

Abstract
Activation of TLRs by components required for pathogen viability results in increased inflammation and an enhanced immune response to infection. Unlike their effects on other immune cells, TLR activation in the absence of T cell antigen receptor (TCR) induction has little effect on T cell activity. Instead, the simultaneous induction of TLR and TCR results in increased cytokine release compared to TCR treatment alone. Thus, the current model states that TLRs alter T cell function only if activated at the same time as the TCR. In this study, we tested the novel hypothesis that prior TLR induction can also alter TCR-mediated functions. We found that human T cells responded to ligands for TLR2 and TLR5. However, only prior TLR5 induction potentiated subsequent TCR-mediated cytokine production in human T cells. This response required at least 24 hours of TLR5 induction and lasted for approximately 24–36 hours after removal of a TLR5 ligand. Interestingly, prior TLR5 induction enhanced TCR-mediated activation of Akt without increasing Lck, LAT or ERK kinase phosphorylation. Together, our studies show that TLR5 induction leads to a transient increase in the sensitivity of T cells to TCR stimulation by selectively enhancing TCR-mediated Akt function, highlighting that timeframe when TLR5 can potentiate TCR-induced downstream functions are significantly longer that previously appreciated.
Keywords: Toll-like receptor 5, T cell receptor, T cells, signal transduction
1. Introduction
For immune cells to respond to pathogens, they must first recognize that an infection is occurring. A critical mechanism for sensing infection is the activation of the TLRs. Humans express 10 different TLRs (TLR1–10) and a non-functional psuedogene (TLR11) (Leulier and Lemaitre, 2008). Each TLR recognizes a different class of pathogen components required for their growth and survival (Brikos and O'Neill, 2008; Leulier and Lemaitre, 2008). For example, TLR2 and its heterodimerization partners TLR1, TLR6 and TLR10 bind to structures from the cell walls of bacteria and fungi (Guan et al., 2010; Leulier and Lemaitre, 2008). TLR5 associates with flagellin found on all motile bacteria and TLR7 is activated by single stranded RNA (Kawai and Akira, 2011; Leulier and Lemaitre, 2008). Activation of TLRs by their ligands leads to localized inflammation, increased function of immune cells, enhanced recruitment of immune cells to sites of infection, and clearance of the pathogen (Drexler and Foxwell, 2010; Kawai and Akira, 2011). Patients with deficiencies or polymorphisms in TLRs or their downstream signaling partners have increased susceptibility to numerous bacterial, viral and fungal infections (Suhir and Etzioni, 2010).
The majority of studies examining TLR-mediated functions have focused on antigen presenting cells (APC), i.e. macrophages, dendritic cells, and B cells. In these cell types, TLR ligands are capable of directly stimulating cytokine production, increasing the surface expression of costimulatory receptor ligands and driving cellular maturation (Kawai and Akira, 2011). These TLR-induced actions in APC indirectly affect T cell activation due to the requirement of activated APCs for the induction of T cell responses. However, human T cell lines and primary peripheral blood T cells express the mRNA for TLRs1–10 and have protein expression of TLR2, TLR3, TLR4, TLR5 and TLR9 (Kabelitz, 2007). In contrast to their function in APCs, the most prominent effect of TLRs in human T cells is their ability to act as classic costimulatory receptors (Kulkarni et al., 2011), where simultaneous stimulation of the TCR and the TLR result in enhanced TCR-induced functions. Ligands for TLR3, TLR4 and TLR9 have no apparent influence on human T cell proliferation and cytokine release, both in the absence and presence of TCR stimulation (Caron et al., 2005; Lancioni et al., 2009; McCarron and Reen, 2009; Merlo et al., 2007). The effects of TLR7 stimulation of T cell activity are controversial. Some studies show that a TLR7 ligand does not affect TCR-mediated proliferation, receptor expression and cytokine production (Lancioni et al., 2009; McCarron and Reen, 2009), whereas other find that TLR7 activation increases TCR-mediated proliferation and IFN-γ release (Caron et al., 2005; Richardt-Pargmann et al., 2011). Interestingly, simultaneous TLR2 or TLR5 activation leads to amplified TCR-mediated proliferation, surface receptor expression and cytokine release (Caron et al., 2005; Crellin et al., 2005; Komai-Koma et al., 2004; Lancioni et al., 2009; McCarron and Reen, 2009; Merlo et al., 2007; Nyirenda et al., 2011; Richardt-Pargmann et al., 2011). Together, these studies suggest that human primary T cells are most potently and consistently activated by ligands for TLR2 and TLR5, but that the enhancements of TCR functions occur only if the TCR and TLRs are activated concurrently.
Although the observed outcome of simultaneous TCR and TLR activation in human T cells is increased cellular responses, only one study has examined whether prior TLR induction alters the responsiveness of the T cell to TCR induction. Okugawa and coworkers found that prior treatment with the TLR5 ligand flagellin resulted in decreased TCR-induced proximal signaling events (Okugawa et al., 2006). However, this study did not investigate the effects of TLR5 induction on TCR-mediated cytokine production or whether other TLRs could also alter TCR-mediated events. We hypothesized that previous TLR activation can modulate TCR-mediated cytokine production, but for a much longer time frame than its similar function as a costimulatory receptor. We observed that previous activation of TLR5, but not TLR2 or TLR7, led to increased cytokine production upon TCR activation that was not attributable to its costimulatory role. The potentiation of TCR function by prior TLR5 activation was transient and required IRAK activity. Previous TLR5 activation both positively and negatively regulated different signaling pathways downstream of the TCR, with the most prominent effect being increased Akt activation. These studies are the first to show that previous TLR5 stimulation leads to a transient increase in the sensitivity of T cells to subsequent TCR activation. Together, this demonstrates that TLR5 can alter T cell function for several days, much longer than thought by the current model where TLR5 acts as classic costimulatory receptors.
2. Materials and Methods
2.1 Reagents
Anti-Erk1/Erk2 pT185/pY187,anti-Akt pS473 antibodies, Fluor-4 combined dyes, Dynabeads, Superscript III one-step RT-PCR system, probenecid, RPMI 1640, RPMI 1640 without phenol red, Iscove's Modified Dulbecco's Media, L-glutamine, penicillin-streptomycin and PBS were purchased from Invitrogen/Life Technologies (Grand Island, NY, USA). Anti-SRC family pY416 was obtained from Cell Signaling Technologies (Danvers, MA, USA). The IRAK1/4 Inhibitor, polyvinylidene difloride membrane, anti-LAT pY191 and anti-actin antibodies were obtained from EMD Millipore (Billerica, MA, USA). Streptavidin-HRP and the anti-mouse and anti-rabbit secondary antibodies were purchased from Jackson Immuno Research Labs (West Grove, PA, USA). The FBS was obtained from Atlanta Biologicals (Lawrenceville, GA, USA). Flagellin, PAM3CSK4 (PAM), and R848 were acquired from InvivoGen (San Diego, CA, USA). Recombinant Flagellin from Salmonella typhimurium that is produced in HEK293 cells were used for all studies, since this flagellin has extremely low contamination from other TLR ligands. The RNeasy Mini Kit was acquired from Qiagen (Venlo, Netherlands). The anti-CD3 antibody (OKT3), anti-CD4 antibody (RPA-T4), anti-CD28 antibody (CD28.2), anti-mouse IgG, recombinant human IFN-γ, purified anti-human IFN-γ and biotin anti-human IFN-γ were obtained from Biolegend (San Diego, CA, USA). Recombinant human IL-2 was acquired from R & D Systems (Minneapolis, MN, USA). Purified anti-human IL-2 and biotin anti-human IL-2 were obtained from eBioscience. Human rIL-2 was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Human rIL-2 from Dr. Maurice Gately, Hoffmann – La Roche Inc. ELISA tetramethylbenzidine peroxidase substrate was purchased from Kirkegaard & Perry Laboratories (Gaithersburg, MD, USA). The Criterion polyacrylamide gels were acquired from Bio-Rad (Hercules, CA, USA). The Supersignal West Pico and Femto Chemiluminescent Substrate and the Restore Western Blot Stripping Buffer were purchased from Pierce (Rockford, IL, USA). All chemicals were research grade and obtained from multiple sources.
