

Abstract
Developmental conservation among related species is a common generalization known as von Baer’s third law and implies that early stages of development are the most refractory to change. The “hourglass model” is an alternative view that proposes that middle stages are the most constrained during development. To investigate this issue, we undertook a genomic approach and provide insights into how natural selection operates on genes expressed during the first 24 h of Drosophila ontogeny in the six species of the melanogaster group for which whole genome sequences are available. Having studied the rate of evolution of more than 2,000 developmental genes, our results showed differential selective pressures at different moments of embryogenesis. In many Drosophila species, early zygotic genes evolved slower than maternal genes indicating that mid-embryogenesis is the stage most refractory to evolutionary change. Interestingly, positively selected genes were found in all embryonic stages even during the period with the highest developmental constraint, emphasizing that positive selection and negative selection are not mutually exclusive as it is often mistakenly considered. Among the fastest evolving genes, we identified a network of nucleoporins (Nups) as part of the maternal transcriptome. Specifically, the acceleration of Nups was driven by positive selection only in the more recently diverged species. Because many Nups are involved in hybrid incompatibilities between species of the Drosophila melanogaster subgroup, our results link rapid evolution of early developmental genes with reproductive isolation. In summary, our study revealed that even within functional groups of genes evolving under strong negative selection many positively selected genes could be recognized. Understanding these exceptions to the broad evolutionary conservation of early expressed developmental genes can shed light into relevant processes driving the evolution of species divergence.
Keywords: embryonic genes, Nups, hourglass model, dosage compensation
Introduction
During ontogeny, most hierarchical features are a consequence of the timing of developmental events. Indeed, as later events depend on earlier ones, developmental constraints during embryonic stages are widespread (Carroll et al. 2001). As a consequence, genes involved in early developmental processes are expected to be under strong negative selection to prevent deleterious cascading effects (Roux and Robinson-Rechavi 2008; Artieri et al. 2009). In Drosophila, it has been recently shown that embryonic genes evolve at a slower pace than postembryonic and adult expressed genes (Artieri et al. 2009). However, the pattern of early conservation has not been supported when embryonic-specific analyses were carried out (Davis et al. 2005; Cruickshank and Wade 2008; Kalinka et al. 2010).
Drosophila development is characterized by a fast segmentation process. Segment determination starts very early in embryogenesis, when approximately 3 h after fertilization the position and identity of all body structures are determined simultaneously during the blastoderm stage (Foe and Alberts 1983). Although not all genes involved in segmentation have an exclusive timing of expression, two hierarchical regulatory layers can be identified: An initial phase of maternal component specification and a succeeding phase involving more complex and interactive zygotic gene expression (Schroeder et al. 2004). Genes in early layers (maternal genes) regulate the expression of genes in subsequent layers (gap, pair-rule, segment polarity, and Hox genes) but not vice versa (Jaeger 2009). In addition, there is cross-regulation among genes in the same hierarchical layer (Manu et al. 2009). Understanding these two phases and their connections is necessary to reconstruct the evolution of the embryonic system (Wilkins 2002).
The developmental stage that is most refractory to evolutionary change is commonly known as the phylotypic stage (Raff 1996). Based on genome-wide expression comparisons (Kalinka et al. 2010) and sequence evolution analyses (Davis et al. 2005; Cruickshank and Wade 2008), the initiation of organogenesis during the burst of expression of segment polarity and Hox genes appears to be the Drosophila phylotypic stage. Instead, earlier embryonic stages, including the maternal component of segmentation, have markedly diverged within and among species (Galis et al. 2002). Thus, given that the highest constraint takes place during middle embryogenesis, a developmental hourglass model likely reflects Drosophila embryonic evolution (Raff 1986).
Despite the strong developmental constraint across phylotypic stages, cases of rapid evolution at embryonic expressed genes were identified. Specifically, the rapid evolution reported for both maternally and zygotically expressed Hox and Hox-derived genes challenges the view of general conservation of embryonic genes (Barker et al. 2005; Casillas et al. 2006). Whether these cases of rapid evolution are driven by positive selection (PS) or just relaxation of selective constraints (RSCs) remains unknown. Moreover, although the incidence of fast evolving genes expressed in early development is expected to be low, there is a lack of studies searching for the signatures of PS and hence fast adaptive change in embryonic genes.
Taking advantage of fine time-course gene expression information (Hooper et al. 2007), the recent burst of Drosophila species whole genomes sequences (Clark et al. 2007) and the development of powerful statistical and bioinformatic tools (Yang 2003, 2007), here we investigate the evolution of tightly regulated groups of embryonic genes in the Drosophila melanogaster species group. Specifically, we studied the evolutionary rates of genes involved in the three major embryonic groups recognized by Hooper et al. (2007): Maternal, early zygotic, and late zygotic genes, all predominantly expressed in a stage-specific fashion during embryogenesis. In addition, we performed specific maximum likelihood (ML) tests to distinguish true cases of PS from likely cases of RSC (Serra et al. 2011). Importantly, our genomic-scale study allowed us not only to dissect the rapidly evolving fraction of the Drosophila embryonic transcriptome but also to contrast the incidence of positively selected genes (PSG) at the phylotypic stage with less constrained periods of development. After identifying rapid evolving genes expressed during embryogenesis, a functional analysis of this particular fraction of the genome was performed to shed light into the processes involved in developmental adaptation.
