

Abstract
The enteric pathogen Salmonella enterica is one of the leading causes of foodborne illness in the world. The species is extremely diverse, containing more than 2,500 named serovars that are designated for their unique antigen characters and pathogenicity profiles—some are known to be virulent pathogens, while others are not. Questions regarding the evolution of pathogenicity, significance of antigen characters, diversity of clustered regularly interspaced short palindromic repeat (CRISPR) loci, among others, will remain elusive until a strong evolutionary framework is established. We present the first large-scale S. enterica subsp. enterica phylogeny inferred from a new reference-free k-mer approach of gathering single nucleotide polymorphisms (SNPs) from whole genomes. The phylogeny of 156 isolates representing 78 serovars (102 were newly sequenced) reveals two major lineages, each with many strongly supported sublineages. One of these lineages is the S. Typhi group; well nested within the phylogeny. Lineage-through-time analyses suggest there have been two instances of accelerated rates of diversification within the subspecies. We also found that antigen characters and CRISPR loci reveal different evolutionary patterns than that of the phylogeny, suggesting that a horizontal gene transfer or possibly a shared environmental acquisition might have influenced the present character distribution. Our study also shows the ability to extract reference-free SNPs from a large set of genomes and then to use these SNPs for phylogenetic reconstruction. This automated, annotation-free approach is an important step forward for bacterial disease tracking and in efficiently elucidating the evolutionary history of highly clonal organisms.
Keywords: H antigens, serovar, O antigens, CRISPR, lineage-through-time plot, comparative method
Introduction
Salmonella enterica is one of the primary causes of foodborne illness in the United States, leading to more deaths than any other food-related pathogen (Scallan et al. 2011). However, using the single label Salmonella for all these cases is somewhat deceptive; this is an extremely diverse species composed of six subspecies and more than 2,500 named serovars (Grimont and Weill 2007). Although the ultimate goal is to recover a fine-scaled, accurate phylogeny of global Salmonella serovar diversity, our efforts are focused on serovar diversity within S. enterica subsp. enterica, a pathogenic lineage that accounts for most of the clinical isolates from human and domestic animals. Past investigations of Salmonella phylogeny have focused primarily on species and subspecies level resolution (Boyd et al. 1996; Brown et al. 2002; McQuiston et al. 2008; Trujillo et al. 2011; Desai et al. 2013) using tools that sampled a subset of the genome, such as multilocus enzyme electrophoresis, pulsed-field gel electrophoresis (PFGE), and targeted protein coding regions. The most recent of these by Desai et al. (2013) used whole genome data from 21 individuals, 11 within S. enterica subsp. enteria, and two from each of the other five S. enterica subspecies, although the phylogenetic discussion focused mainly on subspecies relationships. Unfortunately, these previous studies did not have either the data or taxon sampling required for phylogenetic reconstruction at the interserovar level or intraserovar level in S. enterica subsp. enterica. By contrast, next-generation DNA sequencing (NGS) and computationally efficient analysis methods now enable us to utilize variation across the entire bacterial genome, extracting all of the rare microevolutionary changes useful for investigating evolutionary questions and also for diagnostics and traceback investigations of foodborne outbreaks.
Traditionally, phylogenomic analyses used multilocus sequence alignments, in which orthologous genes were determined across the taxa of interest, and then aligned orthologs were concatenated for downstream analyses. However, this approach leaves at least four areas of uncertainty. First, ortholog determination is a hypothesis of shared ancestry, causing systematic errors if the original determination is incorrect. Second, this step can require many iterations to find the correct number and diversity of loci to include, which can be very time consuming and delay the prompt traceback of contaminated foods or confirmation of an outbreak. Third, when working with diverse taxon sets, multigene alignments often have lots of missing data (i.e., gene presence/absence is highly variable). Finally, multigene alignments can be very long (more than 4 Mb in the case of Salmonella genomes), requiring vast computational resources for analysis and result interpretation.