2.2 Growth and Stimulation of HuT78 Human T cells
HuT78 T cells were used for these studies since these cells have been shown to have similar early signaling and cytokine production to human activated peripheral blood T cells (Bartelt et al., 2009). HuT78 T cells were cultured at 37°C in 5% CO2 in Iscove's Modified Dulbecco's Media supplemented with 20% FBS, 2mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. The cells were grown to a concentration of 2–5 × 105 cells/ml then washed in RPMI 1640 without supplements. They were then resuspended to 5 × 106 cells/ml in RPMI 1640 without supplements and incubated for 10 minutes at 37°C. The cells were stimulated with 10 μg/ml anti-CD3 (OKT3) for various times and lysed with a 4-fold excess of hot 2X lysis buffer (20 mM Tris (pH 8.0), 2 mM EDTA, 2 mM Na3VO4, 20 mM DTT, 2% SDS and 20% glycerol). The lysates were then heated to 95°C for 4 minutes and sonicated to reduce viscosity.
2.3 Growth and Stimulation of Activated Human Peripheral Blood T cells
Activated peripheral blood T cells (APBTs) were obtained from whole blood of healthy anonymous donors. Peripheral blood mononuclear cells (PBMCs) were obtained from anonymous donors from two sources. In the first source, PBMCs were acquired from donors at the DeGowin Blood Center at the University of Iowa who had consented to allow blood cells not used for donation to be used for research by investigators at the University of Iowa. The consent process and consent documents for these donors have been approved by the Institutional Review Board (IRB) for the University of Iowa. Leukocyte reducing cones were used to remove PBMCs from these blood products, and these normally discarded cones were provided to investigators at the University of Iowa. The second source of PBMCs was from participants in IRB approved studies at the University of Iowa. In these studies, the PBMCs were not needed to complete the IRB approved studies and were normally discarded. Because all cells used in these studies were obtained from normally discarded products, the donors had approved for the use of their cells in research projects and the donors were completely de-identified, these studies were exempt from IRB approval.
The PBMCs from the leukocyte reducing cones were flushed from the cone using sterile 1X PBS (Meyer et al., 2005). The PBMCs from both procedures were then isolated by Ficoll density centrifugation and resuspended in RPMI 1640 supplemented with 10% FBS, 2mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 20 ng/ml rIL-2. The PBMCs were activated with magnetic beads coated with anti-CD3 and anti-CD28 for 3–10 days at 37°C to obtain APBTs. By Day 5 after activation, the APBTs were >96% positive for CD3, with <2% contamination from B cells and no detectable myeloid cells (Supplemental Figure 1A). The APBTs were then washed with RPMI 1640 and resuspended to a concentration of 1 × 107 cells/ml. The cells were treated with anti-CD3 (10 μg/ml) and anti-CD4 (2 μg/ml) for 30 minutes on ice. Anti-CD3 and anti-CD4 were used for these studies since previous work has shown that these stimuli provides potent activation of proximal TCR-mediated tyrosine phosphorylation and LAT activation, while anti-CD3 and anti-CD28 have little effect on these events (Houtman et al., 2005b; June et al., 1990). The cells were warmed to 37°C for 10 minutes and stimulated with anti-mouse IgG (25 μg/ml) for various times. The cells were lysed with a 4-fold excess of hot 2X lysis buffer, heated to 95°C for 4 minutes and sonicated to reduce viscosity.
2.4 Immunoblotting
The cellular lysates were separated by polyacrylamide gel electrophoresis (PAGE) using 4–15% Criterion gels and transferred to polyvinylidene difluoride. To analyze site-specific phosphorylation, the membranes were blocked for 1 hour at room temperature in a blocking buffer consisting of TBST (10mM TRIS (pH 8.0), 150mM NaCl, and 0.05% Tween 20) with 5% non-fat dry milk. Primary antibodies were added to blocking buffer at their appropriate dilutions and then incubated with the membrane overnight at 4°C. After washing twice with TBST, the secondary antibody was added in blocking buffer for 30 minutes at room temperature. Antibody binding was identified with Chemiluminescent Substrate and detected using a Fuji Imager. Three independent replicates were performed for each experiment. All membranes were stripped using Restore Western Blot Stripping Buffer, then washed extensively in TBST and reprobed with the anti-actin antibody as described above for the loading control.
2.5 Analysis of Immunoblotting
The intensity of the immunoblotting bands was determined using the gel plotting macro of ImageJ. The immunoblotting exposures were selected such that the maximal response from each individual experiment had approximately equal intensity. The blots were reprobed with the anti-actin antibody to ensure of equivalent loading. The intensity of each time point was normalized to the actin expression and the average time point percentages were calculated by setting the time point with maximal phosphorylation to 100% and the remainder of time points were scaled accordingly. The data was then plotted using the graphing program Prism (Graphpad Software).
2.6 Expression of TLR2, TLR5 and TLR7 mRNA
Total RNA was extracted from 2 × 106 HuT78 or APBTs using RNeasy Mini Kit (Qiagen). The RT-PCR reactions were carried out in the presence of 500 ng template RNA using Superscript III one-step RT-PCR system (Invitrogen). Primers for TLR2, 5, and 7 were designed through PrimerQuest (www.idtdna.com/Primerquest/) at Integrated DNA Technology (Coralville, IA, USA) and subsequently purchased from IDT. The primer sequences were: TLR2 (536 bp, Forward: CCATTGCTCTTTCACTGCTTTC, Reverse: GGTGTCCATATTTCCCACTCTC), TLR5 (774 bp, Forward: TCTCCAGGATGTTGGCTGGTTTCT, Reverse: AAAGTTCTTGGCTCACTAGGGCGA), and TLR7 (481 bp, Forward: TATTCCCACGAACACCACGAACCT, Reverse: AACAGTAGGGACGGCTGTGACATT). The PCR products were visualized using DNA agarose gel electrophoresis.
2.7 Calcium Influx in HuT78 T Cells
HuT78 T cells (0.25 × 106/ml) were pretreated with or without 250ng/ml flagellin for 24 hours at 37°C. The cells were then washed in 5ml RPMI without phenol red and resuspended to 1 × 106 cells/ml. 250 mM probenecid was added to the cells and Fluor-4 combined dyes were added. The cells were incubated for 60 minutes at 37°C. The cells were washed and resupended to 0.5 × 106/ml. 250 mM probenecid was added to the cells again and incubated on ice for 15 minutes. The cells were warmed for 5 minutes and data was collected using the Accuri C6 flow cytometer for 35 seconds. The cells were stimulated with 2 μg/ml anti-CD3 (OKT3) and data was collected for an additional 7 minutes. Ionomycin was then added as a stimulatory control, and the cells were collected for an additional 60 seconds. The data was gated using the software provided by Accuri and the data from the gated cells was exported to Microsoft Excel. The average fluorescence of gated cells collected in a single second was calculated and then was plotted as Fluor-4 Intensity (AU) vs. time.