Having studied the evolution of more than 2,000 embryonic genes, our results are in agreement with the hourglass model of evolution. However, the incidence of PSGs was homogeneous across embryonic development. Among the fastest evolving genes, we identified a network of nucleoporins genes (Nups) as part of the maternal transcriptome. However, these rapidly evolving genes exhibited signatures of adaptive evolution only in the most recently diverged species of the melanogaster species group studied. Because Nups have been shown to be involved in hybrid incompatibilities (Tang and Presgraves 2009; Sawamura et al. 2010), we discuss the possible role of nuclear pore-related developmental processes in species divergence.
Materials and Methods
Data Acquisition
Information on the timing of gene expression was obtained from a previous survey of embryonic gene expression that performed a time-course genome-wide microarray analysis (Hooper et al. 2007). The survey consisted of an extensive analysis of the fly transcriptome obtained in 30 time points, uncovering the entire 24-h period in which the fertilized egg develops into a first-instar larva. By applying convolution methods (e.g., common “sharp” transcript changes among genes), the authors identified three major categories for which all transcript levels increase and/or decrease within a certain time interval, suggesting a common mode of regulation. The first group includes highly expressed genes that encode maternal transcripts that show a subsequent decrease in expression by 12 h after egg-laying. The second group consists of early zygotic genes with high transcription levels starting at 2–3 h after egg laying (embryo stage 5) and that later decrease in midembryogenesis. Finally, late zygotic genes encode transcripts for which we only observe an increase in expression starting 13 h after egg laying (embryo stage 16–17), maintaining high expression levels till the end of embryogenesis. The total number of genes for each embryonic stage included in our analysis along with the proportion of sex-biased expression is given in table 1. Gene orthology relationships among the six Drosophila species studied could be ascertained for only 62% of the genes studied by Hooper et al. (2007). Thus, 999 out of 1,534 of the maternal genes (class I), 496 out of 792 of the early zygotic genes (class II), and 597 out of 1,053 of the late zygotic genes (class III) could be included in the present report. Classes refer to the nomenclature used by Hooper et al. (2007).
Table 1.
Groups of Embryonic Genes and Information of Sex-Biased Expression
| Groups of Genes | No. of Genes | Female-Biased Genes | Male-Biased Genes | Unbiased Genes | Unclassified Genes |
|---|---|---|---|---|---|
| Maternal genes | 999 | 805 (80.6%) | 51 (5.1%) | 99 (10%) | 44 (4.3%) |
| Early zygotic genes | 496 | 151 (30.4%) | 87 (17.5%) | 246 (49.6%) | 12 (2.5%) |
| Late zygotic genes | 597 | 31 (5.2%) | 258 (43.2%) | 237 (39.7%) | 71 (11.9%) |
Evolutionary Rate Estimation
Coding sequences (CDS) data of embryonic genes were obtained from the genomes of 12 Drosophila species available at www.flybase.org (last accessed November 18, 2013; Clark et al. 2007). CDSs were aligned with MUSCLE (Edgar 2004) using predicted amino acid sequences as templates. Aligned columns containing gaps were removed. We only included in the analysis the six species of the D. melanogaster group because saturation in silent site divergence outside the D. melanogaster species group precludes the use of all 12 genomes (Larracuente et al. 2008). For each gene for which orthology could be confidently determined, we calculated the ratio of nonsynonymous (dN) to synonymous (dS) substitutions rates (ω) for each species. Estimates of ω were obtained applying a free ratio branch ML model using CodeML program of the PAML 4 package (Yang 2007). Values of evolutionary rates (dS, dN, and ω) for all genes analyzed are shown in supplementary table S1, Supplementary Material online.
PS was evaluated using two different branch-site models (A and A1) also implemented in CodeML (Yang 2007). Branches in the phylogeny were defined a priori as foreground and background lineages. Under these models, only the foreground lineage may contain events of PS. The test was performed independently for each species by marking its corresponding terminal branch, this mark indicates that a specific evolutionary model may be applied to the flagged branch (either evolving under three main classes of evolutionary rate ω0 < 1, ω1 = 1, ω2a/b > 1; or in the case of A1 model, only under ω0 or ω1). Because the compared models are nested, likelihood ratio tests were performed and likelihood ratio tests statistics {2Δℓ = −2 [ln (likelihood for null model) − ln (likelihood for alternative model)]} were posteriorly transformed into exact P values using the pchisq function of the R statistical package. Likelihood ratio tests were performed using a χ2 distribution with df = 2 for Test A and df = 1 for Test A1, which have been shown to be conservative under conditions of PS (Zhang et al. 2005). P values derived from PS analyses were false discovery rate-adjusted using the method of Benjamini and Hochberg (1995). In contrast to the statistical behavior of previous branch-site tests, the methodology proposed by Zhang et al. (2005) represents an improvement in this kind of test based on the comparison of the ML of different evolutionary models. This approach has proved to be able to successfully differentiate PS from RSCs and weak PS. For further details of the parameters used in ML models, see Lavagnino et al. (2012). All adjusted P values of branch-site models are reported in supplementary table S2, Supplementary Material online. In addition, supplementary table S3, Supplementary Material online contains all genes evolving under PS in all species studied.
