

Abstract
Mitochondria are independent organelles with their own DNA. As a primary function, mitochondria produce the energy for the cell through Oxidative Phosphorylation (OXPHOS) in the Electron Transport Chain (ETC). One of the toxic products of this process is Reactive Oxygen Species (ROS), which can induce oxidative damage in macromolecules like lipids, proteins and DNA. Mitochondrial DNA (mtDNA) is less protected and has fewer reparation mechanisms than nuclear DNA (nDNA), and as such is more exposed to oxidative, mutation-inducing damage. This review analyzes the causes and consequences of mtDNA mutations and their relationship with the aging process. Neurodegenerative diseases, related with the aging, are consequences of mtDNA mutations resulting in a decrease in mitochondrial function. Also described are “mitochondrial diseases”, pathologies produced by mtDNA mutations and whose symptoms are related with mitochondrial dysfunction. Finally, mtDNA haplogroups are defined in this review; these groups are important for determination of geographical origin of an individual. Additionally, different haplogroups exhibit variably longevity and risk of certain diseases. mtDNA mutations in aging and haplogroups are of special interest to forensic science research. Therefore this review will help to clarify the key role of mtDNA mutations in these processes and support further research in this area.
Keywords: Mitochondrial DNA (mtDNA), Electron Transport Chain (ETC), Reactive Oxygen Species (ROS), Aging, Diseases, Forensic Sciences
Mitochondria biology
Mitochondria are organelles that evolved from an ancient endosymbiotic purpurbacteria engulfed by an eukaryotic ancestor approximately 1.5 billion years ago [1]. Due to this origin, mitochondria have a double-membrane structure, consisting of an outer membrane, highly permeable with many pores surrounding the intermembrane space, and an inner membrane, which is impermeable and delimits the internal matrix (Figure 1) [2, 3].
Figure 1.

Mitochondria structure.
Mitochondria are involved in cellular homeostasis. They play a role in intracellular signaling and apoptosis, intermediary metabolism, and in the metabolism of amino acids, lipids, cholesterol, steroids, and nucleotides. The most vital function of the mitochondria is their role in cellular energy metabolism because they generate the majority of the cell’s supply of ATP (Adenosin triphosphate) [4, 5].
Oxidative phosphorylation and Reactive Oxygen Species
Synthesis of ATP occurs via the process of oxidative phosphorylation (OXPHOS) through the respiratory or electron transport chain (ETC), which is located at the inner mitochondrial membrane and consists of five protein complexes (complexes I to V) [5]. Various substrates can be metabolized to produce ATP; reduced cofactors (NADH and FADH2), generated from the intermediary metabolism of carbohydrates (via the citric acid/tricarboxylic, TCA cycle), and proteins and fats (β-oxidation) donate electrons to complex I and complex II. These electrons are passed sequentially to ubiquinone (coenzyme Q or CoQ) to form ubisemiquinone (CoQH) and then ubiquinol (CoQH2). Ubiquinol transfers its electrons to complex III, which transfers them to cytochrome c. From cytochrome c, the electrons flow to complex IV, which donates an electron to oxygen to produce water. Each of these complexes incorporates multiple electron carriers. Complexes I, II and III comprise several ion-sulfur (Fe-S) centers, whereas complexes III and IV include the b+c1 and a+a3 cytochromes. The energy liberated by the flow of electrons is used by complexes I, III and IV to pump protons (H−) out of the mitochondrial inner membrane into the intermembrane space. This proton gradient generates the mitochondrial membrane potential that is coupled with ATP synthesis by complex V from ADP (Adenosin diphosphate) and inorganic phosphate (Pi). ATP is released from the mitochondria in exchange for cytosolic ADP using a carrier, adenine nucleotide translocator (ANT) (Figure 2) [4, 6].
Figure 2.

Oxidative Phosphorylation (OXPHOS). NADH and FADH2 are produced from the intermediary metabolism of carbohydrates, proteins and fats; and they donate electrons to complex I (NADH-ubiquinone oxidoreductase) and complex II (succinate-ubiquinone oxidoreductase). These electrons are passed sequentially to ubiquinone (coenzyme Q or CoQ) to form ubisemiquinone (CoQH) and then ubiquinol (CoQH2). Ubiquinol transfers its electrons to complex III (ubiquinol-cytochrome c oxidase reductase), which transfers them to cytochrome c. From cytochrome c, the electrons flow to complex IV (cytochrome c oxidase or COX), which donates an electron to oxygen to produce water. The energy liberated by the flow of electrons is used by complexes I, III and IV to pump protons (H−) out of the mitochondrial inner membrane into the intermembrane space. This proton gradient generates the mitochondrial membrane potential that is coupled to ATP synthesis by complex V from ADP (Adenosin diphosphate) and inorganic phosphate (Pi). ATP is released from the mitochondria in exchange for cytosolic ADP using a carrier, adenine nucleotide translocator (ANT).
As a by-product of OXPHOS, the mitochondria generate endogenous Reactive Oxygen Species (ROS) (Figure 3). Excess electrons from complex I–III can be transferred directly to O2 to generate superoxide anion (O2−). It is converted into hydrogen peroxide (H2O2) by the matrix enzyme manganese superoxide dismutase (MnSOD or SOD2) or by the mitochondrial intermembrane space and cytosol enzyme copper/zinc SOD (Cu/ZnSOD or SOD1). Hydrogen peroxide is more stable than superoxide anion and can diffuse into the cytosol and nucleus to activate redox-sensitive signaling. Hydrogen peroxide is detoxified in water by glutathione peroxidase (GPx) in mitochondria and cytosol and by catalase (CAT) in peroxisomes. However, in the presence of reduced transition metals (like Fe2+), hydrogen peroxide is converted to hydroxyl radical (OH.) through the Fenton reaction. Hydroxyl radical is the most reactive ROS [6–9]. ROS are highly reactive molecules and behave as oxidants that can extract electrons from DNA, proteins, lipids, and other molecules. ROS activity can result in oxidative damage, which ultimately results in the inactivation of proteins, injury to the integrity of biological membranes and genotoxicity. Sufficiently high levels of ROS induce cell death by apoptotic and/or necrotic mechanisms. Also, low levels of ROS can act as signaling molecules in the cell [10–12].
Figure 3.

Generation of mitochondrial ROS. Superoxide is produced by complex I on the matrix side of the inner mitochondrial membrane and by complex III on both sides of the inner mitochondrial membrane. It is converted to hydrogen peroxide (H2O2) by the matrix enzyme manganese superoxide dismutase (MnSOD or SOD2) or by the mitochondrial intermembrane space and cytosol enzyme copper/zinc SOD (Cu/ZnSOD or SOD1). Hydrogen peroxide can diffuse into the cytosol and nucleus to activate redox-sensitive signaling. Hydrogen peroxide is detoxified in water by glutathione peroxidase (GPx) in the mitochondria and cytosol. In the presence of reduced transition metals (like Fe2+), hydrogen peroxide is converted to hydroxyl radical (OH.) through the Fenton reaction.
Mitochondrial DNA
Mitochondria contain their own DNA, which is inherited through the maternal line. The mitochondrial genome is composed of approximately 16.5 kilobases of circular, negatively supercoiled double-stranded molecules (Figure 4). It contains 37 genes, 12S and 16S rRNAs, 22 tRNAs and 13 proteins, that are structural subunits of OXPHOS enzyme complexes, 7 of Complex I; 3 of Complex IV; 2 of Complex V and 1 of Complex III [5, 13]. Nuclear genes encode approximately 1500 mitochondrial proteins, the majority of mitochondrial respiratory chain polypeptides, including all four subunits of complex II; proteins implicated in replication such as the mitochondrial DNA polymerase γ (DNA pol γ); the mitochondrial RNA polymerase components; the mitochondrial transcription factor (mtTFA); the mitochondrial ribosomal proteins and elongation factors; and the mitochondrial metabolic enzymes [4, 6, 14, 15].
Figure 4.
