

Significance
STAR syndrome is an X-linked dominant developmental disorder caused by mutations in FAM58A, which codes for an orphan cyclin with undescribed functions. Here we demonstrate that cyclin M interacts with CDK10 (one of the last orphan CDKs) to form a novel cyclin-dependent kinase. CDK10 is known to be involved in the control of cell division and in the resistance of certain breast cancers to endocrine therapy. We show that CDK10/cyclin M phosphorylates and positively regulates the degradation of ETS2, a transcription factor that plays key roles in cancer and development. These results shed light on the molecular mechanisms underlying STAR syndrome, and they pave the way for the exploration of the functions of the CDK10/cyclin M kinase.
Abstract
Cyclin-dependent kinases (CDKs) regulate a variety of fundamental cellular processes. CDK10 stands out as one of the last orphan CDKs for which no activating cyclin has been identified and no kinase activity revealed. Previous work has shown that CDK10 silencing increases ETS2 (v-ets erythroblastosis virus E26 oncogene homolog 2)-driven activation of the MAPK pathway, which confers tamoxifen resistance to breast cancer cells. The precise mechanisms by which CDK10 modulates ETS2 activity, and more generally the functions of CDK10, remain elusive. Here we demonstrate that CDK10 is a cyclin-dependent kinase by identifying cyclin M as an activating cyclin. Cyclin M, an orphan cyclin, is the product of FAM58A, whose mutations cause STAR syndrome, a human developmental anomaly whose features include toe syndactyly, telecanthus, and anogenital and renal malformations. We show that STAR syndrome-associated cyclin M mutants are unable to interact with CDK10. Cyclin M silencing phenocopies CDK10 silencing in increasing c-Raf and in conferring tamoxifen resistance to breast cancer cells. CDK10/cyclin M phosphorylates ETS2 in vitro, and in cells it positively controls ETS2 degradation by the proteasome. ETS2 protein levels are increased in cells derived from a STAR patient, and this increase is attributable to decreased cyclin M levels. Altogether, our results reveal an additional regulatory mechanism for ETS2, which plays key roles in cancer and development. They also shed light on the molecular mechanisms underlying STAR syndrome.
Cyclin-dependent kinases (CDKs) play a pivotal role in the control of a number of fundamental cellular processes (1). The human genome contains 21 genes encoding proteins that can be considered as members of the CDK family owing to their sequence similarity with bona fide CDKs, those known to be activated by cyclins (2). Although discovered almost 20 y ago (3, 4), CDK10 remains one of the two CDKs without an identified cyclin partner. This knowledge gap has largely impeded the exploration of its biological functions. CDK10 can act as a positive cell cycle regulator in some cells (5, 6) or as a tumor suppressor in others (7, 8). CDK10 interacts with the ETS2 (v-ets erythroblastosis virus E26 oncogene homolog 2) transcription factor and inhibits its transcriptional activity through an unknown mechanism (9). CDK10 knockdown derepresses ETS2, which increases the expression of the c-Raf protein kinase, activates the MAPK pathway, and induces resistance of MCF7 cells to tamoxifen (6).
Here, we deorphanize CDK10 by identifying cyclin M, the product of FAM58A, as a binding partner. Mutations in this gene that predict absence or truncation of cyclin M are associated with STAR syndrome, whose features include toe syndactyly, telecanthus, and anogenital and renal malformations in heterozygous females (10). However, both the functions of cyclin M and the pathogenesis of STAR syndrome remain unknown. We show that a recombinant CDK10/cyclin M heterodimer is an active protein kinase that phosphorylates ETS2 in vitro. Cyclin M silencing phenocopies CDK10 silencing in increasing c-Raf and phospho-ERK expression levels and in inducing tamoxifen resistance in estrogen receptor (ER)+ breast cancer cells. We show that CDK10/cyclin M positively controls ETS2 degradation by the proteasome, through the phosphorylation of two neighboring serines. Finally, we detect an increased ETS2 expression level in cells derived from a STAR patient, and we demonstrate that it is attributable to the decreased cyclin M expression level observed in these cells.