Heat Map Construction and Clustering Analysis
We employed Ward’s (1963) method for clustering analysis using dN/dS values from free ratio branch ML model. Clusters were defined using the 99.9 percentile of the distance matrix as threshold value. dN/dS values upper than 1 were considered equal to 1 to avoid bias toward infinite values.
Gene-Set Selection Analysis
We performed a gene-set enrichment analysis employing the program BABELOMICS (Al-Shahrour et al. 2007) to study the association of specific Gene Ontology (GO) terms to fast evolving genes in each particular stage studied (maternal, early zygotic, and late zygotic). For this reason, all genes were ranked according to the ω value in D. melanogaster and looked for blocks of functionally related genes in the group of the fast evolving genes. The program also corrected P values for multiple adjustment effects by false discovery rate.
Identification of Networks of PSG
We searched for networks of interacting PSG using R-spider (Antonov et al. 2010). This tool determines whether interactions between input genes are greater than expected by chance. Input data were the complete list of genes classified as PSG in all Drosophila species studied. Finally, employing STRING database (Szklarczyk et al. 2011), the network was built with all Nups and interacting PSG allowing one missing node.
Temporal Specificity of Nups across Development
We adapted the tissue specificity index, τ. to investigate how narrow Nups network genes expression is across development:
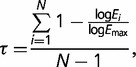 |
where N is the number of stages being compared, Ei is the expression in stage i, and Emax is the maximum expression reached by the gene across stages (Yanai et al. 2005). In our case, we employed expression data from time-course genome-wide microarray analysis of Graveley et al. (2011). τ ranges from 0 to 1, with larger τ values indicating greater temporal specificity.
Results
Rates of Evolution in Coregulated Groups of Genes during Embryogenesis
ML estimates of dN/dS of 2,092 embryonic genes (table 1 and supplementary table S1, Supplementary Material online, for detailed information) using a free ratio branch model revealed significant differences among maternal, early zygotic, and late zygotic groups of genes in all Drosophila species, except D. sechellia (fig. 1). In D. melanogaster and D. simulans, maternal genes evolved significantly faster than early zygotic genes, while maternal genes evolved faster than late zygotic genes in D. melanogaster (fig. 1). By contrast, late zygotic genes evolved significantly faster than maternal and early zygotic genes in D. erecta, D. yakuba, and D. ananassae (fig. 1).
Fig. 1.—

Box plots show the distribution of dN/dS values for maternal (M), early zygotic (EZ), and late zygotic (LZ) genes for the six species of the D. melanogaster group. Each box extends from the first to the third quartile, with the line in the middle of the box indicating the median. Asterisks show a significant difference in dN/dS between different developmental stages in a Kruskal–Wallis test. For D. melanogaster: M versus EZ, P = 0.020, M versus LZ, P = 0.026; for D. simulans: M versus EZ, P = 0.019; for D. yakuba: M versus LZ, P < 0.001, EZ versus LZ, P = 0.006; for D. erecta: M versus LZ, P < 0.001, EZ versus LZ, P < 0.001; for D. ananassae: M versus EZ, P < 0.001, M versus LZ, P < 0.001, EZ versus LZ, P = 0.044. To easier the lecture, the plot was truncated for dN/dS values >0.5.
A clustering analysis using dN/dS values of each gene from the six species identified four different groups of genes (fig. 2). The largest cluster included genes with the lowest dN/dS values and clusters 2, 3, and 4 contained genes with fast evolutionary rates. Interestingly, maternal, early zygotic, and late zygotic genes were not randomly distributed across clusters (χ2 = 10.9, P = 0.0026). Specifically, slow evolving genes (cluster 1) were enriched in early zygotic genes. Such pattern indicates a shared signature of purifying selection during middle embryogenesis across species. On the contrary, fast evolving genes grouped in clusters 3 and 4 exhibited lineage-specific acceleration in D. sechellia and D. simulans, respectively. Finally, a common across-species signature of rapid evolution was detected for genes of cluster 2.
Fig. 2.—

Heat map using dN/dS values for the six species of Drosophila studied. We employed ward method for clustering analysis using dN/dS values derived from free ratio branch ML model.
A gene-set selection analysis (Serra et al. 2011) detected functional sets of rapidly evolving genes in each one of the three embryonic stages of D. melanogaster development (table 2). Notably, we found that rapidly evolving genes of maternal and early zygotic expression are enriched in components of intracellular membranes such as “pore complex” and “organelle envelope” in maternal expressed genes and “intracellular membrane-bound organelle” in the early zygotic genes (table 2).
Table 2.
Overrepresentation of GOs in Fast Evolving Genes
 |
Note.—Red refers to GO biological function terms, blue to GO molecular function terms, and green to GO cellular component terms.