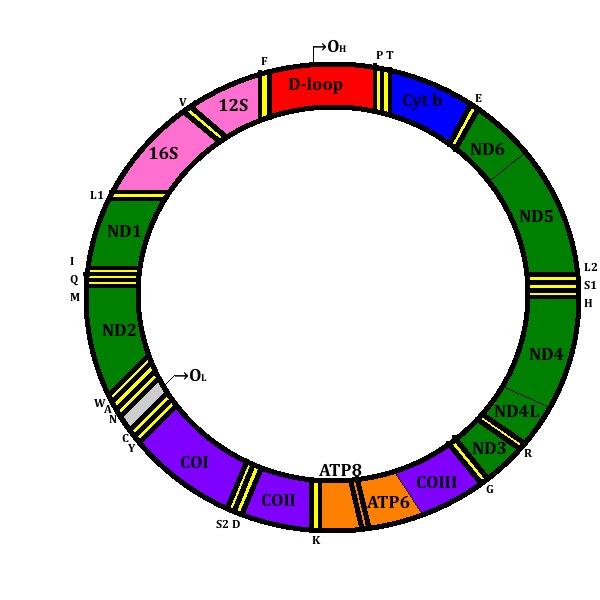
Mitochondrial DNA. D-loop is shown in red. The genes that encode the subunits of complex I (ND1–ND6 and ND4L) are shown in green; cytochrome c oxidase (COI–COIII) is shown in purple; cytochrome b of complex III is shown in blue; and the subunits of the ATP synthase (ATPase 6 and 8) are shown in orange. The genes for the two rRNAs (12S and 16S) are shown in pink and 22 tRNAs (F, V, L1, I, M, W, D, K, G, R, H, S1, L2, T, P, E, S2, Y, C, N, A) are indicated by boxes in yellow. The Origins of Heavy-strand replication (OH) and Light-strand replication (OL) are shown.
mtDNA does not contain introns, has no space between genes and lacks 5′or 3′ non-coding sequences [6, 16]. However, it is organized into protein-DNA complexes, nucleoids, within the mitochondrial matrix, which protects the genome [17]. The two mtDNA strands are called the heavy (purine-rich) strand, in which most of the information is encoded, and the light (pyrimidine-rich) strand, which contains the information for only one polypeptide and 8 tRNAs [18]. mtDNA is replicated by the concerted action of DNA pol γ,its accessory subunit, p55, and replication factors such as the mitochondrial single-stranded DNA binding protein and the mitochondrial DNA helicase (Twinkle) [19]. The replication occurs bi-directionally, initiated at two spatially and temporally distinct origins of replication OH and OL, for the heavy and light strands respectively [20]. However, evidence suggests the presence of conventional duplex mtDNA replication intermediates, indicative of coupled leading and lagging-strand DNA synthesis [21]. mtDNA is exposed to more damage than nuclear DNA. However, it has repair mechanisms. The major DNA repair mechanism is base excision repair (BER), encoded by nuclear DNA, although it is present at lower levels than in nuclear DNA [5, 22]. BER is based on a cascade of reactions, starting with the recognition of damage, followed by enzymatic processing steps that aim to remove the lesion and restore genomic integrity [23]. DNA repair enzymes in the mitochondria include damage-specific DNA glycosylases, such as 8-oxoguanine DNA glycosylase-1 (OGG1); an endonuclease III-like protein (NTH1); apurinic/apyrimidinic endonuclease (APE); and DNA ligase IIIβ. DNA pol γ is implicated in mtDNA BER [24–27]. In addition to BER, there is some evidence that mitochondria possess mismatch repair activities, homologous recombination, and non-homologous end joining [28–30].
mtDNA mutations and aging
The oxidative stress theory of aging
According to the “free radical theory” of aging, aging and associated degenerative diseases can be attributed to deleterious effects of reactive oxygen species [31]. A current version of this theory is the “oxidative stress theory”: “a chronic state of oxidative stress exists in cells of aerobic organisms even under normal physiological conditions because of an imbalance of prooxidants and antioxidants. This imbalance results in a steady-state accumulation of oxidative damage in a variety of macromolecules. Oxidative damage increases during aging, which results in a progressive loss in the functional efficiency of various cellular processes” [32]. A variant of this theory is the “mitochondrial theory”, which predicts that a “vicious cycle” within the mitochondria contributes to the aging process. ROS produced in the mitochondria induce damage to phospholipids, proteins and nucleic acids in the mitochondria; producing mtDNA mutations which lead to the synthesis of functionally impaired respiratory chain subunits, causing respiratory chain dysfunction and augmented reactive oxygen species production, which causes an exponential increase of mtDNA mutations over time, resulting in aging and associated degenerative diseases (Figure 5) [33].
Figure 5.

“Vicious cycle” of mtDNA damage by ROS. ROS can react with mtDNA, inducing mutations. These mutations cause a decrease in the activity of ETC, producing dysfunction in the mitochondria which can lead to cell death.
An important role of mitochondrial ROS production in aging and degenerative diseases is congruent with the life-extending capacity of caloric restriction. Reduction of available calories will starve the mitochondrial ETC for electrons, reducing ROS and protecting the mitochondria and mtDNAs. In rodent studies, mtDNA base oxidation and rearrangement mutation levels have been found to increase with age. Dietary restriction will inhibit the accumulation of both forms of mtDNA damage [34, 35].
Further evidence that ROS toxicity is a limiting factor for life span was directly demonstrated in transgenic Drosophila melanogaster, which express increased Cu/Zn SOD and catalase the longer they live [36]. Also, mice in which MnSOD was genetically inactivated died at a mean age of eight days [37, 38]. More proof that mitochondrial ROS limit mammalian life span has come from the targeting of human catalase to the mouse mitochondria in transgenic mice and observing an increase in life span. Heart mitochondria in these animals showed an increased resistance of mitochondrial aconitase to inactivation by hydrogen peroxide, and the skeletal muscle and heart mtDNAs showed a significant reduction in the age-related accumulation of mtDNA rearrangement mutations [39]. Apart from these studies, a number of transgenic and knock-in mouse models have been developed to test the effects of increased mtDNA mutation accumulation in vivo. Two groups independently generated knock-in mice containing a point mutation in the proof-reading domain of DNA pol γ. The enzyme had a deficient exonuclease activity, with no decrease in DNA polymerase activity. These mtDNA mutant mice resulted in mitochondrial mutation frequencies that were increased by at least 3 to 11 fold in multiple tissues, with accumulation of mtDNA base-substitution mutations, suggesting that the mutations occurs during embryonic and/or fetal development. Deletions of mtDNA can also be detected in these mice. The phenotype resembled many characteristics of accelerating and premature aging: hair graying and loss, reduced bone density and increased incidence of kyphosis, reduced muscle mass, severe reduction in body fat, early loss of fertility, dilated cardiac hypertrophy, accelerated thymic atrophy, presbycusis, anemia, intestinal dysplasia and reduced survival. Also, a decline in respiratory function was observed. In contrast, it did not display signs of increased oxidative damage to proteins, lipids or DNA and antioxidant defense systems were not upregulated. Increase levels of mtDNA mutations were not associated with increased ROS production or increased oxidative stress. These findings imply that there is no vicious cycle leading to increased oxidative damage. In contrast, many tissues in this mouse contained increased levels of caspase-3, a downstream protease activated during the apoptotic pathway. mtDNA mutation accumulation was associated with the activation of apoptosis, leading to cell death [40–42].
mtDNA mutations and aging
mtDNA mutations are accumulated with aging, although it is still unclear whether these alterations are a cause or consequence of the aging process [43]. Somatic mutations in mtDNA lead to a condition called mtDNA heteroplasmy, a mixture of normal and mutant mtDNA molecules in a cell. The typical cell contains hundreds of mitochondria, each housing 2 to 7 mtDNA molecules [44].