By contrast, our approach of building a matrix composed only of the variants, or single nucleotide polymorphisms (SNPs), is relatively new and overcomes these challenges. Preliminary studies reveal this method to be fast and accurate (Snitkin et al. 2012; Allard et al. 2013). Nevertheless, we do foresee a few potential drawbacks for this methodology. One is the unknown effect of applying traditional nucleotide maximum-likelihood (ML) models to SNP matrices. Another is the inability to analyze gene trees separately, reducing the ability to identify and isolate incongruent phylogenetic signals caused by horizontal gene transfer (HGT). Despite these drawbacks, the vast amount of fine-resolution SNP data available holds great promise for shedding light on previously unresolved evolutionary relationships.
A few pioneering studies have shown that serovar-level resolution using whole genome sequence data is possible for this species (den Bakker, Switt, Govoni, et al. 2011; Fricke et al. 2011; Leekitcharoenphon et al. 2012; Desai et al. 2013). Most of the serovar diversity (∼1,500 serovars) lies within one subspecies, S. enterica subsp. enterica. These serovars were originally described based on their unique somatic (O) and flagellar (H) antigenic formulas (Grimont and Weill 2007). As with most historical methods for delimiting taxonomic groups, whether this categorization scheme, which is based on antigenic formulas, reflects evolutionary relatedness is an open question in the field. Preliminary sampling in S. enterica subsp. enterica serovar Newport (S. Newport) reveals at least three independent lineages rendering it polyphyletic (Sangal et al. 2010; Achtman et al. 2012; Cao et al. 2013), but similar taxon sampling in S. Typhimurium (Sangal et al. 2010; Trujillo et al. 2011) and S. Enteritidis (Allard et al. 2013) appears to support those serovars as monophyletic. This early look at the correlation between evolutionary history and O and H antigens suggests both patterns (i.e., monophyly and polyphyly) probably exist among the 1,500 S. enterica subsp. enterica serovars.
Identifying instances of incongruence between antigenic similarities and phylogeny is important for two reasons. First, investigating the cause of incongruence can likely yield important insights into the evolution of bacterial diversity at both the macro- and microevolutionary scale. Second, assigning serovar names that communicate monophyletic lineages is paramount for correctly characterizing and communicating outbreaks. For example, reporting an outbreak of illness as “S. Newport” would not be sufficient to identify the pathogen involved; instead, outbreak data should be described as a specific S. Newport lineage (I, II, or III) to correctly communicate which microorganism is involved (Cao et al. 2013). Thus, a prime focus of this study is assessing the validity of these named categories by thorough taxon sampling across a diversity of S. enterica subsp. enterica serovars.
Once a strong backbone serovar phylogeny is established, there are several hypotheses we can test. For example, does the current taxonomic alignment reflect distinct taxonomic groups as defined under the genealogical species concept (i.e., all individuals assumed to represent the same taxonomic group form a monophyletic lineage to the exclusion of any putative heterospecific samples [Baum and Shaw 1995])? Along similar lines, we can investigate whether the diversity and specificity of O and H antigen characters contain phylogenetic signal or whether they occur independently of the phylogeny. Can we find evidence that one or both of these characters are freely exchanged through HGT? Alternatively, is it possible that the same antigen characters could have evolved through convergent evolution? Recent studies in Escherichia coli suggest that H antigens may be useful for tracking evolutionary history, whereas O antigens are not (Iguchi et al. 2012; Ju et al. 2012). Because we rely on O and H antigens when typing Salmonella serovars, it is important that we understand whether they also allow us to predict evolutionary relationships between serovars.
A second set of questions arise from previous studies that identified a deep split of S. enterica subsp. enterica into two major lineages, Clade A and Clade B (Falush et al. 2006; den Bakker, Switt, Govoni, et al. 2011). Is this split an artifact of the size of earlier data sets, or does this split hold when taxon sampling is substantially increased? If so, can whole genome sequencing enable us to identify which SNPs define each major clade? Extending beyond the two-clade issue, can we uncover historical fluctuations in the rate of diversification? In other words, has the diversification rate been stable over time or is there evidence of radiation bursts early or late in the phylogeny? We can also elucidate the degree to which incomplete lineage sorting and/or introgression may have confounded previous phylogenetic studies by determining the number of SNPs that are shared among the major groups.
