotensin type 1 receptor (AT1R) transactivates the epidermal growth factor receptor (EGFR) to mediate cellular growth, however, the molecular mechanisms involved have not yet been resolved. To address this, we performed a functional siRNA screen of the human kinome in human mammary epithelial cells that demonstrate a robust AT1R–EGFR transactivation. We identified a suite of genes encoding proteins that both positively and negatively regulate AT1R–EGFR transactivation. Many candidates are components of EGFR signalling networks, whereas others, including TRIO, BMX and CHKA, have not been previously linked to EGFR transactivation. Individual knockdown of TRIO, BMX or CHKA attenuated tyrosine phosphorylation of the EGFR by angiotensin II stimulation, but this did not occur following direct stimulation of the EGFR with EGF, indicating that these proteins function between the activated AT1R and the EGFR. Further investigation of TRIO and CHKA revealed that their activity is likely to be required for AT1R–EGFR transactivation. CHKA also mediated EGFR transactivation in response to another G protein-coupled receptor (GPCR) ligand, thrombin, indicating a pervasive role for CHKA in GPCR–EGFR crosstalk. Our study reveals the power of unbiased, functional genomic screens to identify new signalling mediators important for tissue remodelling in cardiovascular disease and cancer.
Key words: Angiotensin; EGFR; G protein-coupled receptor; siRNA, T</emph>ransactivation
Introduction
Apart from its well-studied roles in vasoconstriction, aldosterone release, and fluid balance, angiotensin II (AngII), the principal effector of the renin–angiotensin system (RAS), influences a selection of homeostatic and modulatory processes that can also serve as targets for dysregulation and subsequent development of pathological states. These include cellular growth (hypertrophy) and remodelling (fibrosis); dyslipidaemia; endothelial dysfunction, aneurysms and angiogenesis (Iwai and Horiuchi, 2009; Lu et al., 2008; Mehta and Griendling, 2007; Weiss et al., 2001); pro-inflammatory responses (Rompe et al., 2010; Schiffrin, 2010); stem cell programming and haematopoiesis (Heringer-Walther et al., 2009; Park and Zambidis, 2009; Zambidis et al., 2008); neuromodulation including cognition, memory and dementia (Li et al., 2010; Miners et al., 2009); and cancer, as reviewed by us (George et al., 2010).
AngII acts primarily on the angiotensin type 1 receptor (AT1R), a G protein-coupled receptor (GPCR) encoded by a single gene (AGTR1) in humans and two homologous isoforms in rodents (AT1A and AT1B) (de Gasparo et al., 2000; Hunyady and Catt, 2006; Mehta and Griendling, 2007). Activated AT1Rs couple to the heterotrimeric G proteins Gq/11 (to stimulate phospholipase C β-mediated calcium mobilisation), Gi/o, G12/13 and Gs, as well as the monomeric G proteins (e.g. Rho, Ras and Rac) (de Gasparo et al., 2000; Guilluy et al., 2010). The activated AT1R also couples to soluble and receptor tyrosine kinases, the mitogen-activated protein kinases (MAPK; extracellular regulated kinases, ERK1/2, p38 MAPK and Jun N-terminal kinase), the JAK–STAT pathway, the generation of reactive oxygen species and various ion channels (de Gasparo et al., 2000; Hunyady and Catt, 2006; Mehta and Griendling, 2007). Signal termination is mediated by receptor phosphorylation and the recruitment and binding of arrestins (Qian et al., 2001), which terminate initial signalling, mediate receptor endocytosis and also act as scaffolds to support secondary and tertiary signalling complexes (Shenoy and Lefkowitz, 2011).
Daub and colleagues first reported that the epidermal growth factor receptor (EGFR) and the neu oncoprotein (ErbB2/HER2) are phosphorylated when stimulated with GPCR agonists endothelin 1, lysophosphatidic acid (LPA) and thrombin, which could be inhibited when cells were treated with EGFR antagonist AG1478, suggestive of a role for GPCRs in receptor tyrosine kinase signalling cross-talk (Daub et al., 1996). Since then, we and others have demonstrated that the AT1R can ‘hijack’ EGFR family signalling machinery and this is crucial for the downstream AT1R-mediated growth signalling and functional effects, such as cellular hypertrophy and/or proliferation (Asakura et al., 2002; Eguchi et al., 2001; Mifune et al., 2005; Thomas et al., 2002). Various mechanisms have been described to explain AT1R–EGFR transactivation, including metalloproteinase-mediated EGF ligand shedding (Mifune et al., 2005; Ohtsu et al., 2006), Ca2+-mediated Pyk2 activation (Eguchi et al., 1999), or direct interaction of the EGFR with the activated AT1R, through its tyrosine 319 residue (Seta and Sadoshima, 2003), although this does not appear to be the mechanism in a rat cardiomyocyte model (Smith et al., 2011).
To date, most studies have utilised a candidate approach to identify the proteins that are involved in AT1R-mediated EGFR transactivation in specific cell types. Although this has been informative, more often than not, this approach has failed to give the broader mechanistic picture of the underlying biology. Thus, the exact molecular mechanism linking the AT1R to the EGFR remains poorly defined and much detail is lacking. To address this issue, we generated a robust and tractable human cellular model of AT1R–EGFR transactivation, and used the model to perform functional siRNA screening to unbiasedly identify novel genes involved in the AT1R–EGFR transactivation process.
Results
Generation and characterisation of a stable cellular model of AT1R–EGFR transactivation
In order to examine the molecular mechanisms underlying AT1R–EGFR transactivation, a human cellular model was generated by introducing the AT1R into HMEC-LST cells (HMEC-LST-AT1R) using retroviral delivery of a vector that co-expresses mCherry. The AT1R-expressing cell population was enriched using fluorescence-activated cell sorting (FACS), and a heterogeneous population of cells expressing mCherry (Fig. 1A) was collected and expanded for further analysis. We confirmed expression of the ectopically expressed AT1R protein bearing an N-terminal HA epitope tag using western blot analysis (Fig. 1B). Immunofluorescence detection of the AT1R revealed localisation to the cell surface (Fig. 1C; supplementary material Fig. S1). Competition [125I]AngII radioligand binding assays revealed a Kd for AngII of 1.7 nM and a calculated receptor density of 1.87 pmol receptor/mg cellular protein (Fig. 1D). The AT1R-expressing cells demonstrated a robust, dose-dependent Ca2+ transient upon AngII stimulation, with an EC50 value of 24.8 nM, indicating appropriate coupling to Gq/11 (Fig. 1E).
Fig. 1.

Generation and characterisation of the AT1R–EGFR transactivation model. (A) We stably introduced an N-terminally HA-tagged angiotensin type I receptor (AT1R) into a human mammary epithelial cell line (HMEC-LST) (Elenbaas et al., 2001), and used mCherry expression to FACS enrich a heterogeneous population of cells expressing similar amounts of AT1R. (B) HA-tagged AT1R expression in HMEC-LST cells was determined by western blot analysis of cell lysate. (C) The cells transfected with the HA-tagged AT1R ectopically expressed the protein on the cell surface, but the control HMEC-LST-mCherry-transfected (mCherry) cells showed no such expression, as determined by immunofluorescence when stained with an anti-HA antibody (green) to detect the HA-tagged AT1R and DAPI (blue) to detect nuclei. Scale bars: 50 µm. (For individual confocal images and merged images in supplementary material Fig. S1.) (D) Cells expressing the AT1R readily bound [125I]AngII (EC50 = 1.7 nM) in a radioligand-binding assay; [125I]AngII was not observed to bind to the mCherry cells (n = 4 experiments). (E) When the HMEC-LST-AT1R cells were stimulated with AngII, intracellular Ca2+ was mobilised (EC50 = 24.8 nM); this did not occur</emph> in the mCherry cell line (n = 5 experiments).
Stimulation of the HMEC-LST-AT1R cells with 100 nM AngII led to the robust and reproducible phosphorylation of EGFR (pY1068) and ERK1/2 (p42/44; pERK1/2) (Fig. 2A). Quantification of western blot data revealed a 2-fold activation of EGFR above the level in unstimulated cells (Fig. 2B) and a 2.5-fold activation of ERK1/2 above the unstimulated cells upon AngII treatment (Fig. 2C). Maximal activation of EGFR and ERK1/2 occurred between 5 and 10 minutes (supplementary material Fig. S2), whereas activation of AKT (Ser473), which can also be activated downstream of EGFR and PI3K was not observed, revealing the MAP kinase pathway is a prominent pathway activated in response to AngII stimulation. As is well described, other pathways do exist for ERK1/2 activation from GPCRs (namely Ca2+, PKC and arrestins), therefore, to confirm that activation of the EGFR and downstream ERK1/2 by AngII stimulation was due to transactivation of the EGFR by the AT1R, HMEC-LST-AT1R cells were pretreated for 30 minutes with antagonists to EGFR (AG1478) or AT1R (candesartan cilexetil) prior to stimulation with AngII (Fig. 2D). Phosphorylation of EGFR and ERK1/2 was prevented when cells were pretreated with either inhibitor, demonstrating that activation of EGFR and ERK1/2 is due to the transactivation of the EGFR by ligand (AngII) activation of the AT1R. These findings were also confirmed when the AT1R was introduced into another human mammary epithelial cell line, HMEC-HMLE-AT1R (supplementary material Fig. S3).
Fig. 2.

AT1R–EGFR transactivation occurs in HMEC-LST-AT1R cells. (A) HMEC-LST cells (mCherry or AT1R transduced) were serum starved then stimulated with 100 nM AngII for 5-10 minutes. Robust activation of the EGFR (pY1068) and ERK1/2 was observed in the AT1R-transduced cells. (B,C) Quantification of western blot data using densitometry analysis (ImageJ) illustrates increases in phospho-EGFR (pY1068; B) and phospho-ERK1/2 (C) upon stimulation with AngII (n = 4 experiments; *P = 0.0137, **P = 0.0028, paired two-tailed Student's t-test). (D) HMEC-LST-AT1R cells were treated with 5 µM AG1478 (EGFR inhibitor) or 1 µM candesartan cilexetil (AT1R antagonist) for 30 minutes prior to stimulation with 100 nM AngII for 5-10 minutes. Analysis revealed that inhibition of either EGFR or AT1R activity blocks AngII-mediated AT1R–EGFR transactivation (n = 3 experiments; a representative western blot is shown).
AT1R–EGFR transactivation is inhibited by siRNAs targeting known transactivation components
We performed proof-of-concept experiments to demonstrate that the AT1R–EGFR transactivation response can be inhibited when cells were transfected with siRNAs that target genes involved in the process, specifically those of the AT1R, its cognate G protein (Gq/11) and the EGFR. We used siGENOME SMARTpools targeting Agtr1a to knock down expression of the rat AT1R by 80% (Fig. 3A) and this prevented phosphorylation of both EGFR and ERK1/2 upon AngII stimulation (Fig. 3B). Robust knockdown of Gq/11 (GNAQ), essential for AT1R-mediated cardiomyocyte hypertrophy (Smith et al., 2011) and the development of cardiac hypertrophy in mice (Wettschureck et al., 2001), also abolished EGFR and ERK1/2 phosphorylation following AngII stimulation (Fig. 3C,D). Similarly, knockdown of EGFR using SMARTpool siRNAs prevented EGFR and ERK1/2 phosphorylation upon AngII stimulation (Fig. 3E,F) as well as reducing the total EGFR protein, as expected. Together, these experiments validated this system as suitable for detecting novel genes involved AT1R–EGFR transactivation.