Age-related somatic mtDNA mutations accumulate in postmitotic tissues until a certain tissue-specific threshold in the level of mutant to normal mtDNA molecules is surpassed and cells become energetically compromised [45, 46]. For that reason, the main consequence of mtDNA mutations is an impairment of energy metabolism, inducing aging effects on tissues that display high energetic demands, such as the heart, skeletal muscle, and the brain [47]. The accumulation of mtDNA mutations, including deletions, duplications and point mutations, has been found in a variety of tissues during aging in human, monkeys and rodents [48–53].
The mitochondrial genome contains a non-coding control region, the displacement loop (D-loop), important for mtDNA replication and transcription. Different studies have found an accumulation of point mutations in this region including A189G, T408A, and T414G with aging [54–59]. For example, the A189G mutation was investigated in the muscles of unrelated individuals age 1–97, a higher percentage of mutations exist in older individuals [58]. The age-related accumulation of mutations within this regulatory region of the mitochondrial genome may influence the activity of this tissue.
Apart from point mutations, mtDNA deletions are most likely occurring during the repair of damaged mtDNA. The mechanism by which mtDNA deletions arise during mtDNA repair is through exonuclease activity at double-strand breaks [60]. An age-related accumulation of various deletions has been shown in multiple tissues of different species [61–66]. A number of deletions can occur, however, the most commonly studied in humans is a 4977 bp deletion, also called “common deletion”. This deletion removes all or part of the genes for NADH dehydrogenase subunits, cytochrome c oxidase subunit III, and ATP synthase subunits 6 and 8. An extensive study developed by Meissner et al [66] found an increase in the 4977 bp deletion with aging in the brain, heart and skeletal muscle of 92 individual aged 2 months to 102 years old. However, the levels were highly variable between individuals of the same decade and among different tissues within a single individual. This is in agreement with other studies [61, 62, 65].
The increase in 8-oxo-2′-deoxyguanosine (oxo8G) levels in mtDNA with age appears to be a general phenomenon, probably due to a decline in the reparation mechanism with age [67, 68]. Recent studies have investigated point mutations and deletions in single cells; only one particular point mutation or deletion is found in a single cell, and the percent of mutant DNA molecules within a cell increases with age [69, 70]. Skeletal muscle develops a mosaic pattern of increasing cytochrome c oxidase (COX) deficient muscle fibers with increasing age, and multiple mtDNA alterations (point mutations, length variations, deletions) were found [57, 71]. A study of deletions in COX negative muscle fibers from aged rats showed that >90% of the mtDNA at the site of the electron transport system abnormality contained deletion mutations [72].
mtDNA mutations and diseases
Mitochondrial diseases are a diverse group of human diseases characterized by defects in any aspect of mitochondrial function [73]. It can be inherited, transmitted from the mother to offspring, like patients with Leber Hereditary Optic Neuropathy (LHON); or spontaneous. Mitochondrial diseases can be further divided into diseases related with defects in either mitochondrial DNA or nuclear DNA (Table 1):
Table 1.
Mitochondrial diseases. The classifications in mtDNA mutations and nuclear DNA defects and the mutations in the mitochondrial genes.
| TYPE | SUBTYPE | MUTATION | DISEASE |
|---|---|---|---|
| mtDNA mutations | Rearrangement mutations | 4977 | Progressive external ophtalmoplegia (PEO) |
| Kearns-Sayre Syndrome (KSS) | |||
| Pearson Syndrome (PS) | |||
| Point mutations tRNA or rRNA | A3243G | Mitochondrial Encephalopathy lactic acidosis, and stroke-like episodes (MELAS) | |
| A8344G | Myoclonus Epilepsy and Ragged-Red Fibers (MERRF) | ||
| Point mutations proteins | T8993G | Neurogenic muscle weakness, ataxia and retinitis pigmentosa (NARP) | |
| G11778A | Leber Hereditary Optic Neuropathy (LHON) | ||
| G3460A | Leigh Syndrome | ||
| T14884C | |||
|
| |||
| Nuclear DNA defects | Replication mutations proteins | POLG1 | Chromosome 15-linked Autosomal Dominant Progressive External Ophtalmoplegia (adPEO) |
| Twinkle | Chromosome 10-linked adPEO | ||
| ANT1 | Chromosome 4-linked adPEO | ||
| Mutations in Thymidine Kinase 2 (TK2) and Deoxyguanosine Kinase (dGK) | TK2 | mtDNA depletion Syndrome (MDS) | |
| dGK | |||
| Mutations in Thymidine Phosphorilase (TP) | TP | Mitochondrial Neurogastroinstestinal Encephalomyopathy (MNGIE) | |
Mitochondrial DNA mutations
1. Rearrangement mutations
Rearrangement mutations can be either inherited or spontaneous. Inherited mtDNA rearrangement mutations are primarily insertions, like inherited diabetes and deafness [74, 75].
Spontaneous rearrangement mutations, primarily large deletions, are always heteroplasmic, and always result in the loss of one of the tRNA genes, therefore affecting translation of all 13 mitochondrial genes [76]. The diseases induced by these mutations include progressive external opthalmoplegia (PEO), Kearns-Sayre syndrome (KSS), and Pearson Syndrome (PS). One third of PEO/KSS/PS patients have the common deletion of 4977 bp [77, 78].
2. Point mutations affecting tRNA or rRNA
The most common pathological mtDNA mutations are located in tRNA genes, which produce distinct clinical syndromes: MELAS syndrome (mitochondrial encephalopathy lactic acidosis, and stroke-like episodes), with an A to G transition (A3243G) within the tRNA gene in the mTERF binding site [79]. Another point mutation, A8344G, within the tRNA gene, is associated with MERRF syndrome (myoclonus epilepsy and ragged-red fibers), Ragged-red fibers (RRF) are a key indicator of mitochondrial dysfunction in skeletal muscle [80, 81].
3. Point mutations affecting protein-coding genes
Point mutations like T8993G/C and T9176G/C in human mtDNA convert a conserved leucine to an arginine or proline near the C-terminus of subunit 6 of ATP synthase. If the percentage of T8993G mtDNA in the blood exceeds 70%, NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa) develops [82]. Other point mutations occur in mitochondrial genes encoding subunits of complex I like G11778A in ND4, G3460A in ND1, and T14884C in ND6. These mutations are associated with LHON and Leigh syndrome [83, 84].
Nuclear DNA defects
Most of the 1500 mitochondrial proteins are encoded in nuclear DNA, and for that reason defects in nuclear genes can affect mainly mitochondrial metabolism and OXPHOS [85, 86].
1. Defects in the replication proteins
Autosomal dominant progressive external ophthalmoplegia (adPEO) is characterized by multiple large deletions of mtDNA in the ocular, skeletal muscles as well as brain and heart [87–90].
Mutations in the POLG1 gene produced chromosome 15-linked adPEO. Amino-acid position 955 of POLG1, located in the C-domain, is the most frequent mutation site [90, 91].
Chromosome 10-linked adPEO is caused by mutations in Twinkle, a DNA helicase. Mutations include various missense mutations and an in-frame duplication of 13 amino-acid (dup352–364) [92].
Mutations in adenine nucleotide translocator 1 (ANT1) cause Chromosome 4-linked adPEO. It is produced by a substitution of proline for alanine at position 114 in ANT1 protein [93].
2. Thymidine Kinase 2 (TK2) and Deoxyguanosine Kinase (dGK)
Mutations in the genes encoding these proteins are associated with mtDNA depletion syndrome (MDS). Both TK2 and dGK are required for the synthesis of deoxyribonucleoside triphosphates (dNTPs). Patients develop tissue-specific depletion of mtDNA, with its level in the liver dropping to less than 10% of normal [94, 95].
3. Thymidine Phosphorylase (TP)
Mutations in this gene are associated with depletion and/or multiple deletions of mtDNA in the muscles of patients with MNGIE (Mitochondrial neurogastro-intestinal encephalomyopathy). Although TP is not in the mitochondria, it is implicated in the conversion of thymidine to phosphoribose and thymine. The mutations induce a decrease in enzyme activity, producing an accumulation of thymidine [96, 97].
mtDNA mutations and degenerative diseases
An increased accumulation of mtDNA mutations has also been found in a variety of age-related degenerative diseases, like patients with chronic coronary artery disease [49, 98]. Particularly in the case of neurodegenerative diseases; the central nervous system has intense metabolic requirements, reduced energy production can have a severe impact on neural functioning [99].