It is known that gene expression level and genomic location are among the most important factors affecting evolutionary rates in Drosophila species (Clark et al. 2007). Thus, we investigated the distribution of the genes included in the three major embryonic groups among chromosomes and the relationship between gene expression level and evolutionary rates to rule out the possibility that these factors other than developmental timing of expression are shaping the differential evolutionary rates observed. First, we found that the genes included in the three major embryonic groups are randomly distributed in the genome (χ2 = 7.4, P = 0.285). Second, though a regression analysis of the entire set of genes used in our study indicates that highly expressed genes evolved more slowly than less expressed ones (F1,1740 = 18.09, P < 0.0001), a Kruskal–Wallis analysis of variance (ANOVA) showed that differences in the level of gene expression among genes involved in the different embryonic stages were not significant (P = 0.085). Thus, we can argue that although gene expression level and genomic location are factors known to influence gene sequence evolution in Drosophila species these factors do not affect the comparisons performed in this study.
PSG in the Embryonic Transcriptome
We searched for cases of PSG employing the tests developed by Zhang et al. (2005), which permit to distinguish between cases of PS from false positives due to RSCs (or weak signals of PS). Interestingly, PSGs were found in all stages and species studied (supplementary table S3, Supplementary Material online). Surprisingly, a comparison of the incidence of PS between embryonic and adult stages in the six species of the D. melanogaster group analyzed revealed a higher proportion of PSG in embryonic transcriptomes in D. simulans, D. sechellia, D. erecta, and D. ananassae as shown in table 3. Nevertheless, it is worth mentioning that evolutionary rates of embryonic genes were significantly lower than male-adult expressed genes for all Drosophila species (fig. 3), indicating that the general slower pace of evolution is independent of the high incidence of PS in embryonic genes of the aforementioned species.
Table 3.
Comparison of the Incidence of PSGs in Embryonic and Adult Expressed Genes of the Six Drosophila Species
| Species | Embryonic PSGs | Adult PSGs | χ2 |
|---|---|---|---|
| D. melanogaster | 13 (0.62%) | 9 (0.9%) | ns |
| D. simulans | 61 (2.92%) | 10 (1%) | 10.9*** |
| D. sechellia | 47 (2.25%) | 6 (0.6%) | 10.8*** |
| D. yakuba | 9 (0.43%) | 3 (0.3%) | ns |
| D. erecta | 14 (0.67%) | 1 (0.1%) | 4.5* |
| D. ananassae | 62 (2.96%) | 7 (0.7%) | 15.7*** |
Note.—Number of embryonic expressed genes = 2,092; number of adult expressed genes = 993. *P < 0.05, **P < 0.01, ***P < 0.001. ns: not significant
Fig. 3.—

Box plots show the distribution of dN/dS values for embryonic and adult expressed genes for the six species of the D. melanogaster group. Adult expressed genes evolved significantly faster than embryonic genes in all species (Kolmogorov–Smirnov test, P < 0.001). Adult expressed genes were represented by clusters 18–20 from Graveley et al. (2011). To easier the lecture, the plot was truncated for dN/dS values >0.8.
Network of Embryonic PSG
After the identification of PSG in the embryonic stages of the six species of the D. melanogaster species group sequenced so far, we searched for networks of interacting PSG using the program R-spider (Antonov et al. 2010). Interestingly, only a single network of PSG was identified in the maternal transcriptome (P = 0.025). This module consists of a set of nucleoporins (Nups) genes that encodes proteins involved in the structure of the nuclear pore complex. Within this network, we found cases of PSG in all species studied (table 4). To further investigate the evolution of the Nups network, we divided the sample of embryonic genes into two groups: The Nups network and the rest of embryonic genes and compared their rate of evolution in each species. We found that the Nups network evolved faster than the rest of the embryonic genes in D. sechellia, D. simulans, and D. melanogaster but not in D. erecta, D. yakuba, and D. ananassae (fig. 4). Moreover, to identify at which point in the phylogeny started the acceleration of Nups evolution, we compared the nonsynonymous substitution rates of Nups in each lineage with the rates calculated for the respective most recent common ancestors in the Drosophila phylogeny. These tests also revealed a significant increase in the pace of the evolution of Nups though only in D. sechellia (fig. 4; Wilcoxon-matched pairs test: N = 15, Z = 2.7, P < 0.01). Interestingly, these results are in agreement with a recent report showing nucleoporins as a preferential target of PS in D. mauritiana, in which D. sechellia is a member of the simulans clade (Nolte et al. 2013). All in all, these results suggest that rate of evolution of Nups is only accelerated in recently diverged species of the melanogaster group but not in older lineages.
Table 4.
PS at Nucleoporins and Interacting Partners in Drosophila Species
| Gene | Interactions | Positive Selection |
|---|---|---|
| Pen | 9 | D. simulans and D. yakuba |
| CG4887 | 2 | D. simulans |
| CG7185 | 3 | D. simulans |
| Nup154 | 19 | D. simulans |
| Nup160 | 13 | D. simulans |
| Cpsf160 | 15 | D. simulans |
| dgt5 | 3 | D. sechellia and D. ananassae |
| Rya-R44F | 3 | D. sechellia |
| Nup98 | 13 | D. melanogaster |
| Nup214 | 10 | D. erecta |
| Nup50 | 13 | D. ananassae |
| CG6540 | 13 | D. ananassae |
Note.—Column 2 listed the number of interactions of each gene within Nups genes network.