Results
A yeast two-hybrid (Y2H) screen unveiled an interaction signal between CDK10 and a mouse protein whose C-terminal half presents a strong sequence homology with the human FAM58A gene product [whose proposed name is cyclin M (11)]. We thus performed Y2H mating assays to determine whether human CDK10 interacts with human cyclin M (Fig. 1 A–C). The longest CDK10 isoform (P1) expressed as a bait protein produced a strong interaction phenotype with full-length cyclin M (expressed as a prey protein) but no detectable phenotype with cyclin D1, p21 (CIP1), and Cdi1 (KAP), which are known binding partners of other CDKs (Fig. 1B). CDK1 and CDK3 also produced Y2H signals with cyclin M, albeit notably weaker than that observed with CDK10 (Fig. 1B). An interaction phenotype was also observed between full-length cyclin M and CDK10 proteins expressed as bait and prey, respectively (Fig. S1A). We then tested different isoforms of CDK10 and cyclin M originating from alternative gene splicing, and two truncated cyclin M proteins corresponding to the hypothetical products of two mutated FAM58A genes found in STAR syndrome patients (10). None of these shorter isoforms produced interaction phenotypes (Fig. 1 A and C and Fig. S1A).
Fig. 1.

CDK10 and cyclin M form an interaction complex. (A) Schematic representation of the different protein isoforms analyzed by Y2H assays. Amino acid numbers are indicated. Black boxes indicate internal deletions. The red box indicates a differing amino acid sequence compared with CDK10 P1. (B) Y2H assay between a set of CDK proteins expressed as baits (in fusion to the LexA DNA binding domain) and CDK interacting proteins expressed as preys (in fusion to the B42 transcriptional activator). pEG202 and pJG4-5 are the empty bait and prey plasmids expressing LexA and B42, respectively. lacZ was used as a reporter gene, and blue yeast are indicative of a Y2H interaction phenotype. (C) Y2H assay between the different CDK10 and cyclin M isoforms. The amino-terminal region of ETS2, known to interact with CDK10 (9), was also assayed. (D) Western blot analysis of Myc-CDK10 (wt or kd) and CycM-V5-6His expression levels in transfected HEK293 cells. (E) Western blot analysis of Myc-CDK10 (wt or kd) immunoprecipitates obtained using the anti-Myc antibody. “Inputs” correspond to 10 μg total lysates obtained from HEK293 cells coexpressing Myc-CDK10 (wt or kd) and CycM-V5-6His. (F) Western blot analysis of immunoprecipitates obtained using the anti-CDK10 antibody or a control goat antibody, from human breast cancer MCF7 cells. “Input” corresponds to 30 μg MCF7 total cell lysates. The lower band of the doublet observed on the upper panel comigrates with the exogenously expressed untagged CDK10 and thus corresponds to endogenous CDK10. The upper band of the doublet corresponds to a nonspecific signal, as demonstrated by it insensitivity to either overexpression of CDK10 (as seen on the left lane) or silencing of CDK10 (Fig. S2B). Another experiment with a longer gel migration is shown in Fig. S1D.
Next we examined the ability of CDK10 and cyclin M to interact when expressed in human cells (Fig. 1 D and E). We tested wild-type CDK10 (wt) and a kinase dead (kd) mutant bearing a D181A amino acid substitution that abolishes ATP binding (12). We expressed cyclin M-V5-6His and/or Myc-CDK10 (wt or kd) in a human embryonic kidney cell line (HEK293). The expression level of cyclin M-V5-6His was significantly increased upon coexpression with Myc-CDK10 (wt or kd) and, to a lesser extent, that of Myc-CDK10 (wt or kd) was increased upon coexpression with cyclin M-V5-6His (Fig. 1D). We then immunoprecipitated Myc-CDK10 proteins and detected the presence of cyclin M in the CDK10 (wt) and (kd) immunoprecipitates only when these proteins were coexpressed pair-wise (Fig. 1E). We confirmed these observations by detecting the presence of Myc-CDK10 in cyclin M-V5-6His immunoprecipitates (Fig. S1B). These experiments confirmed the lack of robust interaction between the CDK10.P2 isoform and cyclin M (Fig. S1C). To detect the interaction between endogenous proteins, we performed immunoprecipitations on nontransfected MCF7 cells derived from a human breast cancer. CDK10 and cyclin M antibodies detected their cognate endogenous proteins by Western blotting. We readily detected cyclin M in immunoprecipitates obtained with the CDK10 antibody but not with a control antibody (Fig. 1F). These results confirm the physical interaction between CDK10 and cyclin M in human cells.