Damage to mtDNA could potentially result in bioenergetic dysfunction and consequently aberrant nerve function. Neurodegenerative diseases are associated with a progressive loss of neurons through apoptosis and/or necrosis [5]. An accumulation of mutations and deletions in mtDNA with corresponding defects in energy metabolism have been found in Parkinson’s disease (PD), Alzheimer disease (AD), Amyotrophic Lateral Sclerosis (ALS), and Huntingon’s disease (HD) (Table 2) [3, 6, 100].
Table 2.
Neurodegenerative diseases. Mutated genes related with mitochondrial function in neurodegenerative diseases.
| DISEASE | MUTATION |
|---|---|
| Parkinson | 11778 |
| 10398A | |
| PINK | |
| Parkin | |
| α-synuclein | |
|
| |
| Alzheimer | 4977 |
| 10398A | |
| T414G | |
| T477C | |
| Amyloid Precursor Proteins (APP) | |
| Presenilin | |
| Amyotrophic Lateral Sclerosis | Cu/ZnSOD |
Idiopathic PD is associated with mitochondrial defects, particularly in respiratory complex I. Also, the detection of the common LHON np 11,778 mutation as well as the 10398A mutation indicates a mitochondrial etiology of PD [6, 101, 102]. Loss-of-function mutations in the PTEN-induced kinase 1 (PINK1), or Parkin genes, which encode a mitochondrially localized serine/threonine kinase and an ubiquitin-protein ligase respectively, result in recessive familial forms of Parkinsonism. These proteins maintain mitochondrial integrity by regulating diverse aspects of mitochondrial function [103, 104]. The rare autosomal dominant form of PD is due to mutations in α-synuclein, which produce an alteration in mitochondrial function [105–107].
Similarly to PD, AD also appears to involve mitochondrial dysfunction. The 4977 bp and 10398A common mutations have been shown to be elevated in Alzheimer’s brain tissue [102, 108]. Also, heteroplasmic levels of the T414G and T477C mutations were found in the brain tissues of Alzheimer’s patients [109]. Patients with these mutations exhibit reduction in the mtDNA L-strand ND6 transcript and the mtDNA/nuclear DNA ratio, suggesting that these bases may play an important role in genome maintenance and/or expression. The identification of familial and inherited forms of AD as autosomal-dominant disorders linked to mutations in the gene that encodes the amyloid precursor protein (APP), or genes that encode presenilin proteins [110–115] display a relationship to mitochondrial dysfunctions.
ALS is a neurodegenerative disease that specifically affects the motor neurons. In familial cases, ALS is caused by mutations in the Cu/ZnSOD (SOD1). This familial form appears to be due to a failure in the SOD1’s ability to detoxify mitochondrial superoxide anions, which are released into the mitochondrial inner membrane space from ubisemiquinone bound to the Q0 site of complex III [6, 116].
mtDNA mutations and cancer
Cancer cells produce ATP through glycolysis and lactic acid fermentation rather than oxidative phosphorylation [117]. For that reason, it is suggested that mitochondria are involved in carcinogenesis through respiration alterations [118]. Indeed, an increase in mtDNA mutations has been observed in a variety of cancer types: prostate, thyroid, oral cancer, vulvar cancer, hepatocellular carcinoma [119], colon, bladder, head and neck, lung and a number of blood cancers [120–122]. Mutations are often found in primary tumors, but not in surrounding tissues. The main characteristics of mtDNA mutations common to all tumor types are: the majority of the mutations are base substitutions; mutations occur in all protein-coding mitochondrial genes; the D-loop region is the most frequent site of somatic mutations across tumor types; the mutations are homoplasmic in nature [123].
Deletions, point mutations, insertions, and duplications are reported in many kinds of cancer [124]. The homoplasmic nature of mutated mtDNA suggests the possibility that some mutations are involved in tumorigenesis by affecting energy metabolism and/or ROS production. For example, some studies have observed decreased nuclear and mitochondrial hOGG1 expression in human lung cancer [125]. Moreover, mitochondria play a key role in apoptosis. Another study showed that specific point mutations in mtDNA accelerate growth and reduce apoptosis in a variety of tumors, supporting the idea that some mtDNA mutations in tumors have functional advantages that promote tumor growth [126].
mtDNA mutations in forensic science. Estimation of age-at-death
One of the goals of forensic investigation is to estimate the age-at-death of a single individual in order to identify a victim. However, it is often difficult to determine adult age. Although there are several anthropological methods to determine age-at-death, the new developed methodologies are based in the natural process of aging, which leads to alterations of tissues and organs on different biochemical levels [127]. One of these alterations is the mtDNA mutations.
Few authors tried to find a correlation between mitochondrial mutations and age in order to apply to forensic practice. The studies of Meissner et al. [128, 129] in skeletal muscle showed a correlation between the common mutation 4977 bp and age. Their calculation allowed a rough estimation of the age-at-death and can discriminate between young and elderly persons. Moreover, this methodology, using Polymerase Chain Reaction, can be used in tissue that has undergone extensive putrefaction.
The studies of Theves et al. [58] analyzing the A189G mutations were developed using three different approaches: automated DNA sequencing, Southern blot hybridization using a digoxigenin-labeled oligonucleotide probe, and peptide nucleic acid (PNA)/real-time PCR, due to the fact that this method is more sensitive than DNA sequencing. They demonstrated the accumulation of this mutation in mitotic buccal cells and postmitotic muscle tissue reached very high levels in individual 60 years of age or older. Lacan et al. [130] demonstrated with the same methodology that this somatic point mutation occurs in bone tissue and it is related to age.
Other authors looked for disease mutations like the transition mutation in MERRF syndrome. Munscher et al. [51], using PCR methodology, found an association of this mutation with age in postmortem specimens of extraocular and skeletal muscle from healthy people. Other authors looked for general deletions in mitochondrial DNA. Papiha et al. [131] analyzed blood and bone samples from patients who had undergone orthopaedic surgery. They only found mtDNA deletion in bone from patients up to 70 years old with osteoporosis/rheumatoid arthritis, but not in the blood. In wisdom teeth from healthy subjects, Mornstad et al. [132], used the PCR amplification of hypervariable region 2 (HV2) of the mitochondrial D-loop to demonstrate a decrease in the amount of mtDNA in dentine with age. This decrease is highest in the oldest age groups.
Lacan et al. [133], using capillary electrophoresis, detected three types of miniduplications in bone and muscle samples. Three duplications were never observed in bone in the oldest individuals, but at least one can be detected in individuals over 38 years old. In contrast, the duplicated fragments have been observed in individuals around 20 years old and accumulated in the most aged individuals, carrying several duplications. It seems that strong tissue specificity exists for this type of rearrangement.
Although this methodology could be simple, easy and affordable for forensic laboratories and it is possible to use in tissue that has undergone putrefaction, further research is needed in order to apply this technique to forensic cases.
Haplogroups. Their role in aging, diseases and forensic sciences
A haplogroup is a particular mutation that is well established and widely distributed among individuals of populations [134]. Restriction fragment-length polymorphism studies of mtDNA from a wide range of human populations have revealed sets of ancestral mutations that define these haplogroups that have common ancestry and, because of uniparental inheritance, evolve independently from each other. Each of these haplogroups, which include evolutionary related types of mtDNAs, is defined by specific sets of associated mutations, thus allowing for a quick and precise classification of the mtDNA molecules within a certain population [135]. More than two dozen mtDNA haplogroups are known among human populations around the world [16]. The human mtDNA tree is rooted in Africa, and it has specific arms radiating into different geographic regions [6].