Fig. 4.—

Comparisons of the median dN/dS values between Nups and the rest of embryonic genes for the six species of the D. melanogaster group. Red asterisks show a significant difference in dN/dS between Nups and rest of embryonic genes in a Kolmogorov–Smirnov test, P < 0.05. Black circles indicate ancestral nodes where a comparison between inferred sequences of the respective most recent common ancestors and derived lineages was performed. Red line shows an acceleration of nonsynonymous substitution rate of Nups in D. sechellia linage in comparison with its last common ancestor with D. simulans (Wilcoxon Matched Pairs test: N = 15, Z = 2.7, P < 0.01). The rest of comparisons are not significant.
Temporal Specificity of Nups across Development
Because a positive association between temporal specificity of gene expression and evolutionary rate has been reported (Artieri et al. 2009), we analyzed whether such pattern occurred for Nups network genes. For this reason, we calculated Nups temporal specificity across development using the tissue specificity index (Yanai et al. 2005). The results of this analysis revealed that though Nups exhibited intermediate temporal specificity values with a mean of 0.27, they showed the highest expression during embryogenesis (fig. 5). In this sense, even if we cannot rule out the possibility of Nups function during nonembryonic stages of development, the highest expression level during embryogenesis is a reliable indicator of an important embryonic function.
Fig. 5.—

Relative expression levels of the Nups network genes. All expression data were taken from Graveley et al. (2011). Tau values, τ, expressed temporal specificity of Nups genes across development.
Discussion
The present evolutionary genomics study demonstrates that coexpressed groups of genes across embryogenesis are subject to differential selective pressures. Though we confirm the pervasive role of negative selection in early development, we identified a large number of PSG as part of the embryonic transcriptome. Remarkably, a fast evolving network of nucleoporins stands as an island of rapid embryonic evolution. Altogether, our findings highlight that despite being part of the stage with strongest developmental constraint across Drosophila ontogeny, many embryonic genes show rapid evolutionary change.
Hourglass-Type Embryonic Evolution
Having studied the evolution of 2,092 embryonic genes, we may conclude that early zygotic genes, but not maternal and late zygotic genes, are subject to the strongest evolutionary constraints during embryogenesis in D. melanogaster and D. simulans (fig. 1). Moreover, when analyzing embryonic genes evolution across species, we found a shared signature of purifying selection mainly in early zygotic expressed genes (fig. 2). These results do not support the developmental constraint hypothesis and states that genes involved in early developmental processes are under strong negative selection to prevent deleterious cascading effects (Artieri et al. 2009). On the contrary, our results are in agreement with the embryonic “hourglass” model that posits the onset of segmentation as the Drosophila phylotypic stage (Raff 1986). Such distribution of the rate of evolutionary change of genes expressed at different moments in embryonic development can be mainly explained by two independent causes. First, the strongest refractory period to evolutionary change across ontogeny takes place when organogenesis begins during the early zygotic stage. In addition, the early zygotic transcriptome exhibits the highest number of protein interactions during embryogenesis (Hooper et al. 2007), a fact that may also contribute to the strong functional constraint. Second, the maternal trancriptome is mainly composed of genes with female-biased expression (table 1). Sex-biased and reproduction-related genes are among the fastest evolving genes in animal genomes (Meisel 2011; Assis et al. 2012). Thus, on the one hand, female-biased gene expression may drive maternal transcriptome acceleration, and, on the other hand, the relevance of developmental processes during the onset of segmentation imposes a restriction on evolutionary change in early zygotic genes. Interestingly, another hourglass pattern was recently reported in other stages of Drosophila development. In effect, greater conservation of gene expression levels during the pupal stage was found in comparison with third-instar larvae and adult stages in species of the D. melanogaster subgroup and interspecific hybrids (Artieri and Singh 2010). Such hourglass patterns are not unexpected because many genes share a biphasic expression pattern during development in the early embryo and later in the pupal stage. Strikingly, rounds of extensive organogenesis with regulatory conservation are shared by embryogenesis and the pupal stage (Arbeitman et al. 2002). We may add that though at the level of protein-coding sequence and expression patterns (Kalinka et al. 2010) middle embryogenesis seems to be the most constrained stage of development we cannot assume that at level of regulatory sequences an hourglass model may also fit as well. On the contrary, in vertebrates, the evidence points to the validity of the hourglass model only for regulatory regions but not for protein-coding genes (Piasecka et al. 2013).
Finally, it is worth to mention that both maternal and early zygotic genes evolved at similar rates in D. yakuba and D. erecta (fig. 1). However, these results rest on the assumption that the timing of expression of embryonic genes is conserved across the entire phylogeny of the D. melanogaster group. As a matter of fact, studies of genes with sex-biased expression in D. melanogaster and D. ananassae have shown that about one-third of the genes have either gained or lost sex-biased expression in one species (Grath et al. 2009). These changes in the patterns of gene expression across two distantly related species have likely influenced the evolution of the so-called sex-biased genes.