Fig. 3.
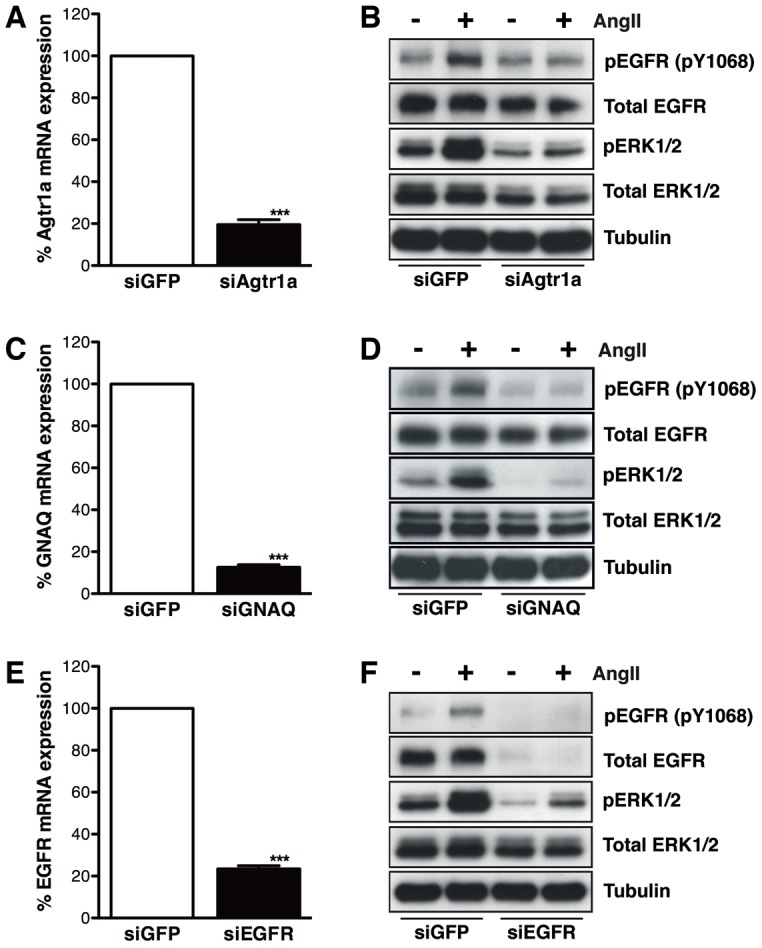
AT1R–EGFR transactivation is reduced when cells are transfected with siRNAs targeting AT1R, Gq/11 and EGFR expression. Cells were transfected with Dharmacon siGENOME SMARTpool siRNAs targeting the ectopically expressed AT1R (siAgtr1a), Gq/11 (GNAQ, siGNAQ) or EGFR (siEGFR) mRNA transcripts. (A,B) Cells transfected with siAgtr1a demonstrated mRNA knockdown of ∼80% at 24 hours (A), and the AT1R–EGFR transactivation at 72 hour knockdown was abolished (B) (n = 3 experiments; ***P = 0.0009, paired two-tailed Student's t-test). (C,D) Transfection with siGNAQ resulted in over an 85% knockdown of GNAQ mRNA at 24 hours (C), and a reduction in EGFR and ERK1/2 phosphorylation post-AngII stimulation (72 hour knockdown; D) (n = 3 experiments; ***P = 0.0002, paired two-tailed Student's t-test). (E,F) Similarly, when cells were transfected with siEGFR, ∼75% knockdown of EGFR mRNA was observed at 24 hours post-transfection (E) and the AT1R–EGFR transactivation response was blunted at 72 hours (F) (n = 3 experiments; ***P = 0.0004, paired two-tailed Student's t-test). Western blot is representative of all of the experiments carried out (n = 3).
Functional siRNA screening of the human kinome reveals candidates that modulate the AT1R–EGFR transactivation response
We used functional genomic screening to identify genes that modulate the AT1R–EGFR transactivation response. Using the HMEC-LST-AT1R cells, we optimised an AT1R–EGFR transactivation assay in microplate format for use in a siRNA screen. We determined that stimulating the HMEC-LST-AT1R cells for 10 minutes with 100 nM AngII in 96-well microplates using the AlphaScreen SureFire phospho-ERK1/2 assay (as a surrogate readout for AT1R–EGFR transactivation) gave a robust 1.5-fold activation of ERK1/2 above that of unstimulated cells (supplementary material Fig. S4). Furthermore, stimulation of cells with 100 ng/ml EGF for 10 minutes led to a 2.8-fold increase in ERK1/2 activation above stimulated, while pretreatment of cells with 5 µM AG1478 30 minutes prior to stimulation blocked AngII and EGF mediated ERK1/2 activation. This method was adapted to an siRNA screening format (supplementary material Fig. S5; Tables S1, S2 and S3).
We performed a primary siRNA screen using the human Dharmacon SMARTpool siRNA kinome library encompassing 720 kinase genes, outlined in Fig. 4A, because we hypothesised that this gene set was likely to encode products that modulate the signalling underlying AT1R–EGFR transactivation and were potentially druggable. Robust Z-score analysis of the data and ranking of each gene from the primary screen were performed (supplementary material Table S2). As expected, and also to validate our screening approach, EGFR present in the siRNA library and independent of the siRNA plate controls, was identified as being one of the top ‘hits’ in the screen.
Fig. 4.

Kinome siRNA screen to determine genes involved in AT1R–EGFR transactivation. (A) Using the HMEC-LST-AT1R cell line, we performed a primary siRNA screen using the Dharmacon siGENOME SMARTpool siRNA library targeting kinase genes (720 in total, 40 nM siRNA). (B) Phospho-ERK1/2 data (SureFire) was normalised to cellular confluency (IncuCyte) for each well, and robust Z-score analysis carried out (for individual gene data and descriptions, see supplementary material Tables S1–S3). Secondary siRNA screening analysis was performed on 50 high-ranking candidates identified from the primary screen using the Dharmacon siGENOME SMARTpool deconvoluted siRNA library (four individual duplexes comprising the SMARTpool; 25 nM siRNA). High (4/4 and 3/4), medium (2/4) and low (1/4, 0/4) confidence candidates were then identified. (B) STRING analysis was performed on the AT1R–EGFR transactivation secondary screen gene set to identify physical and functional associations of the genes in the list. Genes that remained as unconnected ‘nodes’ included FN3KRP, ASK, BMX, CDC2L2, DGKH, FRK, PSKH2, SNRK, STYK1 and TRIO.
We selected 50 highly ranking candidates from the primary screen for further target validation, and performed a secondary siRNA screen using the Dharmacon siGENOME deconvoluted SMARTpool siRNA library (four siRNA duplexes per gene, which comprised the original SMARTpool used in the primary screen). We took an unbiased approach in our candidate gene selection for secondary screening, selecting candidates with the 20 highest and 20 lowest robust Z-scores. In addition, we selected a further 10 highly ranked candidates for analysis based on literature that linked them with AT1R or EGFR signalling, or had demonstrated some association with at least one of the other highest ranked candidates identified by the primary screen.
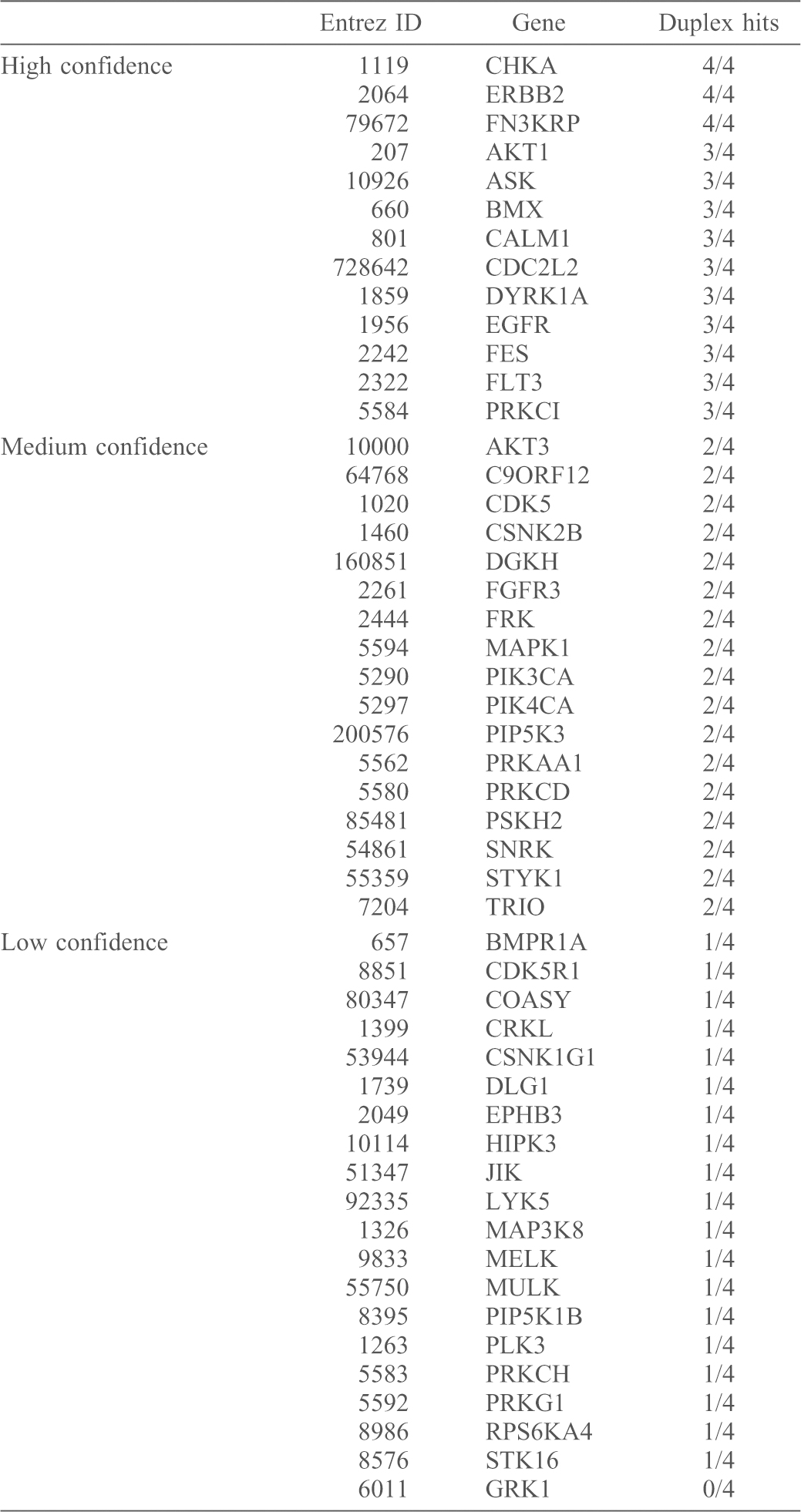
For our secondary screen analysis, we could not use robust Z-scoring to rank hits. This is because the method relies on the assumption that the majority of the siRNAs will not have a biological effect (Birmingham et al., 2009), whereas in the secondary screen, the expectation is that the majority of the siRNAs identified (in the primary screen) are positive hits. Hence, we calculated the average pERK1/2 value for the mock- and siGFP-transfected cells and normalised the pERK1/2 reading of each deconvoluted siRNA duplex to the average of the mock- and siGFP-transfected cells (outlined in supplementary material Table S1). We classified a duplex as being a ‘hit’ if the fold change was greater than 1.5 or less than −1.5 (equating to a 50% or more increase or decrease in AngII-mediated ERK1/2 activation compared to mock and siGFP-stimulated cells). The hit score was identified as the maximum number of hits in one particular direction (either positive or negative; the maximum hit score was 4, the minimum was 0). The duplexes that did not score within our cut-offs were deemed to be ‘unchanged’ and thus did not contribute to the ‘hit’ score. From there, we used hit scoring to identify high (4/4 and 3/4), medium (2/4) and low (1/4, 0/4) confidence targets for their role in AT1R–EGFR transactivation (Fig. 4A). The list of genes analysed in the secondary screen and validation score is given in Table 1.