2.8 TCR-induced Cytokine Production in HuT78 T Cells and APBTs
For simultaneous stimulation, HuT78 T cells (0.5 × 106/ml) were stimulated for 24 hours at 37°C in supplemented RPMI with or without 1 μg/ml plate-bound anti-CD3 and in absence or presence of 1 μg/ml flagellin, 1 μg/ml PAM, or 1 μg/ml R848. For pretreatment experiments, HuT78 T cells (0.5 × 106/ml) or APBTs (1 × 106/ml) were pretreated with flagellin for various times in supplemented RPMI at 37°C. The cells were washed three times with RPMI 1640 without supplements and stimulated overnight with or without 1 μg/ml plate-bound anti-CD3. For the rest experiments, HuT78 T cells were pretreated for 24 hours with or without 250 ng/ml flagellin in supplemented RPMI at 37°C. The cells were washed three times with RPMI 1640 without supplements and then rested for various times in supplemented RPMI at 37°C, counted and then 0.5 × 106/ml cells were stimulated with anti-CD3 for 24 hours. For the IRAK inhibitor experiments, HuT78 T cells (0.5 × 106/ml) were pretreated with or without 250 ng/ml flagellin and various doses of the IRAK inhibitor for 24 hours at 37°C in supplemented RPMI. The cells were washed three times with RPMI 1640 without supplements, resting for 30 minutes between each wash. The cells were then stimulated with or without 1 μg/ml plate-bound anti-CD3 for 24 hours at 37°C. Supernatants for all of the experiments were collected and stored at −20°C. IL-2 and IFN-γ levels in an individual culture supernatants were measured in triplicate by standard TMB based ELISA using an Epoch plate reader at 450 nM (BioTek, Winooski, VT, USA). Three to five independent experiments were completed for each experiment. The values for each experiment were normalized, setting the treatment with the maximal production of the individual cytokine as 100% (see figure legend for details) and the remainder of the values scaled accordingly. The data was then plotted using the graphing program Prism (Graphpad Software, La Jolla, CA, USA).
2.9 Statistical Analysis
All statistical analysis for the ELISA data was performed using using ANOVA with Dunnett's Multiple Comparison Post Test using Prism. The reference treatments were marked with #. The level of significance was identified with * = p<0.05 and ** = p<0.01. Individual timepoints for the immunoblotting data was statistically compared using Student's t test in Prism with the level of significance identified with * = p<0.05 and ** = p<0.01.
3. Results
3.1 TLR2 and TLR5 activation alter TCR-induced cytokine production in HuT78 human T cells
The goal of this study was to examine whether prior TLR induction resulted in increased TCR-mediated signaling and downstream function in human T cells. For these studies, we used a model human CD4+ T cell line, HuT78 T cells, which we have previously shown to have highly similar TCR-induced signaling and cytokine production compared to human APBTs (Bartelt et al., 2009). We then confirmed the results found in HuT78 T cells using isolated human APBTs. Before this could be accomplished, we needed to determine which TLRs are functional in HuT78 T cells. Work by Caron and coworkers have shown that Jurkat E6.1 T cell and human CD4+ peripheral blood T cells both express the mRNA for TLR2, TLR5 and TLR7 (Caron et al., 2005). To confirm these results and examine the expression of the receptors in HuT78 T cells, we used RT-PCR to assess the mRNA expression of TLR2, TLR5 and TLR7. HuT78 T cells and APBTs both express the mRNA for TLR2, TLR5 and TLR7 (Figure 1A). Together, these data show that HuT78 T cells and APBTs are potentially responsive to ligands for these receptors.
Figure 1.

TLR2 and TLR5 activation alter TCR-induced cytokine production in human T cells. (A) The expression of the mRNA for TLR2, TLR5 and TLR7 in HuT78 T cells and human APBTs was assessed by RT-PCR. The expected band sizes were 536 base pairs for TLR2, 744 base pairs for TLR5 and 481 base pairs for TLR7. (B and C) HuT78 T cells were stimulated with 1 μg/ml plate-bound anti-CD3 (OKT3) alone or simultaneously with 1 μg/ml PAM3CSK4 (TLR2), 1μg/ml Flagellin (TLR5), or 1 μg/ml R848 (TLR7) for 24 hours. (D and E) HuT78 T cells were pre-treated for 24 hours with water or the above concentrations of TLR ligands. The cells were then washed and subsequently stimulated with 1 μg/ml plate-bound anti-CD3 for 24 hours in the absence of TLR ligands. The cell supernatants were assayed by ELISA for IL-2 (B and D) and IFN-γ production (C and E). The results for each treatment in an individual experiment was normalized to the amounts produced by cells treated with anti-CD3 alone (B and D) or the water pretreatment subsequently stimulated with anti-CD3 (C and E). The results of four or more experiments were then combined and statistically compared (* = p<0.05 and ** = p<0.01) using ANOVA with Dunnett's Multiple Comparison Post Test with the reference treatment marked with #.
Previous studies have shown that human CD4+ T cells are responsive to ligands for TLR2, TLR5 and potentially TLR7 (Caron et al., 2005; Crellin et al., 2005; Komai-Koma et al., 2004; Lancioni et al., 2009; McCarron and Reen, 2009; Merlo et al., 2007; Nyirenda et al., 2011; Richardt-Pargmann et al., 2011), but the responsiveness of HuT78 T cells to these TLR ligands had yet to be examined. To this end, we stimulated HuT78 T cells for 24 hours with the stimulatory anti-CD3 antibody OKT3 alone or with the simultaneous presence of the TLR2 ligand PAM3CSK4 (PAM), the TLR5 ligand flagellin, or the TLR7 ligand R848. To measure TCR-induced T cell activation, the production the cytokines IL-2 and IFN-γ were measured by ELISA. The concurrent stimulation with TLR2 or TLR5 ligands with TCR resulted in increased TCR-induced production of IL-2 and IFN-γ in HuT78 T cells (Figure 1B and 1C), although only the TLR5-enhance production of IL-2 and IFN-γ was significantly different than TCR stimulation alone. Simultaneous activation of the TCR or TLR7 ligands did not alter cytokine production from HuT78 T cells (Figure 1B and 1C). Our findings show that only TLR2 and TLR5 consistently serve as co-stimulatory receptors in HuT78 T cells.