To unveil a hypothesized CDK10/cyclin M protein kinase activity, we produced GST-CDK10 and StrepII-cyclin M fusion proteins in insect cells, either individually or in combination. We observed that GST-CDK10 and StrepII-cyclin M copurified, thus confirming their interaction in yet another cellular model (Fig. 2A). We then performed in vitro kinase assays with purified proteins, using histone H1 as a generic substrate. Histone H1 phosphorylation was detected only from lysates of cells coexpressing GST-CDK10 and StrepII-cyclin M. No phosphorylation was detected when GST-CDK10 or StrepII-cyclin M were expressed alone, or when StrepII-cyclin M was coexpressed with GST-CDK10(kd) (Fig. 2A). Next we investigated whether ETS2, which is known to interact with CDK10 (9) (Fig. 1C), is a phosphorylation substrate of CDK10/cyclin M. We detected strong phosphorylation of ETS2 by the GST-CDK10/StrepII-cyclin M purified heterodimer, whereas no phosphorylation was detected using GST-CDK10 alone or GST-CDK10(kd)/StrepII-cyclin M heterodimer (Fig. 2B).
Fig. 2.

CDK10 is a cyclin M-dependent protein kinase. (A) In vitro protein kinase assay on histone H1. Lysates from insect cells expressing different proteins were purified on a glutathione Sepharose matrix to capture GST-CDK10(wt or kd) fusion proteins alone, or in complex with STR-CycM fusion protein. Purified protein expression levels were analyzed by Western blots (Top and Upper Middle). The kinase activity was determined by autoradiography of histone H1, whose added amounts were visualized by Coomassie staining (Lower Middle and Bottom). (B) Same as in A, using purified recombinant 6His-ETS2 as a substrate.
CDK10 silencing has been shown to increase ETS2-driven c-RAF transcription and to activate the MAPK pathway (6). We investigated whether cyclin M is also involved in this regulatory pathway. To aim at a highly specific silencing, we used siRNA pools (mix of four different siRNAs) at low final concentration (10 nM). Both CDK10 and cyclin M siRNA pools silenced the expression of their cognate targets (Fig. 3 A and C and Fig. S2) and, interestingly, the cyclin M siRNA pool also caused a marked decrease in CDK10 protein level (Fig. 3A and Fig. S2B). These results, and those shown in Fig. 1D, suggest that cyclin M binding stabilizes CDK10. Cyclin M silencing induced an increase in c-Raf protein and mRNA levels (Fig. 3 B and C) and in phosphorylated ERK1 and ERK2 protein levels (Fig. S3B), similarly to CDK10 silencing. As expected from these effects (6), CDK10 and cyclin M silencing both decreased the sensitivity of ER+ MCF7 cells to tamoxifen, to a similar extent. The combined silencing of both genes did not result in a higher resistance to the drug (Fig. S3C). Altogether, these observations demonstrate a functional interaction between cyclin M and CDK10, which negatively controls ETS2.
Fig. 3.

Cyclin M silencing up-regulates c-Raf expression. (A) Western blot analysis of endogenous CDK10 and cyclin M expression levels in MCF7 cells, in response to siRNA-mediated gene silencing. (B) Western blot analysis of endogenous c-Raf expression levels in MCF7 cells, in response to CDK10 or cyclin M silencing. A quantification is shown in Fig. S3A. (C) Quantitative RT-PCR analysis of CDK10, cyclin M, and c-Raf mRNA levels, in response to CDK10 (Upper) or cyclin M (Lower) silencing. **P ≤ 0.01; ***P ≤ 0.001.
We then wished to explore the mechanism by which CDK10/cyclin M controls ETS2. ETS2 is a short-lived protein degraded by the proteasome (13). A straightforward hypothesis is that CDK10/cyclin M positively controls ETS2 degradation. We thus examined the impact of CDK10 or cyclin M silencing on ETS2 expression levels. The silencing of CDK10 and that of cyclin M caused an increase in the expression levels of an exogenously expressed Flag-ETS2 protein (Fig. S4A), as well as of the endogenous ETS2 protein (Fig. 4A). This increase is not attributable to increased ETS2 mRNA levels, which marginally fluctuated in response to CDK10 or cyclin M silencing (Fig. S4B). We then examined the expression levels of the Flag-tagged ETS2 protein when expressed alone or in combination with Myc-CDK10 or -CDK10(kd), with or without cyclin M-V5-6His. Flag-ETS2 was readily detected when expressed alone or, to a lesser extent, when coexpressed with CDK10(kd). However, its expression level was dramatically decreased when coexpressed with CDK10 alone, or with CDK10 and cyclin M (Fig. 4B). These observations suggest that endogenous cyclin M levels are in excess compared with those of CDK10 in MCF7 cells, and they show that the major decrease in ETS2 levels observed upon CDK10 coexpression involves CDK10 kinase activity. Treatment of cells coexpressing Flag-ETS2, CDK10, and cyclin M with the proteasome inhibitor MG132 largely rescued Flag-ETS2 expression levels (Fig. 4B).
Fig. 4.