Table 1. Genes assayed in the secondary siRNA screen and duplex scores.
The 50-gene list was further analysed using STRING 9.0 (http://string-db.org), an online database of known and predicted physical and functional protein interactions (Szklarczyk et al., 2011) to generate an AT1R–EGFR transactivation ‘interactome’, color-coded to represent the high, medium and low confidence scores for each candidate in the secondary screen analysis (Fig. 4B). Interestingly, we identified 36/50 candidates from the secondary screen that were either functionally or physically connected to at least one other candidate. Validated high and medium confidence targets (30 in total) were implicated in signalling pathways linked to EGFR–ERBB2, PI3K–AKT, MAPK and PKC signalling, which was predicted, as analysis of the literature revealed that these pathways have previously been implicated in EGFR transactivation</emph>.
We subsequently performed validation on a selection of genes from the high and medium hit list to determine (i) if the SMARTpool siRNA was on target, and (ii) how knockdown of the target gene affected transactivation of the EGFR (supplementary material Fig. S6A,B). We focused primarily on three novel genes encoding TRIO [triple functional domain (PTPRF interacting)], a Rho/Rac GEF protein containing a kinase domain; CHKA (choline kinase alpha) and BMX (BMX non-receptor tyrosine kinase), as these have not been previously implicated in GPCR-mediated EGFR transactivation.
TRIO, CHKA and BMX modulate AT1R–EGFR transactivation
We used SMARTpool siRNAs to knock down the expression of TRIO, CHKA and BMX (Fig. 5A,C,E, respectively) and to confirm their contribution to AngII-stimulated activation of EGFR and ERK1/2 (Fig. 5B,D,F). We observed a reduction in AngII-mediated EGFR and ERK1/2 activation with knockdown of TRIO, CHKA and BMX, and a modest reduction in basal ERK1/2 activity with knockdown of all three candidates. We also confirmed that individual siRNA duplexes knocked down the target mRNA transcripts, and assessed their impact on AT1R–EGFR transactivation by western blot analysis (supplementary material Fig. S7A–F).
Fig. 5.
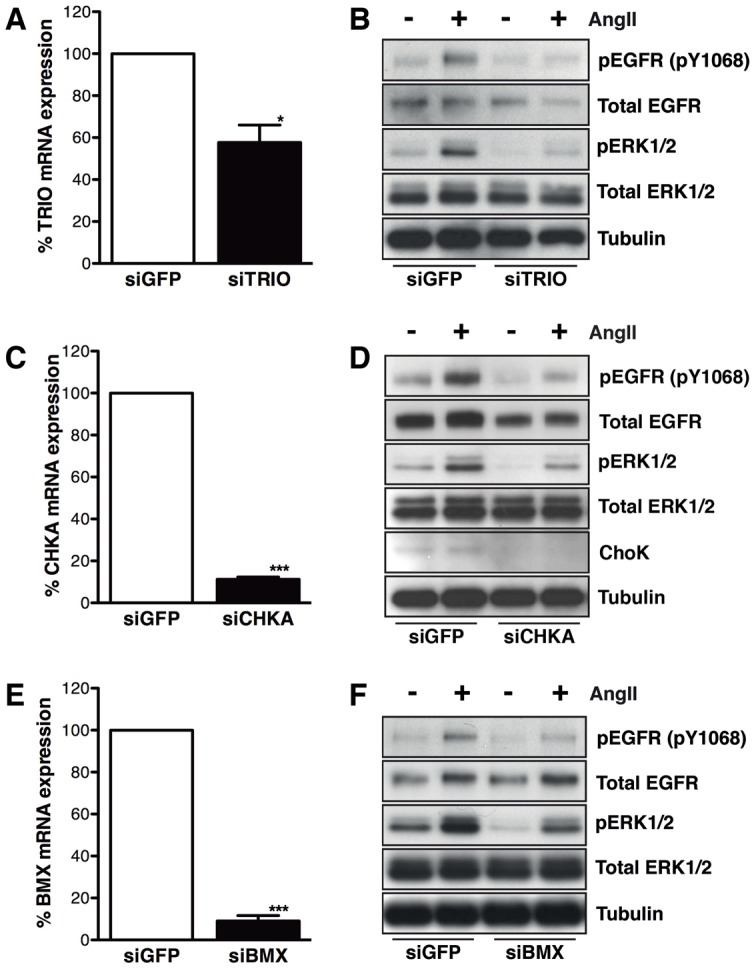
Further validation of TRIO, CHKA and BMX involvement in AT1R–EGFR transactivation. (A,B) HMEC-LST-AT1R cells were reverse transfected with 40 nM Dharmacon siGENOME SMARTpool siRNAs targeting TRIO, CHKA or BMX expression. HMEC-LST-AT1R cells transfected with TRIO siRNA (siTRIO) demonstrated an approximate 45% knockdown of mRNA transcript at 24 hours, by qRT-PCR (A) and at 72 hours post-transfection, knockdown of TRIO reduced AT1R–EGFR transactivation upon stimulation with AngII (B). (C,D) When cells were transfected with CHKA siRNA (siCHKA), an approximate 90% knockdown of CHKA transcript was observed at 24 hours (C), coupled with a reduction in ChoK (CHKA) protein expression and AT1R–EGFR transactivation at 72 hours (D). (E,F) Similarly, cells transfected with BMX siRNA (siBMX) demonstrated an approximate 90% knockdown of mRNA transcript at 24 hours (E), and a reduction in AT1R–EGFR transactivation (as determined by phospho-EGFR and phospho-ERK1/2 abundance) was observed after 72-hour siRNA knockdown (F). n = 3 or 4 experiments for both mRNA and protein analysis; for mRNA analysis *P = 0.0144 (TRIO), ***P = 0.0002 (CHKA) and P = 0.0008 (BMX), paired two-tailed Student's t-test. Western blots are representative of all experiments.
TRIO, CHKA and BMX function between the AT1R and the EGFR
Our data are consistent with TRIO, CHKA and BMX acting downstream of the AT1R, but upstream of the EGFR. To test this, we reasoned that the phosphorylation of the EGFR and ERK1/2, by direct stimulation with EGF ligands to bypass the AT1R (see Fig. 6A), should not be affected by the knockdown of TRIO, CHKA and BMX if they function downstream of the AT1R, but upstream of the EGFR. We therefore knocked down TRIO, CHKA and BMX using SMARTpool siRNAs in HMEC-LST-AT1R cells and stimulated with either 100 nM AngII or 0.5 ng/ml EGF for 10 minutes (Fig. 6B,C,D; supplementary material Fig. S8A–C). Knockdown of TRIO, CHKA or BMX in HMEC-LST-AT1R cells did not prevent activation of EGFR and ERK1/2 by direct stimulation with EGF, indicating that they are likely to sit mechanistically between the AT1R and EGFR in transactivation. Furthermore, our analysis revealed that pre-treatment of cells with 5 µM AG1478 prevented AngII-mediated ERK1/2 activation following TRIO, CHKA and BMX knockdown, implying that the activation of ERK1/2 is primarily EGFR dependent, and that candidate knockdown combined with AG1478 treatment may also eliminate EGFR-independent and -dependent signalling, respectively (supplementary material Fig. S9A–C). Moreover, TRIO, CHKA and BMX knockdown did not have a dramatic impact on Ca2+ mobilisation upon AngII stimulation (supplementary material Fig. S10A,B) suggesting that the mechanism of action of these candidates is unlikely to be through changes in AngII-mediated intracellular Ca2+ levels.
Fig. 6.

The mechanism of action for TRIO, CHKA and BMX lies between the AT1R and EGFR. (A) Our hypothesis is that TRIO, CHKA and BMX lie between the activated AT1R and EGFR and thus modulate the AT1R–EGFR transactivation response. A proposed model for this theory is illustrated. (B) HMEC-LST-AT1R cells were transfected with siGFP, siTRIO, siCHKA or siBMX SMARTpool siRNAs for 72 hours and stimulated with either AngII (100 nM) or EGF ligand (0.5 ng/ml) for 10 minutes. Knockdown of TRIO (B), BMX (C) or CHKA (D) did not alter the response of the cells to 0.5 ng/ml EGF ligand. Data are representative of three independent experiments.
Mechanistic insights into the function of TRIO, CHKA and BMX in GPCR-mediated EGFR transactivation
We further investigated the mechanism(s) by which the three lead candidates identified in our siRNA screen act to modulate AT1R–EGFR transactivation. TRIO contains two guanine nucleotide exchange factor domains that can activate RhoA and Rac1. We took a genetic approach to determine whether RAC1 or RHOA, downstream of TRIO, modulated the AT1R–EGFR transactivation response. We achieved >90% knockdown of RAC1 and RHOA mRNA transcripts at 24 hours using SMARTpool siRNAs (Fig. 7A). Furthermore, we observed a reproducible, robust reduction in EGFR and ERK1/2 activation with RAC1 knockdown (Fig. 7B), although RAC1 knockdown also had a modest effect on total EGFR expression. RHOA knockdown lead to a reduction in EGFR activation, but only a minimal reduction in AngII-mediated ERK1/2 activation (Fig. 7C). To further elucidate whether RAC1 is involved in AT1R–EGFR transactivation, we pre-treated the HMEC-LST-AT1R cells for 30 minutes with 100 µM NSC-23766 (Fig. 7D), which lead to a reduction in AngII-mediated EGFR and ERK1/2 activation. We also examined the requirement of CHKA activity for AngII-mediated EGFR transactivation. Pretreatment of cells with 10 µM CK37, an inhibitor of CHKA activity, for 30 minutes prior to AngII stimulation, also led to a reduction in EGFR and ERK1/2 activation, with no observable effect on total CHKA (ChoK) protein expression (Fig. 7E).
Fig. 7.

Elucidating the function of TRIO, CHKA and BMX in GPCR-mediated EGFR transactivation. RHOA and RAC1, downstream of TRIO, were knocked down in HMEC-LST-AT1R cells to assess their role in AT1R–EGFR transactivation. (A) Greater than 90% knockdown of the RAC1 and RHOA mRNA transcripts were observed at 24 hours. (B,C) At 72 hours post-transfection, knockdown of RAC1 reduced EGFR and ERK1/2 activity upon AngII stimulation, and led to a small reduction in total EGFR protein expression (B), whereas RHOA knockdown affected EGFR but not ERK1/2 activity (C). (D,E) Pre-treatment of cells with NSC-23766 (D) or CK37 (E) revealed a blunting in AT1R–EGFR transactivation upon AngII stimulation. (F) To identify whether TRIO, CHKA and BMX were specific to AT1R–EGFR transactivation, we stimulated the parental HMEC-LST cell line (not containing the ectopically expressed AT1R) with 10 nM thrombin for 10 minutes in the presence or absence of 5 µM AG1478 and evaluated EGFR and ERK1/2 phosphorylation. (G) Knockdown of BMX and TRIO, but not CHKA in the HMEC-LST cells revealed little change in EGFR and ERK1/2 activation post thrombin stimulation (ERK1/2 activation data for three independent experiments in which CHKA was knocked down is given in supplementary material Fig. S11C). n = 3 or 4 experiments for both mRNA and protein analysis; for mRNA analysis **P = 0.0014 (RHOA), ***P = 0.0009 (RAC1), paired two-tailed Student's t-test. Western blots are representative of all experiments performed.