Although numerous previous studies have examined the effects of simultaneous stimulation of TCR and TLRs (Caron et al., 2005; Crellin et al., 2005; Komai-Koma et al., 2004; Lancioni et al., 2009; McCarron and Reen, 2009; Merlo et al., 2007; Nyirenda et al., 2011; Richardt-Pargmann et al., 2011), there is no published information on whether prior activation alters subsequent TCR-mediated cytokine production. To address this knowledge gap, we examined whether the prior activation of the same TLRs resulted in increased TCR-mediated cytokine production. For these studies, HuT78 T cells were incubated for 24 hours with ligands for TLR2, TLR5, or TLR7. After TLR stimulation, the cells were extensively washed and then treated with plate-bound anti-CD3 antibodies. Interestingly, prior treatment with either TLR2 or TLR7 ligand had little effect on subsequent TCR-induced cytokine production in HuT78 T cells (Figure 1D and 1E). Only pretreatment with flagellin resulted in a significant increase in IL-2 and IFN-γ release in TCR-stimulated HuT78 T cells (Figure 1D and 1E). Thus, only prior stimulation with the TLR5 ligand flagellin is capable of enhancing subsequent TCR-mediated cytokine production. This is the first report that pretreatment with flagellin can augment TCR-mediated cytokine production.
3.2 Flagellin pretreatment increases cytokine production in HuT78 human T cells and APBTs
To more closely examine the flagellin-mediated potentiation of T cell activity, we determined the optimal dose of flagellin that increases TCR-induced cytokine production. In these studies, HuT78 cells and APBT were pretreated with various doses of flagellin for 24 hours. The cells were extensively washed and then stimulated with plate-bound anti-CD3. Interestingly, HuT78 cells have a statistically significant increase in IL-2 and IFN-γ production at doses of flagellin of 250 μg/mL and greater when compared to cells with no flagellin pretreatment (Figure 2A and 2B). Pretreatment of APBTs with flagellin also resulted in a significant increase in IL-2 and IFN-γ production in flagellin pretreated cells compared to control cells (Figures 2C and 2D). Interestingly, in APBTs the flagellin-mediated increase in IL-2 production required CD28 costimulation, whereas IFN-γ production was not as dependent on costimulation (Figure 2C and 2D). Together, these studies indicate that human T cells have enhanced cytokine production when treated with flagellin prior to TCR stimulation.
Figure 2.

Flagellin pretreatment increases TCR-induced cytokine production in human T cells. (A and B) HuT78 T cells were treated with 0–1000 ng/ml flagellin for 24 hours. The cells were washed and then stimulated with 1μg/ml plate-bound anti-CD3 (OKT3) for 24 hours. (C and D) APBTs were treated with 0–1000 ng/ml flagellin for 24 hours. The cells were washed and then stimulated with 1μg/ml plate-bound anti-CD3 (OKT3) and/or 2 μg/ml anti-CD28 (CD28.2) for 24 hours. The cell supernatants were assayed by ELISA for IL-2 (A and C) or IFN-γ production (B and D). The results are shown as the average ± SEM of at least three independent experiments normalized to the maximal IL-2 or IFN-γ production in the individual experiment. The data was statistically compared (* = p<0.05 and ** = p<0.01) using ANOVA with Dunnett's Multiple Comparison Post Test with the reference treatment marked with #.
3.3 Flagellin-mediated potentiation of TCR-induced cytokine production in HuT78 T cells is transient
Next, we examined the length of flagellin pretreatment required for enhanced cytokine production, and the how long the flagellin-induced increase in cytokine production lasted in human T cells. These studies were performed using 250 ng/mL flagellin because this dose was the lowest dose of flagellin that consistently provided the maximal increase in TCR-mediated cytokine production (Figure 2). To determine the length of pretreatment needed for enhanced TCR-mediated cytokine production, HuT78 cells were incubated with or without flagellin for various times and then stimulated with plate-bound anti-CD3. As seen in Figure 3A, pretreatment of HuT78 T cells with flagellin for up to eight hours had no significant effect on TCR-induced cytokine production compared to HuT78 T cells that did not receive flagellin pretreatment (the 0 hour time point). In contrast, 24 hours of pretreatment resulted in amplified TCR-induced IL-2 production (Figure 3A). These studies demonstrate that the increased cytokine production seen in Figures 1 and 2 cannot be attributed to the presence of residual flagellin during TCR stimulation. If residual flagellin led to costimulation due to simultaneous TCR and TLR5 induction, then we would observe no differences in cytokine production at different times of flagellin pretreatment. This clearly is not the case; thus, our findings are because flagellin treatment directly alters T cell responsiveness prior to subsequent TCR stimulation.
Figure 3.

Flagellin-mediated potentiation of TCR-induced cytokine production in HuT78 T cells is transient. (A) HuT78 T cells were treated without flagellin (0 hour timepoint) or with 250 ng/ml flagellin for various times. The cells were washed and then stimulated with 1μg/ml plate-bound anti-CD3 (OKT3) overnight. (B) HuT78 T cells pretreated with or without 250 ng/ml flagellin and incubated overnight. The cells were washed, rested for various times, and then stimulated with 1μg/ml plate-bound anti-CD3 for 24 hours. The supernatants were assayed by ELISA for IL-2 production. The results are shown as the average ± SEM of at least three independent experiments normalized to the maximal IL-2 production in the individual experiment. The data was statistically compared (* = p<0.05 and ** = p<0.01) using ANOVA with Dunnett's Multiple Comparison Post Test with the reference treatment marked with #.
We next determined how long after flagellin treatment ended the increases in cytokine production lasted. HuT78 T cells were treated with flagellin for 24 hours. The cells were then extensively washed, rested for various times, and then stimulated with plate-bound anti-CD3. As seen in Figure 3B, resting the cells for up to 24 hours after flagellin pretreatment did not significantly decrease the flagellin-mediated increase in TCR-induced IL-2 production. However, cells rested at least 48 hours between flagellin exposure and TCR stimulation did not have increased IL-2 production compared to untreated cells (Figure 3B). These experiments show that flagellin pretreatment transiently increases TCR-induced cytokine production in human T cells for an approximately 24 hour window that occurs 24 hours after exposure to flagellin.
3.4 IRAK function is required for flagellin-mediated activation of HuT78 T cells
The data described above show conclusively that flagellin pretreatment results in a transient increase in the sensitivity of T cells to TCR induction, but whether this effect is driven specifically by TLR5 induction has not been tested. Flagellin can activate both TLR5(Kawai and Akira, 2011; Leulier and Lemaitre, 2008) and the NOD receptor NLRC4(Sutterwala and Flavell, 2009). To differentiate whether TLR5 or NLRC4 are mediating the effects of flagellin pretreatment on TCR-induced cytokine production, we used a selective inhibitor of the serine/threonine kinases IRAK1 and IRAK4. When tested in vitro against over 200 tyrosine and serine/threonine kinases, this inhibitor substantially inhibited the function of only IRAK1 and IRAK4 (see pubmed compound listing 11983295). This inhibitor will only target TLR5-mediated signaling, since NOD receptors do not utilize IRAKs (Poyet et al., 2001; Sutterwala and Flavell, 2009). For these studies, HuT78 cells were pretreated with 250 ng/mL of flagellin, in the absence or presence of various concentrations of an IRAK1/IRAK4 inhibitor. We did not observe any effects of the IRAK1/IRAK4 inhibitor on cell viability at the concentrations used for these studies (data not shown). The T cells were washed three times, resting 30 minutes between washes to remove the inhibitor, and then similar numbers of live cells were stimulated with plate-bound anti-CD3. As seen in Figure 4, the IRAK1/IRAK4 inhibitor had no significant effect on TCR-induced IL-2 and IFN-γ production in T cells that were not exposed to flagellin. However, the flagellin-mediated enhancement of TCR-induced IL-2 and IFN-γ production were significantly inhibited in a dose dependent manner in T cells, with all doses of the IRAK inhibitor resulting in reduced levels of cytokine production compared to T cells not pretreated with IRAK1/IRAK4 inhibitor. These data suggest that IRAK activity is required for the ability of flagellin to enhance TCR-mediated cytokine production, indicating that TLR5 is driving the augmentation TCR activation.