Third, there is an interest in utilizing clustered regularly interspaced short palindromic repeat (CRISPR) regions in Salmonella as a typing method (Fabre et al. 2012). In contrast to traditional DNA sequence evolution (i.e., base substitutions, insertions and deletions, inversions, etc.), CRISPR’s small RNA-mediated system evolves through the precise incorporation of phage DNA/RNA fragments into the bacterial chromosome (Bhaya et al. 2011; Barrangou and Horvath 2012). This process is thought to confer host immunity, although its exact function in Salmonella is purely conjectural. Because of this unique pattern of evolution, the CRISPR region has been analyzed for use in typing (serovar-level identification; Fricke et al. 2011; Fabre et al. 2012). By examining the level of CRISPR variation across our serovar-level phylogeny, we can test the hypothesis that CRISPR region variations can be used to determine serovar identity with appropriate sensitivity and specificity.
The first step in answering these three questions requires a comprehensive genome-scale analysis using assembled S. enterica genomes, many of which were generated for this study. Our taxon sampling included five of the six Salmonella enterica subspecies: S. enterica subsp. salamae, S. e. diarizonae, S. e. houtenae (two serovars), S. e. indica, and S. e. enterica (78 serovars). We used a nonreference-based approach to extract SNPs. This yielded a large SNP matrix that we used to reconstruct the evolutionary history of this diverse pathogen. Using this phylogeny, we explicitly tested the accuracy of current taxonomic alignments and the phylogenetic signals contained within the O and H antigenic data. We also extracted all CRISPR regions from whole genomes and explored the utility of such regions for typing and subtyping. Aside from our specific research questions, this study accompanies the new release of more than 100 diverse Salmonella genomes to National Center for Biotechnology Information (NCBI), ∼200,000 Salmonella SNPs to NCBI’s dbSNP database, and a rigorous phylogenetic tree deposited at TreeBase.org, study number S14912. These data will benefit public health by drastically increasing publicly available reference genomes and expanding the phylogenetic context for monitoring Salmonella, which will help resolve future outbreaks.
Materials and Methods
Salmonella Strains
A set of 102 S. enterica strains were gathered from in-house strain collections at the Center for Food Safety and Applied Nutrition (FDA-CFSAN) and US Department of Agriculture (USDA) and used for whole genome sequencing. Our sampling focused on S. enterica subsp. enterica with 151 strains spanning the diversity of this subspecies. We gathered four outgroups, one in each of the following subspecies of S. enterica: S. e. diarizonae, S. e. houtenae, S. e. indica, and S. e. salamae. Fifty-four public genomes were also included in the study, producing our final data set of 156 strains that represented 78 S. enterica subsp. enterica serovars. (supplementary data S1, Supplementary Material online)
Growth of Bacterial Strains and Genomic Isolation
For each strain, a pure culture sample was taken from frozen stock, plated on Trypticase Soy Agar, and incubated overnight at 37 °C. The following day, cells were taken from the plate and inoculated into Trypticase Soy Broth culture for DNA extraction. All samples were representative cultures from a full-plate inoculation and were not single colonies. Genomic DNA was extracted using the Qiagen DNeasy kit (Qiagen, Valencia, CA).
Genome Sequencing, Assembly, and Annotation
Most isolates for this study were shotgun sequenced using Roche 454 GS Titanium technology (Roche Diagnostics Corp., Indianapolis, IN). The 454 isolates were run on a quarter of a titanium plate. This produced roughly 250,000 reads per draft genome.
Approximately 20 isolates were prepared using the Nextera Sample Preparation Kit (Illumina, San Diego, CA) and then sequenced on an Illumina MiSeq (Illumina) for 2 × 151 cycles.