CDK10/cyclin M controls ETS2 stability in human cancer derived cells. (A) Western blot analysis of endogenous ETS2 expression levels in MCF7 cells, in response to siRNA-mediated CDK10 and/or cyclin M silencing. A quantification is shown in Fig. S4B. (B) Western blot analysis of exogenously expressed Flag-ETS2 protein levels in MCF7 cells cotransfected with empty vectors or coexpressing Myc-CDK10 (wt or kd), or Myc-CDK10/CycM-V5-6His. The latter cells were treated for 16 h with the MG132 proteasome inhibitor. Proper expression of CDK10 and cyclin M tagged proteins was verified by Western blot analysis. (C and D) Western blot analysis of expression levels of exogenously expressed Flag-ETS2 wild-type or mutant proteins in MCF7 cells, in the absence of (C) or in response to (D) Myc-CDK10/CycM-V5-6His expression. Quantifications are shown in Fig. S4 C and D.
A mass spectrometry analysis of recombinant ETS2 phosphorylated by CDK10/cyclin M in vitro revealed the existence of multiple phosphorylated residues, among which are two neighboring phospho-serines (at positions 220 and 225) that may form a phosphodegron (14) (Figs. S5–S8). To confirm this finding, we compared the phosphorylation level of recombinant ETS2wt with that of ETS2SASA protein, a mutant bearing alanine substitutions of these two serines. As expected from the existence of multiple phosphorylation sites, we detected a small but reproducible, significant decrease of phosphorylation level of ETS2SASA compared with ETS2wt (Fig. S9), thus confirming that Ser220/Ser225 are phosphorylated by CDK10/cyclin M. To establish a direct link between ETS2 phosphorylation by CDK10/cyclin M and degradation, we examined the expression levels of Flag-ETS2SASA. In the absence of CDK10/cyclin M coexpression, it did not differ significantly from that of Flag-ETS2. This is contrary to that of Flag-ETS2DBM, bearing a deletion of the N-terminal destruction (D-) box that was previously shown to be involved in APC-Cdh1–mediated degradation of ETS2 (13) (Fig. 4C). However, contrary to Flag-ETS2 wild type, the expression level of Flag-ETS2SASA remained insensitive to CDK10/cyclin M coexpression (Fig. 4D). Altogether, these results suggest that CDK10/cyclin M directly controls ETS2 degradation through the phosphorylation of these two serines.
Finally, we studied a lymphoblastoid cell line derived from a patient with STAR syndrome, bearing FAM58A mutation c.555+1G>A, predicted to result in aberrant splicing (10). In accordance with incomplete skewing of X chromosome inactivation previously found in this patient, we detected a decreased expression level of cyclin M protein in the STAR cell line, compared with a control lymphoblastoid cell line. In line with our preceding observations, we detected an increased expression level of ETS2 protein in the STAR cell line compared with the control (Fig. 5A and Fig. S10A). We then examined by quantitative RT-PCR the mRNA expression levels of the corresponding genes. The STAR cell line showed a decreased expression level of cyclin M mRNA but an expression level of ETS2 mRNA similar to that of the control cell line (Fig. 5B). To demonstrate that the increase in ETS2 protein expression is indeed a result of the decreased cyclin M expression observed in the STAR patient-derived cell line, we expressed cyclin M-V5-6His in this cell line. This expression caused a decrease in ETS2 protein levels (Fig. 5C).
Fig. 5.
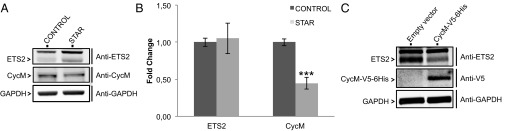
Decreased cyclin M expression in STAR patient-derived cells results in increased ETS2 protein level. (A) Western blot analysis of cyclin M and ETS2 protein levels in a STAR patient-derived lymphoblastoid cell line and in a control lymphoblastoid cell line, derived from a healthy individual. A quantification is shown in Fig. S10A. (B) Quantitative RT-PCR analysis of cyclin M and ETS2 mRNA levels in the same cells. ***P ≤ 0.001. (C) Western blot analysis of ETS2 protein levels in the STAR patient-derived lymphoblastoid cell line transfected with an empty vector or a vector directing the expression of cyclin M-V5-6His. Another Western blot revealing endogenously and exogenously expressed cyclin M levels is shown in Fig. S10B. A quantification of ETS2 protein levels is shown in Fig. S10C.