Figure 4.

IRAK function is required for flagellin-mediated activation of HuT78 T cells. HuT78 T cells treated with or without 250 ng/ml flagellin and with various concentrations of IRAK inhibitor for 24 hours. The cells were washed 3 times, with 30 minute incubation in RPMI between washes. Cells were then stimulated with 1 μg/mL OKT3 overnight. The cell supernatants were collected and assayed for IL-2 (A) and IFN-γ (B) production by ELISA. The results are shown as the average ± SEM of three to four independent experiments normalized to the IL-2 production in untreated T cells. The data was statistically compared (* = p<0.05 and ** = p<0.01) using ANOVA with Dunnett's Multiple Comparison Post Test with the reference treatment marked with #.
3.5 Flagellin pretreatment differentially affects early TCR-induced signaling in HuT78 T cells and APBTs
The previous experiments suggest that TLR5 stimulation leads to increased TCR-mediated cytokine production in human T cells. One potential mechanism for this enhanced function is that TLR5 induction results in the upregulation of TCR expression on the surface of T cells, resulting in increased TCR signaling. To test this possibility, we incubated HuT78 T cells and APBTs with or without flagellin for 24 hours and then assessed differences in steady state TCR expression by flow cytometry. Pretreatment of either HuT78 T cells or APBTs did not result in any substantial changes in the steady state expression of the TCR (Supplemental Figure 1B); thus flagellin treatment has little effect on TCR expression.
A second possible mechanism for the ability of TLR5 stimulation to enhance TCR-mediated cytokine production is that flagellin pretreatment may affect TCR-mediated signaling pathways. To investigate this mechanism, HuT78 cells and APBTs were treated for 24 hours with or without 250 ng/mL flagellin. The cells were stimulated with soluble anti-CD3 for various times and the phosphorylation of intracellular signaling proteins were assessed by immunoblotting. We first examined the phosphorylation of the Src family kinase Lck and the adaptor protein LAT, events that are required for the induction of TCR-mediated signaling pathways (Houtman et al., 2005a; Smith-Garvin et al., 2009; Sommers et al., 2004). Surprisingly, HuT78 T cells and ABPTs pretreated with flagellin had significantly decreased TCR-induced Lck and LAT phosphorylation when compared to T cells not treated with flagellin (Figure 5 and 6). We also examined calcium signaling to determine whether the decrease in the phosphorylation of LAT, which is known to control calcium flux, was also altered by pretreatment with flagellin. As expected, the calcium influx was reduced considerably in cells pretreated with flagellin compared to untreated cells (Figure 5C). However, the decreased phosphorylation of LAT and lower calcium influx did not result in reduced induction of downstream signaling, since Erk1/Erk2 activation was not significantly altered in flagellin treated T cells compared to untreated T cells, except for the 15 minute timepoint in the APBTs (Figure 5 and 6).
Figure 5.

Flagellin pretreatment differentially affects early TCR-induced signaling in HuT78 cells. (A) HuT78 T cells were pretreated for 24 hours with or without 250 ng/ml flagellin. The cells were then stimulated with 10 μg/ml anti-CD3 (OKT3) for various times and lysed. The proteins were separated by PAGE. The phosphorylation of Lck, (top panel) LAT tyrosine 191 (second panel), ERK1/2 (third panel), Akt (fourth panel), and the expression of actin (bottom panel) were examined by immunoblotting. (B) The results were normalized to actin and the maximal level of activation in the individual experiment. The results are an average of three to five independent experiments ± SEM. The data was statistically compared (* = p<0.05 and ** = p<0.01) using Student's t test. (C) HuT78 T cells were pretreated for 24 hours with or without 250 ng/mL flagellin. The cells were then incubated with the calcium sensitive dye Fluor-4. The TCR-induced influx of calcium into the T cells was assessed by flow cytometry with Ionomycin serving as a positive control. The results are representative of three separate experiments.
Figure 6.
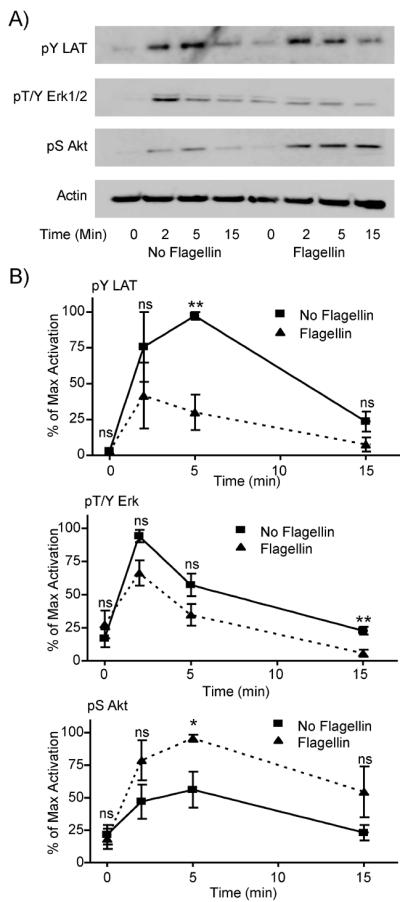
Flagellin pretreatment differentially affects early TCR-induced signaling in APBTs. (A) APBTs were pretreated for 24 hours with or without 250 ng/ml flagellin. The cells were then stimulated with 10 μg/ml anti-CD3 (OKT3) and 2 μg/ml anti-CD4 (RPA-T4) for various times and lysed. The proteins were separated by PAGE. The phosphorylation of LAT (top panel), ERK1/2 (second panel), and Akt (third panel), and the expression of actin (bottom panel) were examined by immunoblotting. (B) The results were normalized to actin and the maximal level of activation in the individual experiment. The results are an average of three to five independent experiments ± SEM. The data was statistically compared (* = p<0.05 and ** = p<0.01) using Student's t test.
We next focused on Akt activation, since this protein is also involved in cytokine release from T cells (Kane and Weiss, 2003). TCR induction alone can lead to the activation of Akt (Chapman et al., 2013; Cruz-Orcutt and Houtman, 2009; Kalland et al., 2011; Kane and Weiss, 2003; Seminario et al., 2004; Shim et al., 2011), but costimulatory receptor activation can enhance TCR-induced Akt induction (Kalland et al., 2011). Interestingly, TCR-induced Akt phosphorylation was significantly greater in HuT78 T cells and APBTs that were pretreated with flagellin compared to untreated cells (Figure 5 and Figure 6). The increased activation of Akt was not due to alterations in the expression of this protein; untreated and pretreated HuT78 T cells had similar levels of LAT, Erk and Akt expression in the samples used in Figure 5 (Supplemental Data Figure 1C and 1D). These results suggest that the mechanism for the potentiation of TCR-mediated cytokine release is due to selectively altered early TCR-mediated signaling through the Akt pathway.