De novo assemblies were generated from all raw sequence data. The 454 reads were assembled using Roche’s Newbler Assembler v. 2.3–2.6 (Margulies et al. 2005). The Illumina reads were assembled with Ray v. 2.2.0 (Boisvert et al. 2010). Default parameters were used in all cases. The contigs for each isolate (draft genomes) were annotated using NCBI’s Prokaryotic Genomes Automatic Annotation Pipeline.
Comparative Genomic and Diversification Analyses
Fifty-nine complete or draft genomes were downloaded from NCBI and included in this study, producing our final data set of 156 annotated genomes. A concatenation of SNPs were gathered in a data matrix (95% majority SNP matrix) using the program kSNP v. 2 ([Gardner and Slezak 2010] http://sourceforge.net/projects/ksnp, last accessed June 3, 2013). The following kSNP parameters were used: k-mer size 25 and SNP locations determined based on the complete annotated genome S. Typhimurium str. LT2 (GenBank: AE006468). One advantage of kSNP is that the putative SNPs are extracted from kmers (one SNP per 25mer in our analysis), which effetely eliminates issues with assembly error in the input draft genomes. Also, raw reads can be used instead of draft assemblies with similar results. Tree inference was performed using RAxML-HPC2 version 7.3.2 (Stamatakis 2006; Stamatakis et al. 2008) under the GTRCAT model for the rapid bootstrapping phase, and GTRGAMMA for the final best scoring ML tree. Bootstrapping was performed under auto Majority Rule Criterion (autoMRE).
We constructed lineage through time plots using the APE (Paradis et al. 2004) package in R (R Core Development Team 2012), which provides information regarding historical fluctuations in the rate of diversification within S. enterica. Because the analysis requires an ultrametric tree, one was constructed using a penalized likelihood method also implemented in ape. We evaluated the sensitivity of the analysis to taxon sampling by constructing plots on both the original data set with 156 tips and on a pruned tree of 126 tips that contained only one representative from each monophyletic serovar group. Results were consistent between the two data sets, and we present results based on the 156 strain data set.
We used GSI (Cummings et al. 2008) to statistically evaluate the degree of genealogical exclusivity among isolates assumed to represent the same serovar. The GSI statistics range from 0 to 1, where 0 means a random distribution of isolates from the same serovar in the phylogeny and 1 represents a monophyletic group. We calculated the weighted statistic that accounts for topological uncertainty by estimating the GSI for each group on each of the 100 randomly selected bootstrap replicates from the phylogenetic analyses. Statistical significance was based on 10,000 random replicates where GSI was calculated for each group after the isolates were randomly assigned to the tips of each bootstrap replicate.
Comparative Method
One goal was to determine if/how targeted characters evolved across the Salmonella phylogeny. We tested several hypotheses, including the following: 1) do the O and H antigen states show phylogenetic signal? 2) are the O and H antigen (Phase_1) characters coevolving? and 3) do the CRIPSR1 and CRISPR2 loci categories show phylogenetic signal or are they distributed randomly across the phylogeny?
Because the O and H antigen characters were collected from the literature (Grimont and Weill 2007), there was no opportunity for strains within a named serovar to have different antigen formulas. For this reason, the 156-taxon ML tree was pruned to allow only one strain per serovar (additional strains were allowed for polyphyletic serovars). Tests for phylogenetic signal were performed using the fitDiscrete function within the Geiger (Harmon et al. 2008) package in R (R Core Development Team 2012). Two ML scores were determined: One with the character mapped onto the pruned ML tree (converted to be ultrametric with a penalized likelihood approach) and the other with the character mapped onto a star phylogeny. The best-fit model was determined by a likelihood ratio test followed by a chi-square. We used the GSI statistic to test the genealogical exclusivity, or degree of monophyly, among the antigen character states for three antigen characters (the O group and two flagellar antigens).