Discussion
In this work, we unveil the interaction between CDK10, the last orphan CDK discovered in the pregenomic era (2), and cyclin M, the only cyclin associated with a human genetic disease so far, and whose functions remain unknown (10). The closest paralogs of CDK10 within the CDK family are the CDK11 proteins, which interact with L-type cyclins (15). Interestingly, the closest paralog of these cyclins within the cyclin family is cyclin M (Fig. S11). The fact that none of the shorter CDK10 isoforms interact robustly with cyclin M suggests that alternative splicing of the CDK10 gene (16, 17) plays an important role in regulating CDK10 functions.
The functional relevance of the interaction between CDK10 and cyclin M is supported by different observations. Both proteins seem to enhance each other’s stability, as judged from their increased expression levels when their partner is exogenously coexpressed (Fig. 1D) and from the much reduced endogenous CDK10 expression level observed in response to cyclin M silencing (Fig. 3A and Fig. S2B). CDK10 is subject to ubiquitin-mediated degradation (18). Our observations suggest that cyclin M protects CDK10 from such degradation and that it is the only cyclin partner of CDK10, at least in MCF7 cells. They also suggest that cyclin M stability is enhanced upon binding to CDK10, independently from its kinase activity, as seen for cyclin C and CDK8 (19). We uncover a cyclin M-dependent CDK10 protein kinase activity in vitro, thus demonstrating that this protein, which was named a CDK on the sole basis of its amino acid sequence, is indeed a genuine cyclin-dependent kinase. Our Y2H assays reveal that truncated cyclin M proteins corresponding to the hypothetical products of two STAR syndrome-associated FAM58A mutations do not produce an interaction phenotype with CDK10. Hence, regardless of whether these mutated mRNAs undergo nonsense-mediated decay (as suggested from the decreased cyclin M mRNA levels in STAR cells, shown in Fig. 5B) or give rise to truncated cyclin M proteins, females affected by the STAR syndrome must exhibit compromised CDK10/cyclin M kinase activity at least in some tissues and during specific developmental stages.
We show that ETS2, a known interactor of CDK10, is a phosphorylation substrate of CDK10/cyclin M in vitro and that CDK10/cyclin M kinase activity positively controls ETS2 degradation by the proteasome. This control seems to be exerted through a very fine mechanism, as judged from the sensitivity of ETS2 levels to partially decreased CDK10 and cyclin M levels, achieved in MCF7 cells and observed in STAR cells, respectively. These findings offer a straightforward explanation for the already reported up-regulation of ETS2-driven transcription of c-RAF in response to CDK10 silencing (6). We bring evidence that CDK10/cyclin M directly controls ETS2 degradation through the phosphorylation of two neighboring serines, which may form a noncanonical β-TRCP phosphodegron (DSMCPAS) (14). Because none of these two serines precede a proline, they do not conform to usual CDK phosphorylation sites. However, multiple so-called transcriptional CDKs (CDK7, -8, -9, and -11) (to which CDK10 may belong; Fig. S11) have been shown to phosphorylate a variety of motifs in a non–proline-directed fashion, especially in the context of molecular docking with the substrate (20). Here, it can be hypothesized that the high-affinity interaction between CDK10 and the Pointed domain of ETS2 (6, 9) (Fig. 1C) would allow docking-mediated phosphorylation of atypical sites. The control of ETS2 degradation involves a number of players, including APC-Cdh1 (13) and the cullin-RING ligase CRL4 (21). The formal identification of the ubiquitin ligase involved in the CDK10/cyclin M pathway and the elucidation of its concerted action with the other ubiquitin ligases to regulate ETS2 degradation will require further studies.
Our results present a number of significant biological and medical implications. First, they shed light on the regulation of ETS2, which plays an important role in development (22) and is frequently deregulated in many cancers (23). Second, our results contribute to the understanding of the molecular mechanisms causing tamoxifen resistance associated with reduced CDK10 expression levels, and they suggest that, like CDK10 (6), cyclin M could also be a predictive clinical marker of hormone therapy response of ERα-positive breast cancer patients. Third, our findings offer an interesting hypothesis on the molecular mechanisms underlying STAR syndrome. Ets2 transgenic mice showing a less than twofold overexpression of Ets2 present severe cranial abnormalities (24), and those observed in STAR patients could thus be caused at least in part by increased ETS2 protein levels. Another expected consequence of enhanced ETS2 expression levels would be a decreased risk to develop certain types of cancers and an increased risk to develop others. Studies on various mouse models (including models of Down syndrome, in which three copies of ETS2 exist) have revealed that ETS2 dosage can repress or promote tumor growth and, hence, that ETS2 exerts noncell autonomous functions in cancer (25). Intringuingly, one of the very few STAR patients identified so far has been diagnosed with a nephroblastoma (26). Finally, our findings will facilitate the general exploration of the biological functions of CDK10 and, in particular, its role in the control of cell division. Previous studies have suggested either a positive role in cell cycle control (5, 6) or a tumor-suppressive activity in some cancers (7, 8). The severe growth retardation exhibited by STAR patients strongly suggests that CDK10/cyclin M plays an important role in the control of cell proliferation.