PS in the Nuclear Pore Gene Network and Implications for Speciation
Despite the pervasive role played by negative selection affecting genes expressed during embryogenesis, we detected cases of PSG that are involved in the three embryonic stages studied and in the six species of the D. melanogaster group (supplementary table S3, Supplementary Material online). Moreover, the incidence of PS is similar, even greater in some cases, than in the sets of genes expressed in postembryonic and adult flies (table 3). Even though we found PSG and overrepresentation of some GO terms among fast evolving genes in all stages of embryogenesis (table 2), only the maternal fraction contained a network of fast evolving genes that consist of a cluster of nuclear pore (Nups) genes. Nups encode components of the nuclear pore complex that form the channels that allow the transport of proteins and RNAs from the nucleus to the cytoplasm and vice versa (Allen et al. 2000; Devos et al. 2006; Tran and Wente 2006). Interestingly, comparative genomics studies indicate that a core of interacting proteins of the nuclear pore have been preserved for at least 1.5 billion years, their association being at least as ancient as the last eukaryotic common ancestor (Bapteste et al. 2005; Neumann et al. 2010). Despite such ancient conservation, and in agreement with many reports, we found that Nups are fast evolving genes in Drosophila (Bapteste et al. 2005; Presgraves and Stephan 2007; Tang and Presgraves 2009; Clark and Aquadro 2010). In any case, rapid evolution of Nups is at odds with the expectation that proteins involved in so relevant cellular mechanisms ought to be highly constrained and under negative selection. Several hypotheses have been proposed to explain Nups rapid evolution. On the one hand, Presgraves (2007) suggested that accelerated evolution of Nups may be related to nuclear transport-related segregation distortion. On the other hand, Sawamura et al. (2010) argued that rapid evolution of Nups may reflect genetic conflicts involving the nuclear entry of retroviruses and retrotransposons. However, it is difficult to reconcile these hypotheses with the novel evidence: The hallmark of PS in Nups is only evident in D. sechellia (present paper) and D. mauritiana (Nolte et al. 2013) which are part of the D. simulans clade, a triad of very recently diverged species (Garrigan et al. 2012) (fig. 4). By contrast, the conflict over nuclear transport-related segregation distortion is thought to be an ancient genetic conflict even predating the D. melanogaster and D. simulans split (Presgraves and Stephan 2007). Likewise, it is difficult to envisage how nuclear entry of retroviruses and retrotransposons would impose a lineage-specific acceleration in Nups only in the recently diverged species. Instead, our proposal is that such lineage-specific acceleration occurring in a short evolutionary timescale is likely a molecular signature of a reproductive isolation-related process framed in the context of early development as it is suggested by their highest expression level during embryogenesis (fig. 5). In this sense, even if we cannot rule out the possibility that Nups acceleration is a consequence of their function during nonembryonic stages of development, the fact that Nups genes present highest expression level during embryogenesis is a reliable indicator of an important embryonic function. Thus, even in the face of pleiotropy our results point out that Nups rapid evolution is related to their expression and role during early development. This explanation has, in addition to the pattern of Nups evolution presented here, empirical and theoretical support. First, it has been shown that many Nups are involved in hybrid incompatibilities between pairs of species of the D. melanogaster subgroup, a feature that places Nups in the selected group of genes involved in early stages of speciation or “speciation genes” (Tang and Presgraves 2009; Sawamura et al. 2010). As proposed in the Dobzhansky–Muller model of evolution of postzyotic barriers to gene flow, independent adaptive fixations in diverging populations can lead to hybrid incompatibilities between interacting genes due to negative epistasis (reviewed in Coyne and Orr 2004). Indeed, coevolution among Nups (Clark and Aquadro 2010) can exacerbate the establishment of hybrid incompatibilities as a consequence of a “contagious” effect of interacting genes, because each substitution in a Nup would trigger a coevolutionary episode of change among other components of the gene network and extend the occurrence of negative epistasis. Second, Nups are involved in transcriptional regulation of a key reproductive trait in early development (Mendjan et al. 2006; Mason and Goldfarb 2009), because the nuclear pore complex provides docking sites for chromatin (Köhler and Hurt 2010) and interacts with the X chromosome as part of the dosage compensation complex (Mendjan et al. 2006). This is particularly interesting because dosage compensation in Drosophila takes place in early development as a key step of male sex determination (Bernstein and Cline 1994; Manu et al. 2013), and several studies have suggested that F1 hybrid lethality in crosses between D. melanogaster and D. simulans is due to dosage compensation failure (Orr 1989). Thus, several features of this scenario lead us to propose that postzygotic isolation between species of the melanogaster subgroup might be partly the result of an improper dosage compensation caused by Nups functional divergence as it also happens for other components of the dosage compensation complex (Rodriguez et al. 2007; Sawamura 2012). If this hypothesis is correct, our study may help to understand how rapid evolving genes involved in the determination of a key reproductive trait in early development affect species divergence.