African mtDNAs are the most diverse and thus most ancient. African mtDNAs fall into four major haplogroups: L0 (oldest), L1, L2, and L3 (youngest). From them, different haplogroups were spawned due to migrations, into Europe and Asia, enriching the haplogroups. This phylogeographic distribution of mtDNA is a useful tool in forensic science to determine the geographic origin since 35% of the mutations were continent-specific and therefore useful for this purpose [6, 136–139].
Also, different studies have associated these variations in mtDNA with human longevity and aging. These studies describe associations between specific inherited mtDNA haplogroups (C150T polymorphism) and extended lifespan in Finish and Japanese subjects [140–142]. Also, haplogroup J is over-represented in the long-living and centenarian northern Italian males [135, 143]. This haplogroup is over-represented in Irish nonagenarians and centenarians and long-living Finish people [141, 144]. In contrast, this haplogroup is underrepresented in Chinese Uygur nonagenarians [145]. Haplogroups are also associated with the risk of diseases. Studies in Japanese centenarians and super-centenarians revealed that these people are resistant against diseases such as type 2 diabetes, myocardial and cerebrovascular infarction, Alzheimer disease (AD) and Parkinson disease (PD). In noncentenarians, mitochondrial haplogroups F and A exhibit risk factors for diabetes and haplogroup N9a was found to be protective against diabetes, especially in females [146, 147].
In Europeans, males with haplogroup U showed an increase in risk of AD, whereas this haplogroup decreased the risk in females. Haplogroup J has a decreased risk of PD versus individuals with the most common haplogroup H [148].
Different studies have described the risk of cancers associated with various haplogroups. Verma et al [149] analyzed 30 haplotypes and their relation with cancer in a Japanese hospital, finding that one of the haplogroups (M7b2) increased the risk for hemopoietic cancer, a risk factor for leukemia. In Northern Americans, the haplogroup U was associated with an increased risk of renal cancer [150]. Also in breast and esophageal cancers, the polymorphism mtG10398A in haplogroup N and its sublineages provides a risk toward this cancer in Indian populations [151].
The results of these studies support the idea that the effect of haplogroups and mtDNA variants on longevity and risk of diseases is population- and possibly sex-dependent, due to differences in genetic background.
Conclusions
Although it is not still clear if mtDNA mutations are the cause or consequence of aging, it is known that these mutations increase with age and play a key role in this process leading to mitochondrial dysfunction. As a result, mtDNA mutations are related to several diseases, particularly neurodegenerative diseases. Also, the alterations in the mitochondrial function due to mtDNA mutations seem to promote tumor growth via inhibition of apoptosis. mtDNA haplogroups can not only be used for the determination of geographical origin, but also different haplogroups can influence longevity and risk of disease. Further research is needed in this field in order to improve the understanding of the role of mtDNA mutation in aging, disease and its application to forensic science.
Acknowledgments
Sara C. Zapico is supported by a Peter Buck Postdoctoral Fellowship from the Smithsonian Institution. For editing the manuscript, the authors acknowledge Christian Thomas of the Smithsonian Institution.
References
- [1].Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome biology. 2001;2 doi: 10.1186/gb-2001-2-6-reviews1018. REVIEWS1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frey TG, Mannella CA. The internal structure of mitochondria. Trends in biochemical sciences. 2000;25:319–324. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- [3].DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. The New England journal of medicine. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- [4].Chinnery PF, Schon EA. Mitochondria. Journal of neurology, neurosurgery, and psychiatry. 2003;74:1188–1199. doi: 10.1136/jnnp.74.9.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mechanisms of ageing and development. 2008;129:383–390. doi: 10.1016/j.mad.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fridovich I. Superoxide radical and superoxide dismutases. Annual review of biochemistry. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- [8].Martin LJ. Biology of mitochondria in neurodegenerative diseases. Progress in molecular biology and translational science. 2012;107:355–415. doi: 10.1016/B978-0-12-385883-2.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends in cardiovascular medicine. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis : an international journal on programmed cell death. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- [11].D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature reviews Molecular cell biology. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- [12].Fogg VC, Lanning NJ, Mackeigan JP. Mitochondria in cancer: at the crossroads of life and death. Chinese journal of cancer. 2011;30:526–539. doi: 10.5732/cjc.011.10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- [14].Schatz G. The protein import system of mitochondria. The Journal of biological chemistry. 1996;271:31763–31766. doi: 10.1074/jbc.271.50.31763. [DOI] [PubMed] [Google Scholar]
- [15].Shoubridge EA. Nuclear genetic defects of oxidative phosphorylation. Human molecular genetics. 2001;10:2277–2284. doi: 10.1093/hmg/10.20.2277. [DOI] [PubMed] [Google Scholar]
- [16].Singh KK, Kulawiec M. Mitochondrial DNA polymorphism and risk of cancer. Methods Mol Biol. 2009;471:291–303. doi: 10.1007/978-1-59745-416-2_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gilkerson RW. Mitochondrial DNA nucleoids determine mitochondrial genetics and dysfunction. The international journal of biochemistry & cell biology. 2009;41:1899–1906. doi: 10.1016/j.biocel.2009.03.016. [DOI] [PubMed] [Google Scholar]
- [18].Kasamatsu H, Vinograd J. Replication of circular DNA in eukaryotic cells. Annual review of biochemistry. 1974;43:695–719. doi: 10.1146/annurev.bi.43.070174.003403. [DOI] [PubMed] [Google Scholar]
- [19].Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chemical reviews. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- [20].Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochimica et biophysica acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- [21].Yang MY, Bowmaker M, Reyes A, Vergani L, Angeli P, Gringeri E, Jacobs HT, Holt IJ. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell. 2002;111:495–505. doi: 10.1016/s0092-8674(02)01075-9. [DOI] [PubMed] [Google Scholar]
- [22].Driggers WJ, LeDoux SP, Wilson GL. Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. The Journal of biological chemistry. 1993;268:22042–22045. [PubMed] [Google Scholar]
- [23].Gredilla R. DNA damage and base excision repair in mitochondria and their role in aging. Journal of aging research. 2010;2011:257093. doi: 10.4061/2011/257093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- [25].Szczesny B, Hazra TK, Papaconstantinou J, Mitra S, Boldogh I. Age-dependent deficiency in import of mitochondrial DNA glycosylases required for repair of oxidatively damaged bases. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10670–10675. doi: 10.1073/pnas.1932854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics. 1996;36:449–458. doi: 10.1006/geno.1996.0490. [DOI] [PubMed] [Google Scholar]
- [27].Lakshmipathy U, Campbell C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Molecular and cellular biology. 1999;19:3869–3876. doi: 10.1128/mcb.19.5.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mason PA, Matheson EC, Hall AG, Lightowlers RN. Mismatch repair activity in mammalian mitochondria. Nucleic acids research. 2003;31:1052–1058. doi: 10.1093/nar/gkg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Thyagarajan B, Padua RA, Campbell C. Mammalian mitochondria possess homologous DNA recombination activity. The Journal of biological chemistry. 1996;271:27536–27543. doi: 10.1074/jbc.271.44.27536. [DOI] [PubMed] [Google Scholar]