We next tested the parental HMEC-LST cell line with a selection of GPCR ligands (endothelin-1, thrombin and 17-β-oestradiol) to determine whether or not we could achieve GPCR-mediated EGFR transactivation with endogenous GPCRs, as a prelude to determining whether our identified screening candidates generically modulate GPCR–EGFR transactivation (supplementary material Fig. S11A). Only thrombin was able to elicit a transactivation response. We next took advantage of our observation that thrombin also promoted robust ERK1/2 signalling through EGFR transactivation in the parental HMEC-LST cell line to determine whether TRIO, CHKA and BMX knockdown modulates GPCR–EGFR transactivation (Fig. 7F). HMEC-LST cells stimulated with 10 nM thrombin for 10 minutes robustly activated the EGFR and ERK1/2, which was blocked when the cells were pretreated for 30 minutes with 5 µM AG1478. We next tested whether the knockdown of TRIO, CHKA or BMX affected thrombin-mediated EGFR transactivation (Fig. 7G; supplementary material Fig. S11B). Thrombin-mediated EGFR and ERK1/2 phosphorylation was reduced with CHKA knockdown, but remained unchanged when TRIO and BMX were silenced, indicating that CHKA may be required for the transactivation of EGFR by GPCR receptors other than AT1R, whereas TRIO and BMX are more selective and may be specific to AT1R-mediated EGFR transactivation. These data provide evidence that both common and distinct mechanisms control GPCR–EGFR transactivation.
Discussion
The development of siRNA platform technologies to permit genome-wide, loss-of-function screening has provided an unprecedented resource for delineating molecular and cellular processes, including, for example, cell migration (Simpson et al., 2008), necrotic cell death (Hitomi et al., 2008) and cell division (Kittler et al., 2007). In the present study, we used RNAi-based screening to unbiasedly identify kinase genes involved in AngII-mediated activation of ERK1/2, specifically those related to AT1R–EGFR transactivation. We developed and characterised a human cellular model of AT1R–EGFR transactivation, then performed a primary kinome screen, comprising SMARTpool siRNAs targeting 720 kinase genes, and a subsequent secondary validation screen of 50 of the highest-ranking candidates using deconvoluted siRNAs. We identified many candidates that modulate the AT1R–EGFR transactivation response; these included molecules clearly related to the EGFR–PKC–PI3K–MAPK signalling axis, as well as novel genes, such as those of TRIO, BMX and CHKA, which appear to act mechanistically between the AT1R and the EGFR. CHKA is likely to be a general mediator of GPCR-induced EGFR transactivation, whereas TRIO and BMX might act more specifically in this process.
The capacity of EGFRs (and other growth factor receptors) to act as important conduits for multiple GPCR-related stimuli that modulate cell growth and tissue remodelling is generally accepted. Many studies, including our own, have linked the activation of the AT1R to the phosphorylation and transactivation of the EGFR in cardiac, renal and vascular tissues (Eguchi et al., 2001; Eguchi et al., 1998; Thomas et al., 2002; Touyz et al., 2002; Uchiyama-Tanaka et al., 2001). In support of the “triple membrane passing signalling paradigm” proposed by Ullrich and colleagues (Daub et al., 1996), some evidence exists for matrix metalloprotease (MMP) and a disintegrin and metalloprotease (ADAM) shedding of EGF ligands as a mechanism for AT1R-mediated EGFR transactivation and tissue remodelling. In the kidney, ADAM17 shedding of TGFα has been associated with renal disease (Shah and Catt, 2006); in heart, ADAM12 shedding of heparin-binding EGF-like growth factor (HB-EGF) reportedly mediates cardiac hypertrophy (Asakura et al., 2002); and ADAM17 inhibition reduces AngII-mediated EGFR phosphorylation and the proliferation of vascular smooth muscle cells (Ohtsu et al., 2006). In contrast, others have suggested alternative mechanisms for EGFR transactivation, such as through intracellular kinases, including Pyk2 and Src, or subsequent to direct interaction between the AT1R and the EGFR (Eguchi et al., 1999; Seta and Sadoshima, 2003; Touyz et al., 2002). So, while many would accept the authenticity and importance of EGFR transactivation by GPCRs, such as the AT1R, the mechanisms remain poorly understood. It was this lack of clarity that compelled us to undertake and unbiased approach to identify novel components involved in transactivation EGFR by AT1R.
To permit a functional siRNA screen, we developed a cellular model of AT1R–EGFR transactivation that met our selection criteria: (1) the cells were of human origin and readily expandable to generate large cell numbers required for screening; (2) they could be efficiently transfected with siRNAs; and (3) they robustly transactivated the EGFR upon stimulation with AngII. Primary cell models of cardiomyocytes, vascular and renal cells used in previous studies to examine AT1R–EGFR transactivation were not amenable to siRNA screening. Therefore, we initially tested 14 human vascular, endothelial and epithelial cell lines for their ability to transactivate the EGFR with either ectopically or endogenously expressed AT1R, under the above criteria. Although several of these cell lines demonstrated AT1R-mediated EGFR transactivation, we ultimately selected an immortalised human mammary epithelial cell line, stably expressing the AT1R (HMEC-LST-AT1R). This cell line displayed high affinity and functional AT1R expression on the cell surface and exhibited appropriate AT1R pharmacology and Gq/11-mediated signalling. Importantly, these cells robustly activated the EGFR and ERK1/2 in response to AngII stimulation and showed efficient siRNA knockdown of known targets, making it suitable for our screen. Additionally, these cells were selected for their functional relevance, where AT1R overexpression has been implicated in breast cancer pathogenesis (De Paepe et al., 2001; Rhodes et al., 2009; Tahmasebi et al., 2006). Furthermore, AT1R–EGFR transactivation could be modulated by siRNA knockdown of either the AT1R or EGFR as well as by targeting Gq/11, an absolute requirement for AT1R–EGFR transactivation in other cell types, including cultured cardiomyocytes (Smith et al., 2011) and vascular smooth muscle cells (VSMC) (Mifune et al., 2005; Ohtsu et al., 2008).
Using our cellular model, we adapted, optimised and developed an AT1R–EGFR transactivation assay in microplate format using the AlphaScreen SureFire phospho-ERK1/2 kit as our readout. We screened using the Dharmacon SMARTpool siRNA kinome library, as we reasoned that kinases were likely to be important for AT1R–EGFR transactivation and any hits would be likely to be druggable in subsequent studies on function. We ranked the candidates from the primary kinome screen on the basis of robust Z-score, and pursued 50 highly ranked candidates for a secondary screen, where we classified ‘hits’ as those with more than a 1.5-fold change (in either direction) compared with the mock- and siGFP-transfected cells. The result was a list of high, medium and low confidence candidates, a number of which we pursued using a candidate-type approach to further validate their role in AT1R–EGFR transactivation.
One of the major outcomes of our screen was the finding that many of the candidates related to the EFGR–ErbB2–PKC–PI3K–MAPK signalling axis. Although in a way expected, this observation provided important confidence and validation that the screen was sufficiently powerful to interrogate AT1R–EGFR transactivation. The finding that ErbB2 (the common dimerising partner for ErbB receptors) modulates the AT1R–EGFR transactivation response in our mammary epithelial cell model is consistent with previous studies suggesting that ErbB2 is required for AngII-mediated EGFR transactivation (Chan et al., 2006; Negro et al., 2006). It also corroborates data from other GPCRs, for example, the thrombin-dependent PAR1 activation in MDA-MB-231 breast cancer cells that transactivates EGFR– ErbB2 and increases cell invasiveness (Arora et al., 2008). In addition to ErbB2, we also identified and validated a number of PKC isoforms (PKC-δ and PKC-ι) as modulators of AT1R–EGFR transactivation, consistent with previous reports of AT1R–EGFR transactivation and PKC-δ translocation in response to AngII in primary breast cancer cells (Greco et al., 2003), the PKC-δ/Pyk2/Src-dependent AT1R–EGFR transactivation observed in hepatic C9 cells (Shah and Catt, 2002) and thromboxane A2 receptor-induced EGFR transactivation, involving Gq/11-mediated PKC-δ and PKC-ε activation (Uchiyama et al., 2009). Moreover, it has not escaped our attention that a number of hits from our screen are proteins that contain pleckstrin homology (PH) domains (including AKT1, AKT3, TRIO, BMX and DGKH), which, along with PI3K and the PKC isoforms, suggests that phosphoinositide biosynthesis, localisation, compartmentalisation and/or signalling are critical factors for AT1R–EGFR transactivation. Interestingly, kinases previously associated with both AT1R and GPCR-mediated EGFR transactivation, including Pyk2 and Src (Andreev et al., 2001; Eguchi et al., 1999; Shah and Catt, 2002) were not strong hits identified by our screen, which could suggest, at least in our breast epithelial cell model, that other signalling candidates may play a more prominent role in AT1R–EGFR transactivation.
When considering potential hits, our primary interest was in candidates that had not been previously (or at least ostensibly) associated with ErbB function. We were also seeking hits where a demonstrable effect on EGFR–ERK activation was direct and not secondary to global effects on cell viability or ‘non-specific’ alterations in total EGFR abundance, both of which might result in apparent reduction in AngII-induced ERK1/2. With this in mind, we assayed a number of the high/medium confidence siRNA screening hits (14 in total) to quantify mRNA knockdown by qRT-PCR and assess their performance in AT1R–EGFR transactivation. For most of these candidates, we confirmed siRNA-mediated knockdown and observed a reduced ERK1/2 activation following AngII stimulation. However, we noted for some candidates, a large, presumably non-specific effect on total EGFR expression (e.g. CDC2L2), or alternatively, little apparent effect on the phosphorylation of the EGFR (e.g. DYRK1A, STYK1), indicating the effect on ERK signalling was probably independent of EGFR transactivation. We dismissed such candidates in favour of others with clear and robust effects on AngII-induced EGFR–ERK1/2 activation, without obvious changes in total ERK–EGFR protein expression. We focused on three leading, proof-of-principle candidates that have not previously been implicated in GPCR-EGFR transactivation, TRIO, CHKA and BMX (a member of the Tec non-receptor tyrosine kinase family). Knockdown of these candidates selectively and strongly prevented AngII-mediated, but not EGF-mediated activation of the EGFR, locating their site of action between the AT1R and the initiation of EGFR activation.