Materials and Methods
Cloning of CDK10 and cyclin M cDNAs, plasmid constructions, tamoxifen response analysis, quantitative RT-PCR, mass spectrometry experiments, and antibody production are detailed in SI Materials and Methods.
Yeast Two-Hybrid Interaction Assays.
We performed yeast interaction mating assays as previously described (27).
Mammalian Cell Cultures and Transfections.
We grew human HEK293 and MCF7 cells in DMEM supplemented with 10% (vol/vol) FBS (Invitrogen), and we grew lymphoblastoid cells in RPMI 1640 GlutaMAX supplemented with 15% (vol/vol) FBS. We transfected HEK293 and MCF7 cells using Lipofectamine 2000 (Invitrogen) for plasmids, Lipofectamine RNAiMAX (Invitrogen) for siRNAs, and Jetprime (Polyplus) for plasmids/siRNAs combinations according to the manufacturers’ instructions. We transfected lymphoblastoid cells by electroporation (Neon, Invitrogen). For ETS2 stability studies we treated MCF7 cells 32 h after transfection with 10 μM MG132 (Fisher Scientific) for 16 h.
Coimmunoprecipitation and Western Blot Experiments.
We collected cells by scraping in PBS (or centrifugation for lymphoblastoid cells) and lysed them by sonication in a lysis buffer containing 60 mM β-glycerophosphate, 15 mM p-nitrophenylphosphate, 25 mM 3-(N-morpholino)propanesulfonic acid (Mops) (pH 7.2), 15 mM EGTA, 15 mM MgCl2, 1 mM Na vanadate, 1 mM NaF, 1mM phenylphosphate, 0.1% Nonidet P-40, and a protease inhibitor mixture (Roche). We spun the lysates 15 min at 20,000 × g at 4 °C, collected the supernatants, and determined the protein content using a Bradford assay. We performed the immunoprecipitation experiments on 500 μg of total proteins, in lysis buffer. We precleared the lysates with 20 μL of protein A or G-agarose beads, incubated 1 h 4 °C on a rotating wheel. We added 5 μg of antibody to the supernatants, incubated 1 h 4 °C on a rotating wheel, added 20 μL of protein A or G-agarose beads, and incubated 1 h 4 °C on a rotating wheel. We collected the beads by centrifugation 30 s at 18,000 × g at 4 °C and washed three times in a bead buffer containing 50 mM Tris (pH 7.4), 5 mM NaF, 250 mM NaCl, 5 mM EDTA, 5 mM EGTA, 0.1% Nonidet P-40, and a protease inhibitor coktail (Roche). We directly added sample buffer to the washed pellets, heat-denatured the proteins, and ran the samples on 10% Bis-Tris SDS/PAGE. We transferred the proteins onto Hybond nitrocellulose membranes and processed the blots according to standard procedures. For Western blot experiments, we used the following primary antibodies: anti-Myc (Abcam ab9106, 1:2,000), anti-V5 (Invitrogen R960, 1:5,000), anti-tubulin (Santa Cruz Biotechnology B-7, 1:500), anti-CDK10 (Covalab pab0847p, 1:500 or Santa Cruz Biotechnology C-19, 1:500), anti-CycM (home-made, dilution 1:500 or Covalab pab0882-P, dilution 1:500), anti-Raf1 (Santa Cruz Biotechnology C-20, 1:1,000), anti-ETS2 (Santa Cruz Biotechnology C-20, 1:1,000), anti-Flag (Sigma F7425, 1:1,000), and anti-actin (Sigma A5060, 1:5,000). We used HRP-coupled anti-goat (Santa Cruz Biotechnology SC-2033, dilution 1:2,000), anti-mouse (Bio-Rad 170–6516, dilution 1:3,000) or anti-rabbit (Bio-Rad 172–1019, 1:5,000) as secondary antibodies. We revealed the blots by enhanced chemiluminescence (SuperSignal West Femto, Thermo Scientific).
Production and Purification of Recombinant Proteins.
GST-CDK10(kd)/StrepII-CycM.