- [30].Coffey G, Lakshmipathy U, Campbell C. Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic acids research. 1999;27:3348–3354. doi: 10.1093/nar/27.16.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- [32].Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Thompson LV. Oxidative stress, mitochondria and mtDNA-mutator mice. Experimental gerontology. 2006;41:1220–1222. doi: 10.1016/j.exger.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Melov S, Hinerfeld D, Esposito L, Wallace DC. Multi-organ characterization of mitochondrial genomic rearrangements in ad libitum and caloric restricted mice show striking somatic mitochondrial DNA rearrangements with age. Nucleic acids research. 1997;25:974–982. doi: 10.1093/nar/25.5.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- [37].Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- [38].Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- [40].Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- [42].Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- [43].Hebert SL, Lanza IR, Nair KS. Mitochondrial DNA alterations and reduced mitochondrial function in aging. Mechanisms of ageing and development. 2010;131:451–462. doi: 10.1016/j.mad.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shuster RC, Rubenstein AJ, Wallace DC. Mitochondrial DNA in anucleate human blood cells. Biochemical and biophysical research communications. 1988;155:1360–1365. doi: 10.1016/s0006-291x(88)81291-9. [DOI] [PubMed] [Google Scholar]
- [45].Rossignol R, Malgat M, Mazat JP, Letellier T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. The Journal of biological chemistry. 1999;274:33426–33432. doi: 10.1074/jbc.274.47.33426. [DOI] [PubMed] [Google Scholar]
- [46].Wallace DC. Mitochondrial DNA mutations in disease and aging. Environmental and molecular mutagenesis. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- [47].Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS genetics. 2007;3:e24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood) 2002;227:671–682. doi: 10.1177/153537020222700901. [DOI] [PubMed] [Google Scholar]
- [49].Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic acids research. 1990;18:6927–6933. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Soong NW, Hinton DR, Cortopassi G, Arnheim N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nature genetics. 1992;2:318–323. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- [51].Munscher C, Rieger T, Muller-Hocker J, Kadenbach B. The point mutation of mitochondrial DNA characteristic for MERRF disease is found also in healthy people of different ages. FEBS letters. 1993;317:27–30. doi: 10.1016/0014-5793(93)81484-h. [DOI] [PubMed] [Google Scholar]
- [52].Schwarze SR, Lee CM, Chung SS, Roecker EB, Weindruch R, Aiken JM. High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mechanisms of ageing and development. 1995;83:91–101. doi: 10.1016/0047-6374(95)01611-3. [DOI] [PubMed] [Google Scholar]
- [53].Khaidakov M, Heflich RH, Manjanatha MG, Myers MB, Aidoo A. Accumulation of point mutations in mitochondrial DNA of aging mice. Mutation research. 2003;526:1–7. doi: 10.1016/s0027-5107(03)00010-1. [DOI] [PubMed] [Google Scholar]
- [54].Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- [55].Calloway CD, Reynolds RL, Herrin GL, Jr, Anderson WW. The frequency of heteroplasmy in the HVII region of mtDNA differs across tissue types and increases with age. American journal of human genetics. 2000;66:1384–1397. doi: 10.1086/302844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Del Bo R, Bordoni A, Martinelli Boneschi F, Crimi M, Sciacco M, Bresolin N, Scarlato G, Comi GP. Evidence and age-related distribution of mtDNA D-loop point mutations in skeletal muscle from healthy subjects and mitochondrial patients. Journal of the neurological sciences. 2002;202:85–91. doi: 10.1016/s0022-510x(02)00247-2. [DOI] [PubMed] [Google Scholar]
- [57].Del Bo R, Crimi M, Sciacco M, Malferrari G, Bordoni A, Napoli L, Prelle A, Biunno I, Moggio M, Bresolin N, Scarlato G, Pietro Comi G. High mutational burden in the mtDNA control region from aged muscles: a single-fiber study. Neurobiology of aging. 2003;24:829–838. doi: 10.1016/s0197-4580(02)00233-6. [DOI] [PubMed] [Google Scholar]
- [58].Theves C, Keyser-Tracqui C, Crubezy E, Salles JP, Ludes B, Telmon N. Detection and quantification of the age-related point mutation A189G in the human mitochondrial DNA. Journal of forensic sciences. 2006;51:865–873. doi: 10.1111/j.1556-4029.2006.00163.x. [DOI] [PubMed] [Google Scholar]
- [59].McInerny SC, Brown AL, Smith DW. Region-specific changes in mitochondrial D-loop in aged rat CNS. Mechanisms of ageing and development. 2009;130:343–349. doi: 10.1016/j.mad.2009.01.008. [DOI] [PubMed] [Google Scholar]
- [60].Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nature genetics. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- [61].Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature genetics. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- [62].Cortopassi GA, Shibata D, Soong NW, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Melov S, Lithgow GJ, Fischer DR, Tedesco PM, Johnson TE. Increased frequency of deletions in the mitochondrial genome with age of Caenorhabditis elegans. Nucleic acids research. 1995;23:1419–1425. doi: 10.1093/nar/23.8.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Melov S, Shoffner JM, Kaufman A, Wallace DC. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic acids research. 1995;23:4122–4126. doi: 10.1093/nar/23.20.4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang C, Bills M, Quigley A, Maxwell RJ, Linnane AW, Nagley P. Varied prevalence of age-associated mitochondrial DNA deletions in different species and tissues: a comparison between human and rat. Biochemical and biophysical research communications. 1997;230:630–635. doi: 10.1006/bbrc.1996.6020. [DOI] [PubMed] [Google Scholar]
- [66].Meissner C, Bruse P, Mohamed SA, Schulz A, Warnk H, Storm T, Oehmichen M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Experimental gerontology. 2008;43:645–652. doi: 10.1016/j.exger.2008.03.004. [DOI] [PubMed] [Google Scholar]
- [67].de Souza-Pinto NC, Hogue BA, Bohr VA. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increase in mitochondrial but not in nuclear extracts. Free radical biology & medicine. 2001;30:916–923. doi: 10.1016/s0891-5849(01)00483-x. [DOI] [PubMed] [Google Scholar]
- [68].Hayakawa M, Torii K, Sugiyama S, Tanaka M, Ozawa T. Age-associated accumulation of 8-hydroxydeoxyguanosine in mitochondrial DNA of human diaphragm. Biochemical and biophysical research communications. 1991;179:1023–1029. doi: 10.1016/0006-291x(91)91921-x. [DOI] [PubMed] [Google Scholar]
- [69].Bodyak ND, Nekhaeva E, Wei JY, Khrapko K. Quantification and sequencing of somatic deleted mtDNA in single cells: evidence for partially duplicated mtDNA in aged human tissues. Human molecular genetics. 2001;10:17–24. doi: 10.1093/hmg/10.1.17. [DOI] [PubMed] [Google Scholar]
- [70].Nekhaeva E, Bodyak ND, Kraytsberg Y, McGrath SB, Van Orsouw NJ, Pluzhnikov A, Wei JY, Vijg J, Khrapko K. Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5521–5526. doi: 10.1073/pnas.072670199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Fayet G, Jansson M, Sternberg D, Moslemi AR, Blondy P, Lombes A, Fardeau M, Oldfors A. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscular disorders : NMD. 2002;12:484–493. doi: 10.1016/s0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- [72].Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. The journals of gerontology Series A, Biological sciences and medical sciences. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mao CC, Holt IJ. Clinical and molecular aspects of diseases of mitochondrial DNA instability. Chang Gung medical journal. 2009;32:354–369. [PubMed] [Google Scholar]
- [74].Ballinger SW, Shoffner JM, Hedaya EV, Trounce I, Polak MA, Koontz DA, Wallace DC. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nature genetics. 1992;1:11–15. doi: 10.1038/ng0492-11. [DOI] [PubMed] [Google Scholar]
- [75].Ballinger SW, Shoffner JM, Gebhart S, Koontz DA, Wallace DC. Mitochondrial diabetes revisited. Nature genetics. 1994;7:458–459. doi: 10.1038/ng0894-458. [DOI] [PubMed] [Google Scholar]