TRIO (also known as UNC-73) is a particularly interesting target with respect to potential AT1R, Gq/11 or EGFR activity. The human full-length cDNA encodes a 2861 amino acid protein, containing a serine/threonine kinase domain and two functional guanine nucleotide exchange factors (GEF) domains, one specific for GDP–GTP exchange on RhoA and the other for Rac1 (Debant et al., 1996). Rho/Rac activity has not, to our knowledge, been directly related to activation of the EGFR, but their capacity to engage downstream kinases (e.g. ROCK) and affect cell growth and function is well described. Although few publications link TRIO activity to AngII stimulation, AngII does productively engage other RhoA GEFs, such as Arhgef1, with important roles in vascular tone and hypertension (Guilluy et al., 2010). Importantly, the TRIO homologue, UNC-73, was revealed in a forward genetic screen in Caenorhabditis elegans as a major mediator of Gq signalling, growth, reproduction and locomotion in worms (Williams et al., 2007). Mechanistically, Gq/11 has been identified to activate the C-terminal Rho-specific DH-PH domain of TRIO, and of the closely related protein, p63RhoGEF (Rojas et al., 2007). Moreover, the crystal structure of activated Gq/11 and p63RhoGEF has been solved, demonstrating that direct binding of activated Gq/11 to p63RhoGEF could specifically induce Rho signalling independently of and in competition to PLCβ activation (Lutz et al., 2005). Intriguingly, a recent study that utilised a genome-wide RNAi screen in Drosophila cells to identify regulators of AP-1 transcription factor complex activation (downstream of Gq) found that TRIO activation is a requirement for mitogenic signalling mediated by Gq, and is part of a ‘hard-wired’ protein–protein-interaction based signalling circuitry that is required for sustained cellular growth signalling and a regulator of normal and aberrant cellular growth (Vaqué et al., 2013). Together with our data, these studies provide robust evidence that TRIO mediates GPCR activation of downstream transcriptional events, in part by EGFR transactivation.
Interestingly, in our HMEC-LST-AT1R cell line, RAC1 knockdown and the NSC-23766 compound (downstream of TRIO) were both able to blunt the AT1R–EGFR transactivation response. An inhibitor of Rac1 binding and activation by the Rac1-specific GEF domain of TRIO (and Tiam1), NSC-23766 is suggested to have no demonstrable effect on RhoA or Cdc42 (Gao et al., 2004). This data suggest that AngII, through AT1R/Gq/11, can enhance TRIO Rac-GEF activity that contributes to activation of signalling pathways and/or cytoskeletal remodelling that is permissive for AT1R–EGFR transactivation.
BMX is a member of the Tec family of non-receptor tyrosine kinases (Tamagnone et al., 1994) and contains Tec homology (TH), PH, SH2, SH3 and kinase domains. BMX is expressed in a variety of human cell types, including bone marrow, haematopoietic and endothelial cells (Tamagnone et al., 1994), and in the endocardium and large arteries of the mouse (Ekman et al., 1997). BMX expression is altered in a number of different cancers, including those of the bladder and prostate (Dai et al., 2006; Guo et al., 2011; Jiang et al., 2007). Tec family kinases have been directly implicated in GPCR signalling, where Gq and/or βγ subunits of the heterotrimeric G protein complex can bind to and activate the kinase (Bence et al., 1997; Langhans-Rajasekaran et al., 1995; Tsukada et al., 1994). BMX, through its PH domain, binds to the thrombin-activated GPCR, PAR1, and this is a requirement for association with Shc and oncogenic activity (Cohen et al., 2010). Together, this evidence suggests that Tec family kinases can modulate GPCR function by acting as signalling scaffolds that allow recruitment of other binding partners and effector proteins, or as second messengers that modulate downstream signalling activities. Indeed, with respect to our observation that AT1R–EGFR transactivation leads to hypertrophic growth in rat cardiomyocytes (Thomas et al., 2002), it is interesting that Bmx knockout mice display reduced cardiac hypertrophy in a model of transverse aortic constriction, suggesting that Bmx is required for the morphological responses to pressure overload in the heart (Mitchell-Jordan et al., 2008). Combined with our finding that BMX knockdown prevents AT1R–EGFR transactivation, this evidence suggests a novel and plausible signalling mechanism underpinning AT1R–EGFR cardiomyocyte hypertrophy, which warrants further investigation.
Choline kinase alpha (CHKA, ChoK) phosphorylates choline to produce phosphocholine (PCho), an important intermediate in the generation of the key membrane phospholipid, phosphatidylcholine (Aoyama et al., 2004). Homozygous knockout of CHKA in mice results in embryonic lethality at the blastocyst stage (Wu et al., 2008), whereas upregulation of CHKA activity, or increased abundance of choline/PCho is commonly observed in cancer (Glunde et al., 2008; Hernando et al., 2009; Miyake and Parsons, 2012). CHKA is required for growth-factor-induced cellular proliferation in primary human mammary epithelial cells (Ramírez de Molina et al., 2004). Inhibition of CHKA in HeLa cells reduces the steady-state levels of phosphatidylcholine and phosphatidic acid, affecting both PI3K and MAPK signalling (Yalcin et al., 2010). Recent evidence suggests that in breast cancer and immortalised mammary epithelial cells, EGFR and c-Src can synergise to regulate CHKA protein expression and activity (Miyake and Parsons, 2012). Furthermore, the same study demonstrated that EGFR and CHKA form a complex in the presence of c-Src, which is required for maximal EGF-dependent cellular growth. Attempts to determine whether CHKA binds to either the HA-tagged AT1R or the EGFR upon AngII stimulation using co-immunoprecipitation were inconclusive (data not shown).
We demonstrate that inhibition of CHKA with CK37, a competitive inhibitor of CHKA activity also blunts the AT1R–EGFR transactivation response. This is particularly interesting because CK37 has been shown to inhibit MAPK and PI3K–AKT signalling, disrupt actin cytoskeleton organisation and reduce membrane ruffling in transformed cells (Clem et al., 2011). Although no published reports link AngII and/or AT1R to CHKA, its role in lipid biosynthesis and its binding to the EGFR and regulation of Src-mediated growth signalling are provocative, especially considering that its knockdown also impeded thrombin-mediated EGFR transactivation, indicating that it affects GPCR–EGFR transactivation more generally than just the AT1R. Furthermore, it may suggest that spatiotemporal changes within a cell upon stimulation with GPCR ligands play an essential role in GPCR-mediated EGFR transactivation, though this will require further investigation.
In conclusion, the present study screened 720 kinase genes to identify critical signalling molecules controlling AT1R–EGFR transactivation that were potentially druggable, which would facilitate subsequent studies to test their importance in pathophysiological states associated with dysregulated AT1R signalling. Using this approach, we have uncovered a suite of new signalling targets involved in AT1R–EGFR transactivation and have also confirmed that the EFGR–ErbB2–PKC–PI3K–MAPK signalling axis is important in AT1R–EGFR transactivation. In particular, this work provides the platform for investigating the molecular basis for TRIO, BMX and CHKA action in AT1R–EGFR transactivation and provides proof that genome-wide approaches can offer powerful new insights into this process and permit the assembly of the AT1R–EGFR transactivation ‘interactome’, which is increasingly appreciated as an important mediator of cell and tissue function.
Materials and Methods
Cell lines and culturing
Human mammary epithelial cells immortalised with human telomerase (hTERT) and transformed with SV40 Large T and small t antigens (HMEC-LST, HMEC-HMLE) were a gift from Professor Robert Weinberg (Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology). HEK293T cells were obtained from Open Biosystems (Thermo Fisher Scientific, Scoresby, Australia). Unless otherwise specified, media and tissue culture consumables were obtained from Gibco BRL (Life Technologies, Mulgrave, Australia). HMEC-LST and HMEC-HMLE cells were maintained in HuMEC ready medium containing HuMEC growth supplements and bovine pituitary extract (no. 12752-010). For serum starvation experiments, cells were washed with Dulbecco's phosphate-buffered saline (PBS, pH 7.4) and serum starved with HuMEC basal medium (no. 12753-018). Unless otherwise indicated, HuMEC media (ready or basal) were supplemented with 50 µg/ml gentamicin. HEK293T cells were grown in DMEM with 20 mM HEPES, 10% FBS and antibiotic-antimycotic solution. HMEC-LST, HMEC-HMLE and HEK293T cell lines were maintained in vented flasks (Becton Dickinson, North Ryde, Australia) in a humidified incubator at 37°C with 5% CO2. Unless otherwise stated, incubation steps for cell culture assays were performed at 37°C with 5% CO2. Cell number was determined using a Z2 Cell and Particle Counter (Beckman Coulter, Lane Cove, Australia). Specific details for passaging the HMEC cell lines are given in supplementary material Table S1.
Retroviral construct generation
The cloning of pRc/CMV/NHA-AT1A, a vector encoding the N-terminally HA-epitope-tagged rat angiotensin type Ia receptor cDNA has been described previously (Thomas et al., 1998). The pRc/CMV/NHA-AT1A plasmid was digested with HindIII and the NHA-AT1A cDNA was subcloned into the HindIII site of the pBluescript KS− plasmid (Stratagene, Agilent Technologies, Mulgrave, Australia). The pBluescript KS-NHA-AT1A plasmid was digested with BamHI and XhoI to liberate the HindIII-flanked NHA-AT1A cDNA, which was inserted into the MSCV-IRES-mCherry vector (a gift from Dr Sarah Russell, Peter MacCallum Cancer Centre, Melbourne, Australia) to generate the MSCV-IRES-NHA-AT1A (AT1R) plasmid.
Generation of stable cell lines
HEK293T cells were co-transfected with MSCV-IRES-mCherry (mCherry, vector control) or receptor-containing MSCV-IRES-NHA-AT1A (AT1R) plasmids in the presence of an amphotrophic packaging vector (Dr Phillip Darcy, Peter MacCallum Cancer Centre, Melbourne, Australia) at a 2∶1 ratio. Viral supernatant was filtered through a 0.45 µm Minisart filter (Sartorius Stedim, Dandenong South, Australia) and Sequabrene (Sigma-Aldrich, Castle Hill, Australia) was added at a final concentration of 4 µg/ml before addition to target cells. Cells were exposed to fresh viral supernatant three times over 24 hours, grown to ∼80% confluency and passaged twice prior to fluorescence-activated cell sorting (FACS) using the FACS Vantage SE Diva (BD Biosciences).
Antibodies, ligands and inhibitors
Angiotensin II (human; no. 2078) and endothelin-1 (no. 2110) was purchased from Auspep (Tullamarine, Australia), and human EGF (no. 100-15) was purchased from Peprotech (Rocky Hill, NJ, USA). Thrombin (human plasma, high activity, no. 605195), CK37 (no. 229103) and AG1478 (no. 658552) were obtained from Calbiochem (Merck Millipore, Kilsyth, Australia). NSC-23766 (no. 2161) was obtained from Tocris Bioscience (Abacus ALS, East Brisbane, Australia). Total EGFR (sc-03), total ERK 1 (sc-93), ChoK (sc-376489) and RhoA (sc-418) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Rac1 antibody (05-389) was purchased from Merck Millipore. Phospho-p44/42 MAPK (ERK1/2; no. 9106), total EGFR (no. 4267), phospho-AKT (Ser473) (no. 4058) and total AKT (no. 9272) antibodies were purchased from Cell Signalling Technologies (Danvers, MA, USA). Phospho-EGFR (pY1068; no. 44-788G) and anti-HA (high affinity, no. 1867423001) were purchased from Life Technologies and Roche (Castle Hill, Australia) respectively. 17-β-oestradiol (no. E8875) and anti-α-tubulin (no. T5168) were obtained from Sigma Aldrich. Goat anti-rabbit IgG (H+L) HRP conjugate (no. 170-6515) and anti-mouse IgG (no. 170-6516) were purchased from Bio-Rad (Gladesville, Australia), and polyclonal rabbit anti-rat immunoglobulin/HRP (P0450) was purchased from DAKO (Campbellfield, Australia). Alexa Fluor 488 goat anti-rat antibody (no. A-11006) was obtained from Molecular Probes (Life Technologies). Candesartan cilexetil was a gift from Astra Zeneca (North Ryde, Australia).