Supplementary Material
Supplementary tables S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Nicolás Frankel and two anonymous reviewers for helpful comments on preliminary versions of this manuscript. J.M., N.L., H.D., and E.H. are members of Carrera del Investigador Científico of CONICET (Argentina). This work was supported by grants from Universidad de Buenos Aires, CONICET, and Agencia Nacional de Promoción Científica y Técnica (MICINN: PIB2010AR-00266).
Literature Cited
- Allen TD, Cronshaw JM, Bagley S, Kiseleva E, Goldberg MW. The nuclear pore complex: mediator of translocation between nucleus and cytoplasm. J Cell Sci. 2000;113:1651–1659. doi: 10.1242/jcs.113.10.1651. [DOI] [PubMed] [Google Scholar]
- Al-Shahrour F, et al. From genes to functional classes in the study of biological systems. BMC Bioinformatics. 2007;8:114. doi: 10.1186/1471-2105-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonov AV, Schmidt EE, Dietmann S, Krestyaninova M, Hermjakob H. R spider: a network-based analysis of gene lists by combining signaling and metabolic pathways from Reactome and KEGG databases. Nucleic Acids Res. 2010;38:W78–83. doi: 10.1093/nar/gkq482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeitman MN, et al. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- Artieri CG, Haerty W, Singh RS. Ontogeny and phylogeny: molecular signatures of selection, constraint, and temporal pleiotropy in the development of Drosophila. BMC Biol. 2009;7:42. doi: 10.1186/1741-7007-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Singh RS. Molecular evidence for increased regulatory conservation during metamorphosis, and against deleterious cascading effects of hybrid breakdown in Drosophila. BMC Biol. 2010;8:26. doi: 10.1186/1741-7007-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Zhou Q, Bachtrog D. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 2012;4:1189–1200. doi: 10.1093/gbe/evs093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, Charlebois RL, MacLeod D, Brochier C. The two tempos of nuclear pore complex evolution: highly adapting proteins in an ancient frozen structure. Genome Biol. 2005;6:R85. doi: 10.1186/gb-2005-6-10-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker MS, Demuth JP, Wade MJ. Maternal expression relaxes constraint on innovation of the anterior determinant, bicoid. PLoS Genet. 2005;1:e57. doi: 10.1371/journal.pgen.0010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Stat Meth. 1995;57:289–300. [Google Scholar]
- Bernstein M, Cline TW. Differential effects of Sex-lethal mutations on dosage compensation early in Drosophila development. Genetics. 1994;136:1051–1061. doi: 10.1093/genetics/136.3.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB, Grenier JK, Weatherbee SD. From DNA to diversity: molecular genetics and the evolution of animal design. Singapore: Blackwell Publishing; 2001. [Google Scholar]
- Casillas S, Negre B, Barbadilla A, Ruiz A. Fast sequence evolution of Hox and Hox-derived genes in the genus Drosophila. BMC Evol Biol. 2006;6:106. doi: 10.1186/1471-2148-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Clark NL, Aquadro CF. A novel method to detect proteins evolving at correlated rates: identifying new functional relationships between coevolving proteins. Mol Biol Evol. 2010;27:1152–1161. doi: 10.1093/molbev/msp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sunderland (MA): Sinauer Associates; 2004. [Google Scholar]
- Cruickshank T, Wade MJ. Microevolutionary support for a developmental hourglass: gene expression patterns shape sequence variation and divergence in Drosophila. Evol Dev. 2008;10:583–590. doi: 10.1111/j.1525-142X.2008.00273.x. [DOI] [PubMed] [Google Scholar]
- Davis JC, Brandman O, Petrov DA. Protein evolution in the context of Drosophila development. J Mol Evol. 2005;60:774–785. doi: 10.1007/s00239-004-0241-2. [DOI] [PubMed] [Google Scholar]
- Devos D, et al. Simple fold composition and modular architecture of the nuclear pore complex. Proc Natl Acad Sci U S A. 2006;103:2172–2177. doi: 10.1073/pnas.0506345103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe VE, Alberts BM. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci. 1983;61:31–70. doi: 10.1242/jcs.61.1.31. [DOI] [PubMed] [Google Scholar]
- Galis F, van Dooren TJ, Metz JA. Conservation of the segmented germband stage: robustness or pleiotropy? Trends Genet. 2002;18:504–509. doi: 10.1016/s0168-9525(02)02739-7. [DOI] [PubMed] [Google Scholar]
- Garrigan D, et al. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 2012;22:1499–1511. doi: 10.1101/gr.130922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Baines JF, Parsch J. Molecular evolution of sex-biased genes in the Drosophila ananassae subgroup. BMC Evol Biol. 2009;9:291. doi: 10.1186/1471-2148-9-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SD, et al. Identification of tightly regulated groups of genes during Drosophila melanogaster embryogenesis. Mol Syst Biol. 2007;3:72. doi: 10.1038/msb4100112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger J. Modelling the Drosophila embryo. Mol Biosyst. 2009;5:1549–1568. doi: 10.1039/b904722k. [DOI] [PubMed] [Google Scholar]
- Kalinka AT, et al. Gene expression divergence recapitulates the developmental hourglass model. Nature. 2010;468:811–814. doi: 10.1038/nature09634. [DOI] [PubMed] [Google Scholar]