Phylogenetic Independent Clustering
To further explore the genomic similarities among isolates, we clustered samples into groups using the model-based Bayesian clustering method implemented in the program STRUCTURE (Falush et al. 2003, 2007). This method does not incorporate any a priori information about group membership but rather assigns samples to “populations” based on similarities in multilocus genotpyes. We used the SNP matrix produced by kSNP as the input and ran the program at values of k (i.e., the number of clusters) 2–5. Given the limitations of the method to infer and graphically represent a large number of clusters, we focused on these values of k to elucidate the coarser genomic similarities among the isolates, such as whether we saw support for the two major clades previously identified. Analyses were run using the admixture model with correlated allele frequencies and consisted of 60,000 generations, the first 10,000 of which served as burnin.
CRISPR Analysis
Salmonella CRISPR loci 1 and 2 were extracted from 128 of our genomes (in-house draft assemblies plus published complete genomes). Spacers and repeats were visualized with the CRISPR DB II Excel Macro (DuPont Inc., Barrangou R, unpublished data), as previously used (Horvath et al. 2008). Repeats were removed to determine the homology of spacers across strains, and the CRISPR spacer array was manually aligned to optimize the homology of spacers across Salmonella strains. This was performed separately for CRISPR 1 and CRISPR 2. To analyze the CRISPR diversity across S. enterica subsp. enterica, we extracted the four most ancestral spacers from the alignment and assigned a category based on their similarity to other strains. Strains within the same category number have the exact same spacer sequence for their first four spacers. The spacer number within each CRISPR locus was collected without reference to the alignment.
Data Deposition
The SNP matrix and phylogenetic trees are available at TreeBase.org, study number S14912. Individual SNPs were deposited at dbSNP database at NCBI under the accession range ss749616252–ss749736198. GenBank accession numbers for the 156 genomes used in this analysis are listed in supplementary table S1, Supplementary Material online. The newly 107 newly sequenced genomes are summarized here in the following format: S. serovar str. ID: WGS accession number.
S. Abaetetuba str. ATCC 35640: APAQ00000000; S. Abony str. 0014: APAB00000000; S. Agona str. 419639 2-1: AOZV00000000; S. Agona str. 632182-2: AOZY00000000; S. Agona str. 648586-1: AOZU00000000; S. Agona str. ATCC 51957: AOZX00000000; S. Albany str. ATCC 51960: AOZW00000000; S. Anatum str. ATCC BAA-1592: AOZZ00000000; S. Anatum str. USDA_100: APAA00000000; S. Bareilly str. 2780: AOZP00000000; S. Bareilly str. ATCC 9115: AOZN00000000; S. Bareilly str. CFSAN000183: AOZT00000000; S. Bareilly str. CFSAN000189: AOZS00000000; S. Bareilly str. CFSAN000197: AOZR00000000; S. Bareilly str. CFSAN000200: AOZQ00000000; S. Berta str. ATCC 8392: AOZO00000000; S. Braenderup str. ATCC BAA-664: AOZM00000000; S. Braenderup str. CFSAN000756: APAP00000000; S. Bredeney str. CFSAN001080: APAJ00000000; S. Cerro str. 818: AOZJ00000000; S. Chester str. ATCC 11997: AOZI00000000; S. Choleraesuis str. 0006: AOZL00000000; S. Choleraesuis