GPCR–EGFR transactivation assays
HMEC-LST cells were seeded into cell culture or microplates and incubated for 24 hours at 37°C with 5% CO2, then serum starved in HuMEC basal medium for 24–48 hours prior to stimulation. For experiments involving inhibitors, cells were pre-treated for 30 minutes at 37°C with either AG1478 (5 µM), candesartan cilexetil (1 µM), CK37 (10 µM) or NSC-23766 (100 µM). Cells were stimulated with 100 nM AngII (or 10 nM thrombin) for specified times at 37°C prior to harvesting. The AlphaScreen SureFire phospho-ERK 1/2 assay (TGR Biosciences no. TGRES10K, Thebarton, Australia; Perkin Elmer no. 658552, Glen Waverley, Australia), a high-throughput microplate-based ERK1/2 activation assay described in the literature (Osmond et al., 2005) was performed as per the standard protocol, details of which can be found in the MIARE (minimum information about an RNAi experiment) in supplementary material Table S1.
siRNA screening and siRNA transfections
A detailed methodology for siRNA screening is outlined in supplementary material Table S1, and primary and secondary siRNA screen data in supplementary material Tables S2 and S3. Data is also publically available in the PubChem BioAssay database (http://pubchem.ncbi.nlm.nih.gov) (Wang et al., 2012). siGENOME SMARTpool siRNAs to rat Agtr1a (M-093349-00), human GNAQ (M-008562-00), EGFR (M-003114-03), BMX (M-003106-04), CHKA, (M-006704-01), TRIO (M-005047-00), RAC1 (M-003560-06), RHOA (M-003860-03) and the GFP duplex (D-001300-01) were obtained from Dharmacon RNAi Technologies (ThermoFisher Scientific) and resuspended in 1× Dharmacon siRNA buffer prior to use. For siRNA validation (quantifying mRNA knockdown and determining the effect of knockdown on transactivation), conditions outlined from the siRNA screen were scaled up to a 12-well plate format to harvest protein and/or RNA. Briefly, 40 nM Dharmacon SMARTpool siRNA or 25 nM Dharmacon SMARTpool deconvoluted siRNA (individual duplex) were complexed for 20 minutes at ambient temperature in a 200 µl volume of HuMEC basal medium with a final concentration of 1 µl per well of DharmaFECT 1 transfection lipid (ThermoFisher Scientific). HMEC-LST-AT1R cells (∼100,000) in antibiotic-free HuMEC complete medium (800 µl volume) were seeded into each well along with the complexed siRNA and incubated for 24 hours. For RNA experiments, RNA was extracted from cells at a 24-hour knockdown (see RNA extraction and cDNA synthesis section for details). For protein analysis, after a 24-hour knockdown, wells were washed once with Dulbecco's PBS (pH 7.4), cells were serum starved for 48 hours in HuMEC basal medium (total 72 hour knockdown) prior to stimulation with AngII for 10 minutes at 37°C, and protein was harvested.
Protein extraction, SDS-PAGE and western blot analysis
Cells were washed twice with ice-cold PBS and harvested in ice-cold RIPA buffer [50 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM EDTA, 50 mM sodium fluoride, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, 10 mM sodium pyrophosphate, 10 mM sodium orthovanadate] containing Complete EDTA-free protease inhibitor cocktail (Roche). Cells were gently lysed for 1 hour at 4°C and centrifuged at 15,000 g for 15 minutes at 4°C. Protein concentration was determined using the DC protein assay kit (Bio-Rad) in microplates as per the manufacturer's instructions. For the resolution of all proteins (apart from the AT1R), protein samples were mixed with Laemmli sample buffer (Laemmli, 1970) containing 8% β-mercaptoethanol and heated at 95°C for 5 minutes. To resolve the AT1R, freshly prepared lysate was mixed at a 1∶1 ratio with 62.5 mM Tris-HCl (pH 6.8), 2% (w/v) SDS, 6 M urea, 10% β-mercaptoethanol and 20% glycerol and heated to 60°C for 15 minutes. Samples were electrophoresed on Tris-glycine SDS-PAGE gels, and protein transferred to PVDF membrane (Immobilon-P, Merck Millipore). Membranes were blocked with 5% low-fat milk (Diploma, Fonterra Foodservices, Mount Waverley, Australia) or 1% BSA (Sigma-Aldrich) in Tris-buffered saline (TBS) pH 7.6 containing 0.05% (v/v) Tween 20 (TBST). Antibodies were prepared in either 5% low-fat milk or 1% BSA in TBST. Membranes were washed with TBST and treated with the relevant secondary antibody in 5% low-fat milk or TBST. Membranes were developed using Western Lightning ECL Plus (Perkin Elmer) to Hyperfilm (GE Healthcare, Rydalmere, Australia).
Immunofluorescence to detect AT1R expression
HMEC-LST cells (150,000) in HuMEC complete medium were seeded into chamber slides (Nunc Lab-Tek II, 4 chamber, BD Biosciences) and incubated for 24 hours. Chambers were washed with PBS, fixed with 4% PFA, blocked in 1% BSA and treated with 40 ng/ml of anti-HA high-affinity antibody in 1% BSA. Chambers were washed with PBS containing 0.05% Tween 20 (PBST). Alexa Fluor 488 goat anti-rat antibody (Molecular Probes) in 1% BSA was added to each well (protected from light). Nuclei and cell membranes were counterstained with DAPI (Sigma-Aldrich) and Cell Mask-Alexa Fluor 647 (Life Technologies), respectively. Slides were mounted in Vectashield (Vector Labs, Burlingame, CA, USA) with a coverslip, and sealed prior to use. Confocal images (Z-stacks at 2 µm intervals) were obtained with the Nikon C2 confocal microscope using the NIS elements AR.3.2 program (Nikon Instruments, Melville, NY, USA) with a 40× objective. Z-stack images were imported into NIH ImageJ (version 1.44o, available online at http://imagej.nih.gov/ij) (Abramoff et al., 2004). The maximum intensity of each stack for each channel (3 channels in total) was obtained, images were smoothed and despeckled and a scale bar added. “RGB channel merge” was used to merge the respective channels. Individual images and a three channel merged image are shown in supplementary material Fig. S1.
Radioligand competition binding assay
For competition radioligand binding assays, 500,000 HMEC-LST-AT1R or control mCherry-expressing cells were seeded into 24-well plates, and allowed to adhere for 24 hours. Culture medium was replaced with unlabelled AngII and 330,000 cpm of [125I]AngII (Prosearch International, Malvern, Australia), diluted in OptiMEM (Life Technologies) supplemented with 1% BSA, was added and incubated at ambient temperature for 1 hour. Wells were washed with PBS and cells solubilised with 500 µl 0.1 M NaOH. Radioactivity was counted using a 2470 Wizard 2 Automatic Gamma Counter (Perkin Elmer). Data for the displacement of bound radiolabelled [125I]AngII by unlabelled AngII was collected. Concentrations of unlabelled AngII were assayed in triplicate wells and averaged for four independent experiments. Relative protein concentration in each well was determined using the DC protein assay kit (Bio-Rad) as per the manufacturer's instructions, and the relative number of binding sites per amount of protein (pmol receptor/mg cellular protein) calculated.
Calcium mobilisation assay
Cells were seeded at a density of 100,000 cells/well into 96-well plates pre-coated with poly-L-lysine, and incubated for ∼5 hours. Cells were washed with PBS and serum starved overnight. The following day, cells were loaded with 2.9 µg/ml of Fluo-4AM (0.3 µg/well) in assay buffer (HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4) for 45 minutes at 37°C, protected from light. Wells were washed once with assay buffer and de-esterified for 30 minutes at ambient temperature, protected from light. Prior to stimulation with AngII, background fluorescence of cells was imaged for 10 seconds on the FLIPR Tetra (Molecular Devices, Sunnyvale, CA, USA). AngII (diluted in the assay buffer at various concentrations) was added using the FLIPR Tetra and fluorescence was measured for a total of 250 seconds/well. Data was analysed using the FLIPR Tetra Software (Screenworks 3.1.0.3, Molecular Devices) to calculate Max-Min (10–250 seconds) values, then data plotted into GraphPad Prism 5.0d (Graphpad Software, La Jolla, CA, USA) to fit curves using non-linear regression. Concentrations of AngII were assayed in triplicate wells in each independent experiment. To perform calcium mobilisation with siRNA knockdown, the protocol was performed as described above with the following modifications: HMEC-LST-AT1R cells (50,000 cells/well) were reverse transfected with 40 nM Dharmacon SMARTpool siRNA and 0.1 µl DharmaFECT 1 (per well; duplicate plates for each condition) and incubated for 24 hours. At 24 hours, RNA was extracted from eight wells of one plate and pooled to assess target knockdown (described below). The second plate was serum starved for 48 hours (72 hour knockdown) prior to Fluo-4AM loading and proceeding with the assay as described. Data are displayed as the percentage change in fluorescence (ΔF) over baseline fluorescence (F0).
RNA extraction and cDNA synthesis
Cell monolayers were washed with ice-cold PBS, and RNA extracted using the Bioline Isolate RNA mini kit (Bioline, Alexandria, Australia) as per manufacturer's instructions. RNA was eluted from columns with 40 µl of RNase-free H2O and the concentration determined using the Nanodrop ND-1000 spectrophotometer (Thermo Scientific). RNA was stored at −80°C until use. For cDNA synthesis, up to 400 ng RNA was treated with 1 Unit of RQ1 RNase-free DNase (Promega, Alexandria, Australia) for 30 minutes at 37°C, then reverse transcribed for 1 hour at 50°C using the Superscript III kit (Invitrogen, Life Technologies) with random primers (Promega). cDNA was stored at −20°C prior to use.
Primers and real-time quantitative PCR
Primers for real-time quantitative PCR were designed over exon–exon junctions (where possible) using NCBI Primer Blast (Primer3) (http://www.ncbi.nlm.nih.gov/tools/primer-blast) (Rozen and Skaletsky, 2000) with predicted amplicon sizes ranging from 110–160 bp. Sequences are listed in supplementary material Table S4. Primers were obtained from Geneworks (Hindmarsh, Australia), resuspended in sterile H2O and stored at −20°C prior to use. For real-time PCR analysis, Fast SYBR Green Master Mix (Applied Biosystems, Life Technologies), cDNA template and 300 nM of forward and reverse primers in a final reaction volume of 20 µl was assayed. Samples were run on the Applied Biosystems StepOne Real-Time PCR System for 40 cycles using the default machine settings with a melt curve ramp of 0.7°C. Data was analysed using the 7000 SDS 1.1 RQ Software (Applied Biosystems) where relative quantification of gene expression was performed and gene expression was normalised to GAPD expression.
STRING analysis
STRING 9.0 (http://string-db.org), a publically available online database of functional protein interactions (Szklarczyk et al., 2011) was used to generate an AT1R–EGFR transactivation ‘interactome’. The secondary siRNA screen gene list was submitted to the database using the ‘high confidence interactions’ option. Data were redrawn in Adobe Illustrator CS5 (Adobe Systems, San Jose, CA, USA) to incorporate the confidence level of each target identified from the siRNA screen. Thicker connecting lines (linking the nodes) represent increasing confidence of interactions.