- [76].Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, Nakase H, Bonilla E, Werneck LC, Servidei S, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. The New England journal of medicine. 1989;320:1293–1299. doi: 10.1056/NEJM198905183202001. [DOI] [PubMed] [Google Scholar]
- [77].Schon EA, Rizzuto R, Moraes CT, Nakase H, Zeviani M, DiMauro S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science. 1989;244:346–349. doi: 10.1126/science.2711184. [DOI] [PubMed] [Google Scholar]
- [78].Mita S, Rizzuto R, Moraes CT, Shanske S, Arnaudo E, Fabrizi GM, Koga Y, DiMauro S, Schon EA. Recombination via flanking direct repeats is a major cause of large-scale deletions of human mitochondrial DNA. Nucleic acids research. 1990;18:561–567. doi: 10.1093/nar/18.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- [80].Hammans SR, Sweeney MG, Brockington M, Morgan-Hughes JA, Harding AE. Mitochondrial encephalopathies: molecular genetic diagnosis from blood samples. Lancet. 1991;337:1311–1313. doi: 10.1016/0140-6736(91)92981-7. [DOI] [PubMed] [Google Scholar]
- [81].Silvestri G, Ciafaloni E, Santorelli FM, Shanske S, Servidei S, Graf WD, Sumi M, DiMauro S. Clinical features associated with the A-->G transition at nucleotide 8344 of mtDNA (“MERRF mutation”) Neurology. 1993;43:1200–1206. doi: 10.1212/wnl.43.6.1200. [DOI] [PubMed] [Google Scholar]
- [82].Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. American journal of human genetics. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- [83].Solano A, Roig M, Vives-Bauza C, Hernandez-Pena J, Garcia-Arumi E, Playan A, Lopez-Perez MJ, Andreu AL, Montoya J. Bilateral striatal necrosis associated with a novel mutation in the mitochondrial ND6 gene. Annals of neurology. 2003;54:527–530. doi: 10.1002/ana.10682. [DOI] [PubMed] [Google Scholar]
- [84].Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- [85].Sue CM, Schon EA. Mitochondrial respiratory chain diseases and mutations in nuclear DNA: a promising start? Brain Pathol. 2000;10:442–450. doi: 10.1111/j.1750-3639.2000.tb00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Smeitink J, van den Heuvel L. Human mitochondrial complex I in health and disease. American journal of human genetics. 1999;64:1505–1510. doi: 10.1086/302432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Suomalainen A, Majander A, Haltia M, Somer H, Lonnqvist J, Savontaus ML, Peltonen L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. The Journal of clinical investigation. 1992;90:61–66. doi: 10.1172/JCI115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zeviani M, Bresolin N, Gellera C, Bordoni A, Pannacci M, Amati P, Moggio M, Servidei S, Scarlato G, DiDonato S. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: a new autosomal dominant disease. American journal of human genetics. 1990;47:904–914. [PMC free article] [PubMed] [Google Scholar]
- [89].Suomalainen A, Majander A, Wallin M, Setala K, Kontula K, Leinonen H, Salmi T, Paetau A, Haltia M, Valanne L, Lonnqvist J, Peltonen L, Somer H. Autosomal dominant progressive external ophthalmoplegia with multiple deletions of mtDNA: clinical, biochemical, and molecular genetic features of the 10q-linked disease. Neurology. 1997;48:1244–1253. doi: 10.1212/wnl.48.5.1244. [DOI] [PubMed] [Google Scholar]
- [90].Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature genetics. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- [91].Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, Comi GP, Zeviani M. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Annals of neurology. 2002;52:211–219. doi: 10.1002/ana.10278. [DOI] [PubMed] [Google Scholar]
- [92].Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nature genetics. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- [93].Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- [94].Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nature genetics. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- [95].Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nature genetics. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- [96].Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- [97].Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, Pela I, Zammarchi E, Donati MA, Oliver JA, Hirano M. Altered thymidine metabolism due to defects of thymidine phosphorylase. The Journal of biological chemistry. 2002;277:4128–4133. doi: 10.1074/jbc.M111028200. [DOI] [PubMed] [Google Scholar]
- [98].Corral-Debrinski M, Stepien G, Shoffner JM, Lott MT, Kanter K, Wallace DC. Hypoxemia is associated with mitochondrial DNA damage and gene induction. Implications for cardiac disease. JAMA : the journal of the American Medical Association. 1991;266:1812–1816. [PubMed] [Google Scholar]
- [99].Kennedy SR, Loeb LA, Herr AJ. Somatic mutations in aging, cancer and neurodegeneration. Mechanisms of ageing and development. 2012;133:118–126. doi: 10.1016/j.mad.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kang D, Hamasaki N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Current medicinal chemistry. 2005;12:429–441. doi: 10.2174/0929867053363081. [DOI] [PubMed] [Google Scholar]
- [101].Simon DK, Pulst SM, Sutton JP, Browne SE, Beal MF, Johns DR. Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology. 1999;53:1787–1793. doi: 10.1212/wnl.53.8.1787. [DOI] [PubMed] [Google Scholar]
- [102].van der Walt JM, Nicodemus KK, Martin ER, Scott WK, Nance MA, Watts RL, Hubble JP, Haines JL, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, Hiner BC, Jankovic J, Ondo WG, Allen FH, Jr, Goetz CG, Small GW, Mastaglia F, Stajich JM, McLaurin AC, Middleton LT, Scott BL, Schmechel DE, Pericak-Vance MA, Vance JM. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. American journal of human genetics. 2003;72:804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: a mitochondrial quality control system? Journal of bioenergetics and biomembranes. 2009;41:499–503. doi: 10.1007/s10863-009-9253-3. [DOI] [PubMed] [Google Scholar]
- [105].Orth M, Tabrizi SJ, Schapira AH, Cooper JM. Alpha-synuclein expression in HEK293 cells enhances the mitochondrial sensitivity to rotenone. Neuroscience letters. 2003;351:29–32. doi: 10.1016/s0304-3940(03)00941-8. [DOI] [PubMed] [Google Scholar]
- [106].Lee HJ, Lee SJ. Characterization of cytoplasmic alpha-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. The Journal of biological chemistry. 2002;277:48976–48983. doi: 10.1074/jbc.M208192200. [DOI] [PubMed] [Google Scholar]
- [107].Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. The American journal of pathology. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, Graham BH, Wallace DC. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23:471–476. doi: 10.1006/geno.1994.1525. [DOI] [PubMed] [Google Scholar]
- [109].Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- [111].Tilley L, Morgan K, Kalsheker N. Genetic risk factors in Alzheimer’s disease. Molecular pathology : MP. 1998;51:293–304. doi: 10.1136/mp.51.6.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- [113].Naruse S, Igarashi S, Kobayashi H, Aoki K, Inuzuka T, Kaneko K, Shimizu T, Iihara K, Kojima T, Miyatake T, et al. Mis-sense mutation Val----Ile in exon 17 of amyloid precursor protein gene in Japanese familial Alzheimer’s disease. Lancet. 1991;337:978–979. doi: 10.1016/0140-6736(91)91612-x. [DOI] [PubMed] [Google Scholar]
- [114].Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, Martin C, Moreau V, Charbonnier F, Didierjean O, Tardieu S, et al. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Human molecular genetics. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- [115].Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- [116].Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta neuropathologica. 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- [117].Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- [118].Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- [119].Czarnecka AM, Bartnik E. The role of the mitochondrial genome in ageing and carcinogenesis. Journal of aging research. 2011;2011:136435. doi: 10.4061/2011/136435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Copeland WC, Wachsman JT, Johnson FM, Penta JS. Mitochondrial DNA alterations in cancer. Cancer investigation. 2002;20:557–569. doi: 10.1081/cnv-120002155. [DOI] [PubMed] [Google Scholar]