4. Discussion
In these studies, we have shown for the first time that prior stimulation of TLR5, but not TLR2 or TLR7, potently amplified subsequent TCR-induced IL-2 and IFN-γ release from human T cells (Figure 1 and Figure 2). An interesting observation from this study is that the effects on TCR-mediated functions are localized to TLR5. Even though the mRNA for TLR7 is expressed in HuT78 T cells, a potent TLR7 ligand have no effect on TCR-mediated cytokine production, either when give prior to or concurrent with TCR induction (Figure 1). These data match well with more recent studies that human T cells do not have functional TLR7 (Lancioni et al., 2009; McCarron and Reen, 2009). In contrast, TLR2 is a functional costimulatory receptor for human T cells but prior activation of this receptor does not potentiate TCR function (Figure 1). We have recently shown that concurrent activation of the TCR and TLR2 results in enhanced Erk1/Erk2 and AKT phosphorylation compared to TCR stimulation alone, but had no effect on NF-κB activation, either alone or in conjunction with TCR induction (Chapman et al., 2013). The lack of NF-κB activation downstream of TLR2 in human T cells appears to be due to the expression of TLR10, a heterodimerization partner for TLR2 that does not signal through NF-κB(Guan et al., 2010). The mechanism for how simultaneous activation of the TCR and TLR5 result in increased TCR-mediated functions is unknown, but induction of TLR5 results in the activation of NF-κB in multiple cell types (Eaves-Pyles et al., 2001a; Eaves-Pyles et al., 2001b; Lee et al., 2010; Smith et al., 2003; Tallant et al., 2004). Thus, the selective ability of TLR5 to modulate subsequent TCR-mediated functions may be due to its ability to induce NF-κB function in human T cells compared to TLR2.
The findings in this paper are distinct from previous studies examining the costimulatory function of TLRs where simultaneous TCR and TLR stimulation enhance TCR-mediated downstream events in T cells. In the previous studies, signaling pathways driven by the simultaneous induction of the TCR and TLR2 or TLR5 converge to synergistically enhance cytokine production. In contrast to the previous studies that examined the costimulatory function of TLR5, our data suggests that TLR5 induction enhances the responsiveness and/or sensitivity of the T cell to TCR-mediated signals. This highlights that the “window” when TLR5 can enhance TCR functions is much longer than previously appreciated. Two recent studies have also observed that prior stimulation of T cells by IL-7, IL-15, IL-12 or type I interferons enhanced the responsiveness and/or sensitivity of T cells to subsequent TCR stimulation (Deshpande et al., 2013; Richer et al., 2013). In contrast to our studies, prior activation of T cells by these cytokines resulted in enhanced TCR-mediated Erk1/Erk2 phosphorylation (Deshpande et al., 2013; Richer et al., 2013). Additionally, IL-12 or type I interferons appear to have no effect on Lck phosphorylation but increase TCR-induced phosphorylation of ZAP-70, JNK and PLC-γ1 (Richer et al., 2013). Together, our data and the studies published by Richer and coworkers and Deshpande and coworkers highlight a novel function of inflammatory cytokines and ligands for TLR5, specifically to transiently enhance the sensitivity of T cells to subsequent TCR induction. However, the molecular mechanism that drives the potentiation of TCR signals is distinct for each receptor, suggesting that prior induction of TLR5 and inflammatory cytokines could result in subtlety different outcomes when activated alone. What is unknown is how a T cell will respond when it encounters multiple inflammatory signals. The different signaling patterns suggest that TLR5 ligands and inflammatory cytokines may have antagonistic or synergistic effects when given together. An interesting question for future studies will be to address how signals from multiple inflammatory mediators integrate to regulate the function of human T cells.
This effect of flagellin had several interesting features. First, it required more than 8 hours of stimulation before TCR-mediated signals were augmented (Figure 3A). The most probable explanation for this observation is that flagellin activation leads to alterations in gene transcription, and ultimately protein expression, which are driving altered TCR responsiveness. This could be due to the release of cytokines, the upregulation of surface receptors, and/or changes in the expression of proteins that control intracellular signaling. Our data point to alterations in signaling as the most likely mechanism, since flagellin pretreatment has differential effects on signaling pathways downstream of the TCR. It is possible that TLR5 induction results in transcriptional changes in genes that control the activation of Akt and other TCR-mediated signaling pathways. Previous studies have shown that flagellin pretreatment in human T cells results in increased SOCS1 expression, which then inhibits TCR-mediated ZAP-70 phosphorylation (Okugawa et al., 2006). It is also possible the TLR5 controls the expression of microRNAs. Several microRNAs, including miRs-181a, -214, -19b, -21, -30b and -155, have been shown to regulate the expression of PTEN, which controls PI3 kinase function, and the phosphatases, dual specificity phosphatases 5, 6, and 10, SHP-2 and PTPN-22 (Chang et al., 2012; Jiang et al., 2011; Jindra et al., 2010; Li et al., 2012; Li et al., 2007). Future studies will elucidate the exact mechanism of the augmentation of TCR-induced functions by TLR5 activation.
A second interesting feature of the ability of TLR5 stimulation to increase TCR-induced cytokine production is that the effect is transient. The increased TCR-mediated cytokine production is maximal at 24 hours but by 48 hours post treatment this effect is greatly diminished (Figure 3A). Similarly, the effects of flagellin on TCR-induced NFAT activation required at least 6 hours of prior treatment (Okugawa et al., 2006). Additionally, Richer and coworkers observed that the enhanced TCR sensitivity due to inflammatory cytokines was transient, with increased potentiation occurring at five days post infection compared to eight days post infection (Richer et al., 2013). Additionally, if flagellin is withdrawn from the cells, the boosting of TCR-mediated cytokine production lasts only for 24–36 hours (Figure 3B). This observation is in direct contrast to the effects of IL-7 and IL15 on TCR sensitivity, whose effects only lasted for approximately two hours (Deshpande et al., 2013). These data suggest that the presence of flagellin provides a transient window of 24–48 hours after treatment where T cells are more sensitive to TCR-mediated activation.
The effect of prior flagellin treatment on TCR-induced cytokine production appears to require the enzymatic function of IRAK1 and IRAK4 (Figure 4). Since these kinases are required for signaling downstream of TLR5 (Flannery and Bowie, 2010), this supports the role of TLR5 in the increased TCR function upon flagellin pretreatment. It is possible that flagellin treatment could also lead to the release of other cytokines, such as IL-1β, whose receptors utilize IRAK1 and IRAK4. This is not likely the case since neither HuT78 T cells nor human APBTs release IL-1β upon TCR, TLR or costimulatory receptor stimulation in our hands (data not shown and (Bartelt et al., 2009)). It is also possible that NOD receptors that are activated by flagellin, most notably NLRC4, could be mediating this effect of flagellin. Again, this is unlikely since NLRC4 does not appear to utilize IRAKs (Sutterwala and Flavell, 2009). In the end, these studies strongly suggest that signals emanating from TLR5 via IRAK1 and IRAK4 potentiate TCR functions.