Densitometry analysis
Densitometry analysis of western blot data (scanned to TIFF format) was performed using NIH ImageJ 1.44o software (Abramoff et al., 2004). The density of phospho-EGFR (pY1068) or phospho-ERK1/2 bands were normalised to tubulin band density for each sample. Data was imported into Microsoft Excel (Microsoft, Redmond, WA, USA), and graphs drawn in GraphPad Prism 5.0d.
Statistical analyses
The statistical analyses performed for the siRNA screen are outlined in supplementary material Table S1. Data was analysed using paired two-tailed Student's t-tests within the GraphPad Prism 5.0d program. Unless otherwise described, data presented graphically are the means ± standard error of the mean (s.e.m.), with statistical significance set at P<0.05.
Supplementary Material
Acknowledgments
Our thanks to Mr Ralph Rossi and Ms Viki Milovak (FACS), Dr Jillian Danne (confocal microscopy), Ms Alison Boast, Ms Analia Lesmana and Ms Anna-Kristen Szubert (technical assistance), Associate Professor Phillip Darcy and Dr Sarah Russell (plasmid constructs) and Dr Katherine Hannan (proofreading manuscript) (all at Peter MacCallum Cancer Centre, Melbourne, Australia). Thanks to Professor Robert Weinberg (Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, USA) for providing HMEC cell lines.
The Victorian Centre for Functional Genomics is funded by the Australian Cancer Research Foundation (ACRF), the Victorian Department of Industry, Innovation and Regional Development (DIIRD), the Australian Phenomics Network supported by funding from the Australian Government's Education Investment Fund through the Super Science Initiative, the Australasian Genomics Technologies Association (AMATA) and the Brockoff Foundation.
Footnotes
Author contributions
A.J.G designed, performed and analysed the siRNA screening and validation experiments and wrote the manuscript; B.W.P performed the radioligand binding and intracellular calcium mobilisation assays and reviewed the manuscript; C.M.G performed the informatics analyses; D.W.T and Y.H assisted with assay design and siRNA screening; G.A.Q-R assisted with siRNA knockdown and calcium mobilisation assays; K.A.M and H.Q assisted with the generation and testing of cell lines; K.J.S assisted with the design and execution of the siRNA screen, discussed data and reviewed the manuscript; W.G.T and R.D.H designed the conceptual framework of the study and experiments, analysed and discussed data, obtained funding for this study and wrote the manuscript.
Funding
This work was supported by the Australian National Health and Medical Research Council project grants [grant numbers 472640, 1024726 to W.G.T. and R.D.H]; and a project grant awarded to R.D.H, funded in Australia by the Captain Courageous Foundation (http://www.captaincourageousfoundation.com). R.D.H also holds an NHMRC senior research fellowship [grant number 1022402].
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.128280/-/DC1
References
- Abramoff M. D., Magalhaes P. J., Ram S. J. (2004). Image Processing with ImageJ. Biophotonics Intern. 11, 36–42 [Google Scholar]
- Andreev J., Galisteo M. L., Kranenburg O., Logan S. K., Chiu E. S., Okigaki M., Cary L. A., Moolenaar W. H., Schlessinger J. (2001). Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J. Biol. Chem. 276, 20130–20135 10.1074/jbc.M102307200 [DOI] [PubMed] [Google Scholar]
- Aoyama C., Liao H., Ishidate K. (2004). Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res. 43, 266–281 10.1016/j.plipres.2003.12.001 [DOI] [PubMed] [Google Scholar]
- Arora P., Cuevas B. D., Russo A., Johnson G. L., Trejo J. (2008). Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 27, 4434–4445 10.1038/onc.2008.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura M., Kitakaze M., Takashima S., Liao Y., Ishikura F., Yoshinaka T., Ohmoto H., Node K., Yoshino K., Ishiguro H. et al. (2002). Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat. Med. 8, 35–40 10.1038/nm0102-35 [DOI] [PubMed] [Google Scholar]
- Bence K., Ma W., Kozasa T., Huang X. Y. (1997). Direct stimulation of Bruton's tyrosine kinase by G(q)-protein alpha-subunit. Nature 389, 296–299 10.1038/38520 [DOI] [PubMed] [Google Scholar]
- Birmingham A., Selfors L. M., Forster T., Wrobel D., Kennedy C. J., Shanks E., Santoyo-Lopez J., Dunican D. J., Long A., Kelleher D. et al. (2009). Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods 6, 569–575 10.1038/nmeth.1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. W., Jenkins A., Pipolo L., Hannan R. D., Thomas W. G., Smith N. J. (2006). Effect of dominant-negative epidermal growth factor receptors on cardiomyocyte hypertrophy. J. Recept. Signal Transduct. Res. 26, 659–677 10.1080/10799890600923187 [DOI] [PubMed] [Google Scholar]
- Clem B. F., Clem A. L., Yalcin A., Goswami U., Arumugam S., Telang S., Trent J. O., Chesney J. (2011). A novel small molecule antagonist of choline kinase-α that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene 30, 3370–3380 10.1038/onc.2011.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I., Maoz M., Turm H., Grisaru-Granovsky S., Maly B., Uziely B., Weiss E., Abramovitch R., Gross E., Barzilay O. et al. (2010). Etk/Bmx regulates proteinase-activated-receptor1 (PAR1) in breast cancer invasion: signaling partners, hierarchy and physiological significance. PLoS ONE 5, e11135 10.1371/journal.pone.0011135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai B., Kim O., Xie Y., Guo Z., Xu K., Wang B., Kong X., Melamed J., Chen H., Bieberich C. J. et al. (2006). Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 66, 8058–8064 10.1158/0008-5472.CAN-06-1364 [DOI] [PubMed] [Google Scholar]
- Daub H., Weiss F. U., Wallasch C., Ullrich A. (1996). Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557–560 10.1038/379557a0 [DOI] [PubMed] [Google Scholar]
- de Gasparo M., Catt K. J., Inagami T., Wright J. W., Unger T. (2000). International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472 [PubMed] [Google Scholar]
- De Paepe B., Verstraeten V. L., De Potter C. R., Vakaet L. A., Bullock G. R. (2001). Growth stimulatory angiotensin II type-1 receptor is upregulated in breast hyperplasia and in situ carcinoma but not in invasive carcinoma. Histochem. Cell Biol. 116, 247–254 [DOI] [PubMed] [Google Scholar]
- Debant A., Serra-Pagès C., Seipel K., O'Brien S., Tang M., Park S. H., Streuli M. (1996). The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 93, 5466–5471 10.1073/pnas.93.11.5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi S., Numaguchi K., Iwasaki H., Matsumoto T., Yamakawa T., Utsunomiya H., Motley E. D., Kawakatsu H., Owada K. M., Hirata Y. et al. (1998). Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J. Biol. Chem. 273, 8890–8896 10.1074/jbc.273.15.8890 [DOI] [PubMed] [Google Scholar]
- Eguchi S., Iwasaki H., Inagami T., Numaguchi K., Yamakawa T., Motley E. D., Owada K. M., Marumo F., Hirata Y. (1999). Involvement of PYK2 in angiotensin II signaling of vascular smooth muscle cells. Hypertension 33, 201–206 10.1161/01.HYP.33.1.201 [DOI] [PubMed] [Google Scholar]
- Eguchi S., Dempsey P. J., Frank G. D., Motley E. D., Inagami T. (2001). Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J. Biol. Chem. 276, 7957–7962 10.1074/jbc.M008570200 [DOI] [PubMed] [Google Scholar]
- Ekman N., Lymboussaki A., Västrik I., Sarvas K., Kaipainen A., Alitalo K. (1997). Bmx tyrosine kinase is specifically expressed in the endocardium and the endothelium of large arteries. Circulation 96, 1729–1732 10.1161/01.CIR.96.6.1729 [DOI] [PubMed] [Google Scholar]
- Elenbaas B., Spirio L., Koerner F., Fleming M. D., Zimonjic D. B., Donaher J. L., Popescu N. C., Hahn W. C., Weinberg R. A. (2001). Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65 10.1101/gad.828901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Dickerson J. B., Guo F., Zheng J., Zheng Y. (2004). Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 101, 7618–7623 10.1073/pnas.0307512101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A. J., Thomas W. G., Hannan R. D. (2010). The renin-angiotensin system and cancer: old dog, new tricks. Nat. Rev. Cancer 10, 745–759 10.1038/nrc2945 [DOI] [PubMed] [Google Scholar]
- Glunde K., Shah T., Winnard P. T., Jr, Raman V., Takagi T., Vesuna F., Artemov D., Bhujwalla Z. M. (2008). Hypoxia regulates choline kinase expression through hypoxia-inducible factor-1 alpha signaling in a human prostate cancer model. Cancer Res. 68, 172–180 10.1158/0008-5472.CAN-07-2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco S., Muscella A., Elia M. G., Salvatore P., Storelli C., Mazzotta A., Manca C., Marsigliante S. (2003). Angiotensin II activates extracellular signal regulated kinases via protein kinase C and epidermal growth factor receptor in breast cancer cells. J. Cell. Physiol. 196, 370–377 10.1002/jcp.10313 [DOI] [PubMed] [Google Scholar]
- Guilluy C., Brégeon J., Toumaniantz G., Rolli-Derkinderen M., Retailleau K., Loufrani L., Henrion D., Scalbert E., Bril A., Torres R. M. et al. (2010). The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 16, 183–190 10.1038/nm.2079 [DOI] [PubMed] [Google Scholar]
- Guo S., Sun F., Guo Z., Li W., Alfano A., Chen H., Magyar C. E., Huang J., Chai T. C., Qiu S. et al. (2011). Tyrosine kinase ETK/BMX is up-regulated in bladder cancer and predicts poor prognosis in patients with cystectomy. PLoS ONE 6, e17778 10.1371/journal.pone.0017778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heringer-Walther S., Eckert K., Schumacher S. M., Uharek L., Wulf-Goldenberg A., Gembardt F., Fichtner I., Schultheiss H. P., Rodgers K., Walther T. (2009). Angiotensin-(1-7) stimulates hematopoietic progenitor cells in vitro and in vivo. Haematologica 94, 857–860 10.3324/haematol.2008.000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando E., Sarmentero-Estrada J., Koppie T., Belda-Iniesta C., Ramírez de Molina V., Cejas P., Ozu C., Le C., Sánchez J. J., González-Barón M. et al. (2009). A critical role for choline kinase-alpha in the aggressiveness of bladder carcinomas. Oncogene 28, 2425–2435 10.1038/onc.2009.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J., Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., Yuan J. (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 10.1016/j.cell.2008.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunyady L., Catt K. J. (2006). Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 20, 953–970 10.1210/me.2004-0536 [DOI] [PubMed] [Google Scholar]
- Iwai M., Horiuchi M. (2009). Role of renin-angiotensin system in adipose tissue dysfunction. Hypertens. Res. 32, 425–427 10.1038/hr.2009.55 [DOI] [PubMed] [Google Scholar]
- Jiang X., Borgesi R. A., McKnight N. C., Kaur R., Carpenter C. L., Balk S. P. (2007). Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J. Biol. Chem. 282, 32689–32698 10.1074/jbc.M703412200 [DOI] [PubMed] [Google Scholar]