- [121].Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287:2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- [122].Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nature genetics. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- [123].Carew JS, Huang P. Mitochondrial defects in cancer. Molecular cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, Wu CW, Chi CW, Tam TN, Wei YH. Mitochondrial genome instability and mtDNA depletion in human cancers. Annals of the New York Academy of Sciences. 2005;1042:109–122. doi: 10.1196/annals.1338.011. [DOI] [PubMed] [Google Scholar]
- [125].Mambo E, Chatterjee A, de Souza-Pinto NC, Mayard S, Hogue BA, Hoque MO, Dizdaroglu M, Bohr VA, Sidransky D. Oxidized guanine lesions and hOgg1 activity in lung cancer. Oncogene. 2005;24:4496–4508. doi: 10.1038/sj.onc.1208669. [DOI] [PubMed] [Google Scholar]
- [126].Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer research. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- [127].S CZ, Ubelaker DH. Applications of physiological bases of ageing to forensic sciences. Estimation of age-at-death. Ageing research reviews. 2013;12:605–617. doi: 10.1016/j.arr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- [128].Meissner C, von Wurmb N, Oehmichen M. Detection of the age-dependent 4977 bp deletion of mitochondrial DNA. A pilot study International journal of legal medicine. 1997;110:288–291. doi: 10.1007/s004140050089. [DOI] [PubMed] [Google Scholar]
- [129].Meissner C, von Wurmb N, Schimansky B, Oehmichen M. Estimation of age at death based on quantitation of the 4977-bp deletion of human mitochondrial DNA in skeletal muscle. Forensic science international. 1999;105:115–124. doi: 10.1016/s0379-0738(99)00126-7. [DOI] [PubMed] [Google Scholar]
- [130].Lacan M, Theves C, Amory S, Keyser C, Crubezy E, Salles JP, Ludes B, Telmon N. Detection of the A189G mtDNA heteroplasmic mutation in relation to age in modern and ancient bones. International journal of legal medicine. 2009;123:161–167. doi: 10.1007/s00414-008-0266-y. [DOI] [PubMed] [Google Scholar]
- [131].Papiha SS, Rathod H, Briceno I, Pooley J, Datta HK. Age related somatic mitochondrial DNA deletions in bone. Journal of clinical pathology. 1998;51:117–120. doi: 10.1136/jcp.51.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Mornstad H, Pfeiffer H, Yoon C, Teivens A. Demonstration and semi-quantification of mtDNA from human dentine and its relation to age. International journal of legal medicine. 1999;112:98–100. doi: 10.1007/s004140050209. [DOI] [PubMed] [Google Scholar]
- [133].Lacan M, Theves C, Keyser C, Farrugia A, Baraybar JP, Crubezy E, Ludes B. Detection of age-related duplications in mtDNA from human muscles and bones. International journal of legal medicine. 2011;125:293–300. doi: 10.1007/s00414-010-0440-x. [DOI] [PubMed] [Google Scholar]
- [134].Malhi RS, Mortensen HM, Eshleman JA, Kemp BM, Lorenz JG, Kaestle FA, Johnson JR, Gorodezky C, Smith DG. Native American mtDNA prehistory in the American Southwest. American journal of physical anthropology. 2003;120:108–124. doi: 10.1002/ajpa.10138. [DOI] [PubMed] [Google Scholar]
- [135].De Benedictis G, Rose G, Carrieri G, De Luca M, Falcone E, Passarino G, Bonafe M, Monti D, Baggio G, Bertolini S, Mari D, Mattace R, Franceschi C. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1999;13:1532–1536. doi: 10.1096/fasebj.13.12.1532. [DOI] [PubMed] [Google Scholar]
- [136].Quintans B, Alvarez-Iglesias V, Salas A, Phillips C, Lareu MV, Carracedo A. Typing of mitochondrial DNA coding region SNPs of forensic and anthropological interest using SNaPshot minisequencing. Forensic science international. 2004;140:251–257. doi: 10.1016/j.forsciint.2003.12.005. [DOI] [PubMed] [Google Scholar]
- [137].Nelson TM, Just RS, Loreille O, Schanfield MS, Podini D. Development of a multiplex single base extension assay for mitochondrial DNA haplogroup typing. Croatian medical journal. 2007;48:460–472. [PMC free article] [PubMed] [Google Scholar]
- [138].Nunez C, Sosa C, Baeta M, Geppert M, Turnbough M, Phillips N, Casalod Y, Bolea M, Roby R, Budowle B, Martinez-Jarreta B. Genetic analysis of 7 medieval skeletons from the Aragonese Pyrenees. Croatian medical journal. 2011;52:336–343. doi: 10.3325/cmj.2011.52.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Mielnik-Sikorska M, Daca P, Malyarchuk B, Derenko M, Skonieczna K, Perkova M, Dobosz T, Grzybowski T. The history of Slavs inferred from complete mitochondrial genome sequences. PloS one. 2013;8:e54360. doi: 10.1371/journal.pone.0054360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Niemi AK, Moilanen JS, Tanaka M, Hervonen A, Hurme M, Lehtimaki T, Arai Y, Hirose N, Majamaa K. A combination of three common inherited mitochondrial DNA polymorphisms promotes longevity in Finnish and Japanese subjects. European journal of human genetics : EJHG. 2005;13:166–170. doi: 10.1038/sj.ejhg.5201308. [DOI] [PubMed] [Google Scholar]
- [141].Niemi AK, Hervonen A, Hurme M, Karhunen PJ, Jylha M, Majamaa K. Mitochondrial DNA polymorphisms associated with longevity in a Finnish population. Human genetics. 2003;112:29–33. doi: 10.1007/s00439-002-0843-y. [DOI] [PubMed] [Google Scholar]
- [142].Tanaka M, Takeyasu T, Fuku N, Li-Jun G, Kurata M. Mitochondrial genome single nucleotide polymorphisms and their phenotypes in the Japanese. Annals of the New York Academy of Sciences. 2004;1011:7–20. doi: 10.1007/978-3-662-41088-2_2. [DOI] [PubMed] [Google Scholar]
- [143].Chen A, Raule N, Chomyn A, Attardi G. Decreased reactive oxygen species production in cells with mitochondrial haplogroups associated with longevity. PloS one. 2012;7:e46473. doi: 10.1371/journal.pone.0046473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ross OA, McCormack R, Curran MD, Duguid RA, Barnett YA, Rea IM, Middleton D. Mitochondrial DNA polymorphism: its role in longevity of the Irish population. Experimental gerontology. 2001;36:1161–1178. doi: 10.1016/s0531-5565(01)00094-8. [DOI] [PubMed] [Google Scholar]
- [145].Ren WH, Li XH, Zhang HG, Deng FM, Liao WQ, Pang Y, Liu YH, Qiu MJ, Zhang GY, Zhang YG. Mitochondrial DNA haplogroups in a Chinese Uygur population and their potential association with longevity. Clinical and experimental pharmacology & physiology. 2008;35:1477–1481. doi: 10.1111/j.1440-1681.2008.05028.x. [DOI] [PubMed] [Google Scholar]
- [146].Fuku N, Park KS, Yamada Y, Nishigaki Y, Cho YM, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Nozawa Y, Lee HK, Tanaka M. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. American journal of human genetics. 2007;80:407–415. doi: 10.1086/512202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Alexe G, Fuku N, Bilal E, Ueno H, Nishigaki Y, Fujita Y, Ito M, Arai Y, Hirose N, Bhanot G, Tanaka M. Enrichment of longevity phenotype in mtDNA haplogroups D4b2b, D4a, and D5 in the Japanese population. Human genetics. 2007;121:347–356. doi: 10.1007/s00439-007-0330-6. [DOI] [PubMed] [Google Scholar]
- [148].van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, Schmechel DE, Murali Doraiswamy P, Gilbert JR, Haines JL, Vance JM, Pericak-Vance MA. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neuroscience letters. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- [149].Verma M, Naviaux RK, Tanaka M, Kumar D, Franceschi C, Singh KK. Meeting report: mitochondrial DNA and cancer epidemiology. Cancer research. 2007;67:437–439. doi: 10.1158/0008-5472.CAN-06-4119. [DOI] [PubMed] [Google Scholar]
- [150].Booker LM, Habermacher GM, Jessie BC, Sun QC, Baumann AK, Amin M, Lim SD, Fernandez-Golarz C, Lyles RH, Brown MD, Marshall FF, Petros JA. North American white mitochondrial haplogroups in prostate and renal cancer. The Journal of urology. 2006;175:468–472. doi: 10.1016/S0022-5347(05)00163-1. discussion 472–463. [DOI] [PubMed] [Google Scholar]
- [151].Darvishi K, Sharma S, Bhat AK, Rai E, Bamezai RN. Mitochondrial DNA G10398A polymorphism imparts maternal Haplogroup N a risk for breast and esophageal cancer. Cancer letters. 2007;249:249–255. doi: 10.1016/j.canlet.2006.09.005. [DOI] [PubMed] [Google Scholar]