We generated recombinant bacmids in DH10Bac Escherichia coli and baculoviruses in Sf9 cells using the Bac-to-Bac system, as described by the provider (Invitrogen). We infected Sf9 cells with GST-CDK10- (or GST-CDK10kd)-producing viruses, or coinfected the cells with StrepII-CycM–producing viruses, and we collected the cells 72 h after infection. To purify GST-fusion proteins, we spun 250 mL cells and resuspended the pellet in 40 mL lysis buffer (PBS, 250 mM NaCl, 0.5% Nonidet P-40, 50 mM NaF, 10 mM β-glycerophosphate, and 0.3 mM Na-vanadate) containing a protease inhibitor mixture (Roche). We lysed the cells by sonication, spun the lysate 30 min at 15,000 × g, collected the soluble fraction, and added it to a 1-mL glutathione-Sepharose matrix. We incubated 1 h at 4 °C, washed four times with lysis buffer, one time with kinase buffer A (see below), and finally resuspended the beads in 100 μL kinase buffer A containing 10% (vol/vol) glycerol for storage.
6His-ETS2.
We transformed Origami2 DE3 (Novagen) with the 6His-ETS2 expression vector. We induced expression with 0.2 mM isopropyl-β-d-1-thiogalactopyranoside for 3 h at 22 °C. To purify 6His-ETS2, we spun 50 mL cells and resuspended the pellet in 2 mL lysis buffer (PBS, 300 mM NaCl, 10 mM Imidazole, 1 mM DTT, and 0.1% Nonidet P-40) containing a protease inhibitor mixture without EDTA (Roche). We lysed the cells at 1.6 bar using a cell disruptor and spun the lysate 10 min at 20,000 × g. We collected the soluble fraction and added it to 200 μL Cobalt beads (Thermo Scientific). After 1 h incubation at 4 °C on a rotating wheel, we washed four times with lysis buffer. To elute, we incubated beads 30 min with elution buffer (PBS, 250 mM imidazole, pH 7.6) containing the protease inhibitor mixture, spun 30 s at 10,000 × g, and collected the eluted protein.
Protein Kinase Assays.
We mixed glutathione-Sepharose beads (harboring GST-CDK10 wt or kd, either monomeric or complexed with StrepII-CycM), 22.7 μM BSA, 15 mM DTT, 100 μM ATP, 5 μCi ATP[γ-32P], 7.75 μM histone H1, or 1 μM 6His-ETS2 and added kinase buffer A (25 mM Tris·HCl, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, and 3.7 μM heparin, pH 7.5) up to a total volume of 30 μL. We incubated the reactions 30 min at 30 °C, added Laemli sample buffer, heat-denatured the samples, and ran 10% Bis-Tris SDS/PAGE. We cut gel slices to detect GST-CDK10 and StrepII-CycM by Western blotting. We stained the gel slices containing the substrate with Coomassie (R-250, Bio-Rad), dried them, and detected the incorporated radioactivity by autoradiography.
Supplementary Material
Acknowledgments
We thank Jens Reinhardt, Sébastien Colin, Dianne Baunbaek, and Nathalie Desban for early contributions to the project; Jean-Yves Thuret, François Leteurtre, and Irina Lassot for the gift of plasmids; Geert Mortier for the gift of a STAR patient-derived cell line; Frédéric Lesourd and Xavier Fant for their advice on RT-PCR and immuno-detection, respectively; Alexandre Reymond for his guidance on the design of FAM58A STAR mutated forms; Olivier Coux for his advice on protein degradation studies; Bertrand Cosson, Régis Giet, Pascal Loyer, and Sandrine Ruchaud for useful discussions; and Mark Molloy and Judith Nicholson for having hosted and guided C.G. in their laboratory and for having analyzed the mass spectrometry data. This project was funded by the Région Bretagne, the Ligue Nationale contre le Cancer Grand Ouest, the Département Finistère, the Association pour la Recherche contre le Cancer, and the Fondation Jérôme Lejeune. V.J.G. is a recipient of a doctoral fellowship from the French Ministry of Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306814110/-/DCSupplemental.