- Kittler R., Pelletier L., Heninger A. K., Slabicki M., Theis M., Miroslaw L., Poser I., Lawo S., Grabner H., Kozak K. et al. (2007). Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat. Cell Biol. 9, 1401–1412 10.1038/ncb1659 [DOI] [PubMed] [Google Scholar]
- Laemmli U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- Langhans-Rajasekaran S. A., Wan Y., Huang X. Y. (1995). Activation of Tsk and Btk tyrosine kinases by G protein beta gamma subunits. Proc. Natl. Acad. Sci. USA 92, 8601–8605 10.1073/pnas.92.19.8601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N. C., Lee A., Whitmer R. A., Kivipelto M., Lawler E., Kazis L. E., Wolozin B. (2010). Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 340, b5465 10.1136/bmj.b5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Rateri D. L., Cassis L. A., Daugherty A. (2008). The role of the renin-angiotensin system in aortic aneurysmal diseases. Curr. Hypertens. Rep. 10, 99–106 10.1007/s11906-008-0020-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S., Freichel-Blomquist A., Yang Y., Rümenapp U., Jakobs K. H., Schmidt M., Wieland T. (2005). The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J. Biol. Chem. 280, 11134–11139 10.1074/jbc.M411322200 [DOI] [PubMed] [Google Scholar]
- Mehta P. K., Griendling K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. 292, C82–C97 10.1152/ajpcell.00287.2006 [DOI] [PubMed] [Google Scholar]
- Mifune M., Ohtsu H., Suzuki H., Nakashima H., Brailoiu E., Dun N. J., Frank G. D., Inagami T., Higashiyama S., Thomas W. G. et al. (2005). G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J. Biol. Chem. 280, 26592–26599 10.1074/jbc.M502906200 [DOI] [PubMed] [Google Scholar]
- Miners S., Ashby E., Baig S., Harrison R., Tayler H., Speedy E., Prince J. A., Love S., Kehoe P. G. (2009). Angiotensin-converting enzyme levels and activity in Alzheimer's disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl Res. 1, 163–177 [PMC free article] [PubMed] [Google Scholar]
- Mitchell-Jordan S. A., Holopainen T., Ren S., Wang S., Warburton S., Zhang M. J., Alitalo K., Wang Y., Vondriska T. M. (2008). Loss of Bmx nonreceptor tyrosine kinase prevents pressure overload-induced cardiac hypertrophy. Circ. Res. 103, 1359–1362 10.1161/CIRCRESAHA.108.186577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake T., Parsons S. J. (2012). Functional interactions between Choline kinase α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene 31, 1431–1441 10.1038/onc.2011.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro A., Brar B. K., Gu Y., Peterson K. L., Vale W., Lee K. F. (2006). erbB2 is required for G protein-coupled receptor signaling in the heart. Proc. Natl. Acad. Sci. USA 103, 15889–15893 10.1073/pnas.0607499103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu H., Dempsey P. J., Frank G. D., Brailoiu E., Higuchi S., Suzuki H., Nakashima H., Eguchi K., Eguchi S. (2006). ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 26, e133–e137 [DOI] [PubMed] [Google Scholar]
- Ohtsu H., Higuchi S., Shirai H., Eguchi K., Suzuki H., Hinoki A., Brailoiu E., Eckhart A. D., Frank G. D., Eguchi S. (2008). Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology 149, 3569–3575 10.1210/en.2007-1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmond R. I., Sheehan A., Borowicz R., Barnett E., Harvey G., Turner C., Brown A., Crouch M. F., Dyer A. R. (2005). GPCR screening via ERK 1/2: a novel platform for screening G protein-coupled receptors. J. Biomol. Screen. 10, 730–737 10.1177/1087057105277968 [DOI] [PubMed] [Google Scholar]
- Park T. S., Zambidis E. T. (2009). A role for the renin-angiotensin system in hematopoiesis. Haematologica 94, 745–747 10.3324/haematol.2009.006965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H., Pipolo L., Thomas W. G. (2001). Association of beta-Arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol. Endocrinol. 15, 1706–1719 10.1210/me.15.10.1706 [DOI] [PubMed] [Google Scholar]
- Ramírez de Molina A., Báñez-Coronel M., Gutiérrez R., Rodríguez-González A., Olmeda D., Megías D., Lacal J. C. (2004). Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 64, 6732–6739 10.1158/0008-5472.CAN-04-0489 [DOI] [PubMed] [Google Scholar]
- Rhodes D. R., Ateeq B., Cao Q., Tomlins S. A., Mehra R., Laxman B., Kalyana-Sundaram S., Lonigro R. J., Helgeson B. E., Bhojani M. S. et al. (2009). AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc. Natl. Acad. Sci. USA 106, 10284–10289 10.1073/pnas.0900351106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R. J., Yohe M. E., Gershburg S., Kawano T., Kozasa T., Sondek J. (2007). Galphaq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J. Biol. Chem. 282, 29201–29210 10.1074/jbc.M703458200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompe F., Unger T., Steckelings U. M. (2010). The angiotensin AT2 receptor in inflammation. Drug News Perspect. 23, 104–111 10.1358/dnp.2010.23.2.1475901 [DOI] [PubMed] [Google Scholar]
- Rozen S., Skaletsky H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386 [DOI] [PubMed] [Google Scholar]
- Schiffrin E. L. (2010). T lymphocytes: a role in hypertension? Curr. Opin. Nephrol. Hypertens. 19, 181–186 10.1097/MNH.0b013e3283360a2e [DOI] [PubMed] [Google Scholar]
- Seta K., Sadoshima J. (2003). Phosphorylation of tyrosine 319 of the angiotensin II type 1 receptor mediates angiotensin II-induced trans-activation of the epidermal growth factor receptor. J. Biol. Chem. 278, 9019–9026 10.1074/jbc.M208017200 [DOI] [PubMed] [Google Scholar]
- Shah B. H., Catt K. J. (2002). Calcium-independent activation of extracellularly regulated kinases 1 and 2 by angiotensin II in hepatic C9 cells: roles of protein kinase Cdelta, Src/proline-rich tyrosine kinase 2, and epidermal growth receptor trans-activation. Mol. Pharmacol. 61, 343–351 10.1124/mol.61.2.343 [DOI] [PubMed] [Google Scholar]
- Shah B. H., Catt K. J. (2006). TACE-dependent EGF receptor activation in angiotensin-II-induced kidney disease. Trends Pharmacol. Sci. 27, 235–237 10.1016/j.tips.2006.03.010 [DOI] [PubMed] [Google Scholar]
- Shenoy S. K., Lefkowitz R. J. (2011). β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 10.1016/j.tips.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson K. J., Selfors L. M., Bui J., Reynolds A., Leake D., Khvorova A., Brugge J. S. (2008). Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 10, 1027–1038 10.1038/ncb1762 [DOI] [PubMed] [Google Scholar]
- Smith N. J., Chan H. W., Qian H., Bourne A. M., Hannan K. M., Warner F. J., Ritchie R. H., Pearson R. B., Hannan R. D., Thomas W. G. (2011). Determination of the exact molecular requirements for type 1 angiotensin receptor epidermal growth factor receptor transactivation and cardiomyocyte hypertrophy. Hypertension 57, 973–980 10.1161/HYPERTENSIONAHA.110.166710 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P. et al. (2011). The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 10.1093/nar/gkq973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasebi M., Barker S., Puddefoot J. R., Vinson G. P. (2006). Localisation of renin-angiotensin system (RAS) components in breast. Br. J. Cancer 95, 67–74 10.1038/sj.bjc.6603213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnone L., Lahtinen I., Mustonen T., Virtaneva K., Francis F., Muscatelli F., Alitalo R., Smith C. I., Larsson C., Alitalo K. (1994). BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene 9, 3683–3688 [PubMed] [Google Scholar]
- Thomas W. G., Motel T. J., Kule C. E., Karoor V., Baker K. M. (1998). Phosphorylation of the angiotensin II (AT1A) receptor carboxyl terminus: a role in receptor endocytosis. Mol. Endocrinol. 12, 1513–1524 10.1210/me.12.10.1513 [DOI] [PubMed] [Google Scholar]
- Thomas W. G., Brandenburger Y., Autelitano D. J., Pham T., Qian H., Hannan R. D. (2002). Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ. Res. 90, 135–142 10.1161/hh0202.104109 [DOI] [PubMed] [Google Scholar]
- Touyz R. M., Wu X. H., He G., Salomon S., Schiffrin E. L. (2002). Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 39, 479–485 10.1161/hy02t2.102909 [DOI] [PubMed] [Google Scholar]
- Tsukada S., Simon M. I., Witte O. N., Katz A. (1994). Binding of beta gamma subunits of heterotrimeric G proteins to the PH domain of Bruton tyrosine kinase. Proc. Natl. Acad. Sci. USA 91, 11256–11260 10.1073/pnas.91.23.11256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama K., Saito M., Sasaki M., Obara Y., Higashiyama S., Nakahata N. (2009). Thromboxane A2 receptor-mediated epidermal growth factor receptor transactivation: involvement of PKC-delta and PKC-epsilon in the shedding of epidermal growth factor receptor ligands. Eur. J. Pharm. Sci. 38, 504–511 10.1016/j.ejps.2009.09.016 [DOI] [PubMed] [Google Scholar]
- Uchiyama-Tanaka Y., Matsubara H., Nozawa Y., Murasawa S., Mori Y., Kosaki A., Maruyama K., Masaki H., Shibasaki Y., Fujiyama S. et al. (2001). Angiotensin II signaling and HB-EGF shedding via metalloproteinase in glomerular mesangial cells. Kidney Int. 60, 2153–2163 10.1046/j.1523-1755.2001.00067.x [DOI] [PubMed] [Google Scholar]
- Vaqué J. P., Dorsam R. T., Feng X., Iglesias-Bartolome R., Forsthoefel D. J., Chen Q., Debant A., Seeger M. A., Ksander B. R., Teramoto H. et al. (2013). A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol. Cell 49, 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Xiao J., Suzek T. O., Zhang J., Wang J., Zhou Z., Han L., Karapetyan K., Dracheva S., Shoemaker B. A. et al. (2012). PubChem's BioAssay Database. Nucleic Acids Res 40, D400–D412 10.1093/nar/gkr1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss D., Sorescu D., Taylor W. R. (2001). Angiotensin II and atherosclerosis. Am. J. Cardiol. 87, Suppl., 25C-32C 10.1016/S0002-9149(01)01539-9 [DOI] [PubMed] [Google Scholar]
- Wettschureck N., Rütten H., Zywietz A., Gehring D., Wilkie T. M., Chen J., Chien K. R., Offermanns S. (2001). Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 7, 1236–1240 10.1038/nm1101-1236 [DOI] [PubMed] [Google Scholar]
- Williams S. L., Lutz S., Charlie N. K., Vettel C., Ailion M., Coco C., Tesmer J. J., Jorgensen E. M., Wieland T., Miller K. G. (2007). Trio's Rho-specific GEF domain is the missing Galpha q effector in C. elegans. Genes Dev. 21, 2731–2746 10.1101/gad.1592007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Aoyama C., Young S. G., Vance D. E. (2008). Early embryonic lethality caused by disruption of the gene for choline kinase alpha, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 283, 1456–1462 10.1074/jbc.M708766200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin A., Clem B., Makoni S., Clem A., Nelson K., Thornburg J., Siow D., Lane A. N., Brock S. E., Goswami U. et al. (2010). Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene 29, 139–149 10.1038/onc.2009.317 [DOI] [PubMed] [Google Scholar]
- Zambidis E. T., Park T. S., Yu W., Tam A., Levine M., Yuan X., Pryzhkova M., Péault B. (2008). Expression of angiotensin-converting enzyme (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells. Blood 112, 3601–3614 10.1182/blood-2008-03-144766 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.