- Köhler A, Hurt E. Gene regulation by nucleoporins and links to cancer. Mol Cell. 2010;38:6–15. doi: 10.1016/j.molcel.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Larracuente AM, et al. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008;24:114–123. doi: 10.1016/j.tig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lavagnino N, Serra F, Arbiza L, Dopazo H, Hasson E. Evolutionary genomics of genes involved in olfactory behavior in the Drosophila melanogaster species group. Evol Bioinform Online. 2012;8:89–104. doi: 10.4137/EBO.S8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manu, et al. 2009. Canalization of gene expression in the Drosophila blastoderm by gap gene cross regulation. PLoS Biol. 7(3):e1000049. [DOI] [PMC free article] [PubMed]
- Manu, Ludwig MZ, Kreitman M. 2013. Sex-specific pattern formation during early Drosophila development. Genetics 194:163–173. [DOI] [PMC free article] [PubMed]
- Mason DA, Goldfarb DS. The nuclear transport machinery as a regulator of Drosophila development. Semin Cell Dev Biol. 2009;20:582–589. doi: 10.1016/j.semcdb.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Meisel RP. Towards a more nuanced understanding of the relationship between sex-biased gene expression and rates of protein-coding sequence evolution. Mol Biol Evol. 2011;28:1893–1900. doi: 10.1093/molbev/msr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan S, et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell. 2006;21:811–823. doi: 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Neumann N, Lundin D, Poole AM. Comparative genomic evidence for a complete nuclear pore complex in the last eukaryotic common ancestor. PLoS One. 2010;5(10):e13241. doi: 10.1371/journal.pone.0013241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte V, Pandey RV, Kofler R, Schlötterer C. Genome-wide patterns of natural variation reveal strong selective sweeps and ongoing genomic conflict in Drosophila mauritiana. Genome Res. 2013;23:99–110. doi: 10.1101/gr.139873.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. Does postzygotic isolation result from improper dosage compensation? Genetics. 1989;122:891–894. doi: 10.1093/genetics/122.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecka B, Lichocki P, Moretti S, Bergmann S, Robinson-Rechavi M. The hourglass and the early conservation models—co-existing patterns of developmental constraints in vertebrates. PLoS Genet. 2013;9(4):e1003476. doi: 10.1371/journal.pgen.1003476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC. Does genetic conflict drive rapid molecular evolution of nuclear transport genes in Drosophila? Bioessays. 2007;29:386–391. doi: 10.1002/bies.20555. [DOI] [PubMed] [Google Scholar]
- Presgraves DC, Stephan W. Pervasive adaptive evolution among interactors of the Drosophila hybrid inviability gene, Nup96. Mol Biol Evol. 2007;24:306–314. doi: 10.1093/molbev/msl157. [DOI] [PubMed] [Google Scholar]
- Raff RA. The shape of life: genes, development, and the evolution of animal form. Chicago: The University of Chicago Press; 1996. [Google Scholar]
- Rodriguez MA, Vermaak D, Bayes JJ, Malik HS. Species-specific positive selection of the male-specific lethal complex that participates in dosage compensation in Drosophila. Proc Natl Acad Sci U S A. 2007;104:15412–15417. doi: 10.1073/pnas.0707445104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux J, Robinson-Rechavi M. Developmental constraints on vertebrate genome evolution. PLoS Genet. 2008;4:e1000311. doi: 10.1371/journal.pgen.1000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamura K. Chromatin evolution and molecular drive in speciation. Int J Evol Biol. 2012;2012:1–9. doi: 10.1155/2012/301894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamura K, et al. Introgression of Drosophila simulans nuclear pore protein 160 in Drosophila melanogaster alone does not cause inviability but does cause female sterility. Genetics. 2010;186:669–676. doi: 10.1534/genetics.110.119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder MD, et al. Transcriptional control in the segmentation gene network of Drosophila. PLoS Biol. 2004;2(9):E271. doi: 10.1371/journal.pbio.0020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra F, Arbiza L, Dopazo J, Dopazo H. Natural selection on functional modules, a genome-wide analysis. PLoS Comput Biol. 2011;7(3):e1001093. doi: 10.1371/journal.pcbi.1001093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Presgraves DC. Evolution of the Drosophila nuclear pore complex results in multiple hybrid incompatibilities. Science. 2009;323:779–82. doi: 10.1126/science.1169123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell. 2006;125:1041–1053. doi: 10.1016/j.cell.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Ward JH. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963;58:236–244. [Google Scholar]
- Wilkins AS. The evolution of developmental pathways. China: Sinauer Associates, Inc.; 2002. Publishers. [Google Scholar]
- Yanai I, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yang Z. Adaptive molecular evolution. In: Balding D, Bishop M, Cannings C, editors. Handbook of statistical genetics. New York: John Wiley; 2003. [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.