We have also characterized the molecular mechanism for how prior exposure to flagellin leads to increased TCR-mediated cytokine production. Interestingly, pretreatment with flagellin results in the decreased ability of the TCR to induce Lck and LAT phosphorylation and calcium influx compared to control T cells, while having no effect on TCR-induced Erk1/Erk2 phosphorylation (Figures 5 and 6). These findings are similar to work by Okugawa and coworkers, who observed that flagellin pretreatment resulted in decreased TCR-mediated ZAP-70 phosphorylation and NFAT induction (Okugawa et al., 2006). Our work is in contrast to priming by IL-12 and type I interferons, which enhance TCR-induced ZAP-70, PLC-γ1, Erk1/Erk2 and JNK phosphorylation (Richer et al., 2013). This suggests that prior TLR5 induction has a distinct molecular mechanism that decreases the activation of signaling complexes at the TCR and/or LAT but is still sufficient to drive relatively normal downstream signaling. Instead, we observed that TCR-mediated Akt phosphorylation was substantially increased in HuT78 T cells and APBTs pretreated with flagellin. Akt activation plays a critical role in multiple T cell events, including thymocyte and mature T cell differentiation, cellular metabolism, cytokine release and apoptosis (Juntilla and Koretzky, 2008; Kane and Weiss, 2003; Pierau et al., 2009). We have observed that a potent Akt inhibitor (BML 257) markedly reduces TCR-mediated IL-2 production, indicating the critical role this kinase plays in cytokine production in human T cells (data not shown). Together, our findings suggest that flagellin pretreatment increases the ability of the TCR to stimulate Akt function leading to increased T cell activation and function. We plan to actively pursue the exact mechanism for the flagellin-mediated increase of TCR-induced Akt function in future studies.
Ultimately, these studies point to a critical question, what is the physiological function of the priming of T cells by flagellin? The simple answer is that flagellin primes T cells for activation as a method of sensing infection by bacterial pathogens for a longer timeframe than would occur if it could only function as a costimulatory receptor. Flagellin flowing into the secondary lymphoid tissues from the lymph and blood may prime resident T cells to respond to APCs that migrate into these tissues from sites of infection. IL-2 production, the cytokine critical for T cell activation and expansion, is stimulated by prior TLR5 induction best in the presence of both TCR and CD28 stimulation. Therefore, the effect of flagellin on T cells appears to enhance, not replace, CD28-mediated costimulation. Flagellin may also enhance the ability of activated T cells to respond to weak TCR signals at sites of infection and inflammation. We would predict this to be independent of costimulation, since flagellin pretreatment affects the release of the effector cytokine IFN-γ occurs in the presence and absence of costimulation. Additionally, this effect is transient so that once the infection is cleared, T cells would rapidly lose their increased sensitivity to TCR signals. This would be critical to suppressing autoimmunity, which several studies have suggested is due to aberrant activation of TLRs in T cells (Marsland and Kopf, 2007). In fact, priming of TCR sensitivity by IL-7 and IL-15 increases the ability of T cells to respond to auto-antigens (Deshpande et al., 2013). Finally, the potentiation of T cell responsiveness would be antigen independent. Infection by one pathogen would greatly enhance the T cell responses to other infections. T cells that are primed by IL-12 or type I interferons from one infection do have enhanced TCR sensitivity to other pathogens (Richer et al., 2013). Thus, T cells would respond better to simultaneous infections by multiple pathogens.
5. Conclusion
These studies are the first to show that exposure of human T cells to flagellin leads to a transient TLR5-induced increase in the sensitivity of these cells to subsequent TCR activation. This allows T cells to sense the presence of pathogenic bacteria and/or other pathogens and then respond for a longer timeframe when activated by APCs expressing TCR-specific antigen/MHC complexes. These experiments provide novel insight into the basic mechanism that human T cells use to respond to infection and indicate that TLR5 priming plays a critical role in the early events leading to T cell activation.
Supplementary Material
Highlights
Activation of TLR5, but not TLR2, potentiates ensuing TCR-induced cytokine release
The effects of TLR5 on TCR-mediated cytokine production lasts for 24–36 hours
Prior TLR5 induction enhances TCR-mediated AKT activation
TLR5 pretreatments suppresses TCR-induced Lck and LAT induction
Acknowledgements
We want to thank Michaela Collins for technical support and Nicole Chapman, Rebekah Bartelt, and Jayakumar Poovassery for helpful discussions and critical reading of the manuscript. This work was supported by the Biological Sciences Funding Program from the Office of the Vice President for Research at the University of Iowa, the American Heart Association (Grant number 0830244N to J.C.D.H.) and the National Institutes of Health (R01 CA136729 to J.C.D.H. and T32 AI007485 to M.Y.B.).
Abbreviations
- APBTs
activated peripheral blood T cells
- APC
antigen presenting cells
- IFN-γ
Interferon-γ
- PAM
PAM3CSK4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartelt RR, Cruz-Orcutt N, Collins M, Houtman JC. Comparison of T cell receptor-induced proximal signaling and downstream functions in immortalized and primary T cells. PLoS ONE. 2009;4:e5430. doi: 10.1371/journal.pone.0005430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brikos C, O'Neill LA. Signalling of toll-like receptors. Handb Exp Pharmacol. 2008;183:21–50. doi: 10.1007/978-3-540-72167-3_2. [DOI] [PubMed] [Google Scholar]
- Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551–7. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- Chang CC, Zhang QY, Liu Z, Clynes RA, Suciu-Foca N, Vlad G. Downregulation of inflammatory microRNAs by Ig-like transcript 3 is essential for the differentiation of human CD8(+) T suppressor cells. J Immunol. 2012;188:3042–52. doi: 10.4049/jimmunol.1102899. [DOI] [PubMed] [Google Scholar]
- Chapman NM, Bilal MY, Cruz-Orcutt N, Knudson C, Madinaveitia S, Light J, Houtman JC. Distinct signaling pathways regulate TLR2 co-stimulatory function in human T cells. Cell Signal. 2013;25:639–50. doi: 10.1016/j.cellsig.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051–9. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- Cruz-Orcutt N, Houtman JCD. PI3 kinase function is vital for the function but not formation of LAT-mediated signaling complexes. Mol Immunol. 2009;46:2274–83. doi: 10.1016/j.molimm.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Deshpande P, Cavanagh MM, Le Saux S., Singh K, Weyand CM, Goronzy JJ. IL-7- and IL-15-mediated TCR sensitization enables T cell responses to self-antigens. J Immunol. 2013;190:1416–23. doi: 10.4049/jimmunol.1201620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler SK, Foxwell BM. The role of toll-like receptors in chronic inflammation. Int J Biochem Cell Biol. 2010;42:506–18. doi: 10.1016/j.biocel.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Eaves-Pyles T, Murthy K, Liaudet L, Virag L, Ross G, Soriano FG, Szabo C, Salzman AL. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J Immunol. 2001a;166:1248–60. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]
- Eaves-Pyles TD, Wong HR, Odoms K, Pyles RB. Salmonella flagellin-dependent proinflammatory responses are localized to the conserved amino and carboxyl regions of the protein. J Immunol. 2001b;167:7009–16. doi: 10.4049/jimmunol.167.12.7009. [DOI] [PubMed] [Google Scholar]
- Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80:1981–91. doi: 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Guan Y, Ranoa DR, Jiang S, Mutha SK, Li X, Baudry J, Tapping RI. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J Immunol. 2010;184:5094–103. doi: 10.4049/jimmunol.0901888. [DOI] [PubMed] [Google Scholar]
- Houtman JCD, Barda-Saad M, Samelson LE. Examining multiprotein signaling complexes from all angles: the use of complementary techniques to characterize the complex formation at the adapter protein LAT. FEBS J. 2005a;272:5426–35. doi: 10.1111/j.1742-4658.2005.04972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtman JCD, Houghtling RA, Barda-Saad M, Toda Y, Samelson LE. Early phosphorylation kinetics of proteins involved in proximal TCR-mediated signaling pathways. J Immunol. 2005b;175:2449–58. doi: 10.4049/jimmunol.175.4.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li C, Olive V, Lykken E, Feng F, Sevilla J, Wan Y, He L, Li QJ. Molecular dissection of the miR-17–92 cluster's critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood. 2011;118:5487–97. doi: 10.1182/blood-2011-05-355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindra PT, Bagley J, Godwin JG, Iacomini J. Costimulation-dependent expression of microRNA-214 increases the ability of T cells to proliferate by targeting Pten. J Immunol. 2010;185:990–7. doi: 10.4049/jimmunol.1000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, Fletcher MC, Ledbetter JA, Samelson LE. Increases in tyrosine phosphorylation are detectable before phospholipase C activation after T cell receptor stimulation. J Immunol. 1990;144:1591–9. [PubMed] [Google Scholar]
- Juntilla MM, Koretzky GA. Critical roles of the PI3K/Akt signaling pathway in T cell development. Immunol Lett. 2008;116:104–10. doi: 10.1016/j.imlet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Kalland ME, Oberprieler NG, Vang T, Tasken K, Torgersen KM. T cell-signaling network analysis reveals distinct differences between CD28 and CD2 costimulation responses in various subsets and in the MAPK pathway between resting and activated regulatory T cells. J Immunol. 2011;187:5233–45. doi: 10.4049/jimmunol.1101804. [DOI] [PubMed] [Google Scholar]
- Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029–34. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni R, Behboudi S, Sharif S. Insights into the role of Toll-like receptors in modulation of T cell responses. Cell Tissue Res. 2011;343:141–52. doi: 10.1007/s00441-010-1017-1. [DOI] [PubMed] [Google Scholar]
- Lancioni CL, Thomas JJ, Rojas RE. Activation requirements and responses to TLR ligands in human CD4+ T cells: comparison of two T cell isolation techniques. J Immunol Methods. 2009;344:15–25. doi: 10.1016/j.jim.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim H, Kim S, Kim KH, Chung JH. Activation of toll-like receptors 2, 3 or 5 induces matrix metalloproteinase-1 and -9 expression with the involvement of MAPKs and NF-kappaB in human epidermal keratinocytes. Exp Dermatol. 2010;19:e44–9. doi: 10.1111/j.1600-0625.2009.00963.x. [DOI] [PubMed] [Google Scholar]
- Leulier F, Lemaitre B. Toll-like receptors--taking an evolutionary approach. Nat Rev Genet. 2008;9:165–78. doi: 10.1038/nrg2303. [DOI] [PubMed] [Google Scholar]
- Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, Goronzy JJ. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. 2012;18:1518–24. doi: 10.1038/nm.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–61. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Marsland BJ, Kopf M. Toll-like receptors: paving the path to T cell-driven autoimmunity? Curr Opin Immunol. 2007;19:611–4. doi: 10.1016/j.coi.2007.07.022. [DOI] [PubMed] [Google Scholar]
- McCarron M, Reen DJ. Activated human neonatal CD8+ T cells are subject to immunomodulation by direct TLR2 or TLR5 stimulation. J Immunol. 2009;182:55–62. doi: 10.4049/jimmunol.182.1.55. [DOI] [PubMed] [Google Scholar]
- Merlo A, Calcaterra C, Menard S, Balsari A. Cross-talk between toll-like receptors 5 and 9 on activation of human immune responses. J Leukoc Biol. 2007;82:509–18. doi: 10.1189/jlb.0207100. [DOI] [PubMed] [Google Scholar]
- Meyer TP, Zehnter I, Hofmann B, Zaisserer J, Burkhart J, Rapp S, Weinauer F, Schmitz J, Illert WE. Filter Buffy Coats (FBC): a source of peripheral blood leukocytes recovered from leukocyte depletion filters. J Immunol Methods. 2005;307:150–66. doi: 10.1016/j.jim.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Nyirenda MH, Sanvito L, Darlington PJ, O Brien K., Zhang GX, Constantinescu CS, Bar-Or A, Gran B. TLR2 Stimulation Drives Human Naive and Effector Regulatory T Cells into a Th17-Like Phenotype with Reduced Suppressive Function. J mmunol. 2011 doi: 10.4049/jimmunol.1003715. [DOI] [PubMed] [Google Scholar]
- Okugawa S, Yanagimoto S, Tsukada K, Kitazawa T, Koike K, Kimura S, Nagase H, Hirai K, Ota Y. Bacterial flagellin inhibits T cell receptor-mediated activation of T cells by inducing suppressor of cytokine signalling-1 (SOCS-1) Cell Microbiol. 2006;8:1571–80. doi: 10.1111/j.1462-5822.2006.00731.x. [DOI] [PubMed] [Google Scholar]
- Pierau M, Engelmann S, Reinhold D, Lapp T, Schraven B, Bommhardt UH. Protein kinase B/Akt signals impair Th17 differentiation and support natural regulatory T cell function and induced regulatory T cell formation. J Immunol. 2009;183:6124–34. doi: 10.4049/jimmunol.0900246. [DOI] [PubMed] [Google Scholar]
- Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001;276:28309–13. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- Richardt-Pargmann D, Wechsler M, Krieg AM, Vollmer J, Jurk M. Positive T cell co-stimulation by TLR7/8 ligands is dependent on the cellular environment. Immunobiology. 2011;216:12–23. doi: 10.1016/j.imbio.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer MJ, Nolz JC, Harty JT. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity. 2013;38:140–52. doi: 10.1016/j.immuni.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminario M-C, Precht P, Bunnell SC, Warren SE, Morris CM, Taub D, Wange RL. PTEN permits acute increases in D3-phosphoinositide levels following TCR stimulation but inhibits distal signaling events by reducing the basal activtiy of Akt. Eur. J. Immunol. 2004;34 doi: 10.1002/eji.200425206. In Press. [DOI] [PubMed] [Google Scholar]
- Shim EK, Jung SH, Lee JR. Role of two adaptor molecules SLP-76 and LAT in the PI3K signaling pathway in activated T cells. J Immunol. 2011;186:2926–35. doi: 10.4049/jimmunol.1001785. [DOI] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MF, Jr., Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, Crowe S, Goldberg JB. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem. 2003;278:32552–60. doi: 10.1074/jbc.M305536200. [DOI] [PubMed] [Google Scholar]
- Sommers CL, Samelson LE, Love PE. LAT: a T lymphocyte adapter protein that couples the antigen receptor to downstream signaling pathways. Bioessays. 2004;26:61–7. doi: 10.1002/bies.10384. [DOI] [PubMed] [Google Scholar]
- Suhir H, Etzioni A. The role of Toll-like receptor signaling in human immunodeficiencies. Clin Rev Allergy Immunol. 2010;38:11–9. doi: 10.1007/s12016-009-8135-0. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Flavell RA. NLRC4/IPAF: a CARD carrying member of the NLR family. Clin Immunol. 2009;130:2–6. doi: 10.1016/j.clim.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallant T, Deb A, Kar N, Lupica J, de Veer MJ, DiDonato JA. Flagellin acting via TLR5 is the major activator of key signaling pathways leading to NF-kappa B and proinflammatory gene program activation in intestinal epithelial cells. BMC Microbiol. 2004;4:33. doi: 10.1186/1471-2180-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.