References
- 1.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: A changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, et al. Cyclin-dependent kinases: A family portrait. Nat Cell Biol. 2009;11(11):1275–1276. doi: 10.1038/ncb1109-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graña X, Claudio PP, De Luca A, Sang N, Giordano A. PISSLRE, a human novel CDC2-related protein kinase. Oncogene. 1994;9(7):2097–2103. [PubMed] [Google Scholar]
- 4.Brambilla R, Draetta G. Molecular cloning of PISSLRE, a novel putative member of the cdk family of protein serine/threonine kinases. Oncogene. 1994;9(10):3037–3041. [PubMed] [Google Scholar]
- 5.Li S, et al. The cdc-2-related kinase, PISSLRE, is essential for cell growth and acts in G2 phase of the cell cycle. Cancer Res. 1995;55(18):3992–3995. [PubMed] [Google Scholar]
- 6.Iorns E, et al. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell. 2008;13(2):91–104. doi: 10.1016/j.ccr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Yu JH, et al. CDK10 functions as a tumor suppressor gene and regulates survivability of biliary tract cancer cells. Oncol Rep. 2012;27(4):1266–1276. doi: 10.3892/or.2011.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong XY, et al. Clinical and biological significance of Cdk10 in hepatocellular carcinoma. Gene. 2012;498(1):68–74. doi: 10.1016/j.gene.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Kasten M, Giordano A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene. 2001;20(15):1832–1838. doi: 10.1038/sj.onc.1204295. [DOI] [PubMed] [Google Scholar]
- 10.Unger S, et al. Mutations in the cyclin family member FAM58A cause an X-linked dominant disorder characterized by syndactyly, telecanthus and anogenital and renal malformations. Nat Genet. 2008;40(3):287–289. doi: 10.1038/ng.86. [DOI] [PubMed] [Google Scholar]
- 11.Murray AW, Marks D. Can sequencing shed light on cell cycling? Nature. 2001;409(6822):844–846. doi: 10.1038/35057033. [DOI] [PubMed] [Google Scholar]
- 12.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262(5142):2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 13.Li M, et al. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat Cell Biol. 2008;10(9):1083–1089. doi: 10.1038/ncb1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28(5):730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Loyer P, et al. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: Influence of cyclin L isoforms on splice site selection. J Biol Chem. 2008;283(12):7721–7732. doi: 10.1074/jbc.M708188200. [DOI] [PubMed] [Google Scholar]
- 16.Crawford J, et al. The PISSLRE gene: Structure, exon skipping, and exclusion as tumor suppressor in breast cancer. Genomics. 1999;56(1):90–97. doi: 10.1006/geno.1998.5676. [DOI] [PubMed] [Google Scholar]
- 17.Sergère JC, Thuret JY, Le Roux G, Carosella ED, Leteurtre F. Human CDK10 gene isoforms. Biochem Biophys Res Commun. 2000;276(1):271–277. doi: 10.1006/bbrc.2000.3395. [DOI] [PubMed] [Google Scholar]
- 18.Khanal P, et al. Proyl isomerase Pin1 facilitates ubiquitin-mediated degradation of cyclin-dependent kinase 10 to induce tamoxifen resistance in breast cancer cells. Oncogene. 2012;31(34):3845–3856. doi: 10.1038/onc.2011.548. [DOI] [PubMed] [Google Scholar]
- 19.Barette C, Jariel-Encontre I, Piechaczyk M, Piette J. Human cyclin C protein is stabilized by its associated kinase cdk8, independently of its catalytic activity. Oncogene. 2001;20(5):551–562. doi: 10.1038/sj.onc.1204129. [DOI] [PubMed] [Google Scholar]
- 20.Larochelle S, et al. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13(1):55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- 21.Emanuele MJ, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147(2):459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raouf A, Seth A. Ets transcription factors and targets in osteogenesis. Oncogene. 2000;19(55):6455–6463. doi: 10.1038/sj.onc.1204037. [DOI] [PubMed] [Google Scholar]
- 23.Dwyer JM, Liu JP. Ets2 transcription factor, telomerase activity and breast cancer. Clin Exp Pharmacol Physiol. 2010;37(1):83–87. doi: 10.1111/j.1440-1681.2009.05236.x. [DOI] [PubMed] [Google Scholar]
- 24.Sumarsono SH, et al. Down’s syndrome-like skeletal abnormalities in Ets2 transgenic mice. Nature. 1996;379(6565):534–537. doi: 10.1038/379534a0. [DOI] [PubMed] [Google Scholar]
- 25.Threadgill DW. Down’s syndrome: Paradox of a tumour repressor. Nature. 2008;451(7174):21–22. doi: 10.1038/451021a. [DOI] [PubMed] [Google Scholar]
- 26.Rakenius A, et al. STAR syndrome and nephroblastoma-FAM58A linking organogenesis and tumorigenesis? Eur J Pediatr. 2010;169:389. [Google Scholar]
- 27.Bickle MBT, Dusserre E, Moncorgé O, Bottin H, Colas P. Selection and characterization of large collections of peptide aptamers through optimized yeast two-hybrid procedures. Nat Protoc. 2006;1(3):1066–1091. doi: 10.1038/nprot.2006.32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.