str. ATCC 10708: AOZK00000000; S. Cubana str. CFSAN001083: APAG00000000; S. Derby str. 626: AOZH00000000; S. Dublin str. HWS51: AHUK00000000; S. Dublin str. SL1438: AHUJ00000000; S. Eastbourne str. CFSAN001084: APAF00000000; S. Enteritidis str. 436: AHUO00000000; S. Enteritidis str. 81-2625: ALIB00000000; S. Gallinarum str. 9184: AHUH00000000; S. Gaminara str. ATCC BAA-711: AOZF00000000; S. Give str. 564: AOZG00000000; S. Hadar str. ATCC 51956: AOZE00000000; S. Hartford str. CFSAN001075: APAO00000000; S. Havana str. CFSAN001082: APAH00000000; S. Heidelberg str. 82-2052: AMMX00000000; S. Heidelberg str. SARA35: AMLT00000000; S. Indiana str. ATCC 51959: AOZC00000000; S. Inverness str. ATCC 10720: AOZD00000000; S. Javiana str. 10721: AOZA00000000; S. Javiana str. PRS_2010_0720: AOZB00000000; S. Kentucky str. 5349: AOYZ00000000; S. Kentucky str. ATCC 9263: AOYY00000000; S. Litchfield str. CFSAN001076: APAN00000000; S. London str. CFSAN001081: APAI00000000; S. Manhattan str. CFSAN001078: APAL00000000; S. Mbandaka str. ATCC 51958: AOYR00000000; S. Meleagridis str. 0047: AOYN00000000; S. Miami str. 1923: AOYS00000000; S. Minnesota str. ATCC 49284: AOYO00000000; S. Montevideo str. 8387: AOYQ00000000; S. Muenchen str. ATCC 8388: AOXN00000000; S. Muenchen str. baa1594: AOYV00000000; S. Muenchen str. baa1674: AOYT00000000; S. Muenster str. 0315: AOYX00000000; S. Muenster str. 420: AOYW00000000; S. Nchanga str. CFSAN001091: APAE00000000; S. Nchanga str. CFSAN001092: APAD00000000; S. Norwich str. CFSAN001077 Serovar:APAM00000000; S. Ohio str. CFSAN001079: APAK00000000; S. Oranienburg str. 0250: AOYM00000000; S. Oranienburg str. 701: AOYL00000000; S. Panama str. ATCC 7378: AOYJ00000000; S. Paratyphi A str. ATCC 11511: AOYH00000000; S. Paratyphi B str. ATCC 10719: AOYF00000000; S. Paratyphi B str. ATCC 19940: AOYD00000000; S. Paratyphi B str. ATCC 51962: AOYC00000000; S. Paratyphi B str. ATCC 8759: AOYE00000000; S. Paratyphi B str. ATCC BAA-1585: AOYG00000000; S. Paratyphi B str. SARA42: AOYB00000000; S. Paratyphi B str. SARA56: AOXH00000000; S. Paratyphi B str. SARA62: AOXG00000000; S. Poona str. ATCC BAA-1673: AOYK00000000; S. Pullorum str. 19945: AOYI00000000; S. Pullorum str. 9120: AMYM00000000; S. Rubislaw str. ATCC 10717: AOYA00000000; S. Saintpaul str. JO2008: AOXY00000000; S. Saintpaul str. SARA26: AOXF00000000; S. Senftenberg str. 316235162: AOYU00000000; S. Senftenberg str. 423984–2: AOYP00000000; S. Senftenberg str. 604314: AOXW00000000; S. Senftenberg str. ATCC 43845: AOXX00000000; S. Senftenberg str. ATCC 8400: AOXU00000000; S. Sloterdijk str. ATCC 15791: AOXT00000000; S. Soerenga str. 695: AOXZ00000000; S. Stanley str. ATCC 7308: AOXV00000000; S. Stanleyville str. CFSAN000624: APAR00000000; S. subsp. diarizonae ser. 60:r:e,n,x,z15 str. 01-0170: APAC00000000; S. subsp. houtenae ser. 50:g,z51:- str. 01-0133: AOXJ00000000; S. subsp. indica ser. 6,14,25:z10:1,(2),7 str. 1121: AOXI00000000; S. subsp. salamae ser. 58:l,z13,z28:z6 str. 00-0163: AOXE00000000; S. Tallahassee str. 0012: AOXS00000000; S. Tennessee str. TXSC_TXSC08-19: AOXR00000000; S. Tennessee str. TXSC_TXSC08-21: AOXQ00000000; S. Thompson str. ATCC 8391: AOXP00000000; S. Typhimurium str. AZ 057: AOXC00000000; S. Typhimurium str. SARA13: AOXO00000000; S. Typhimurium str. ST4581: AOXD00000000; S. Urbana str. ATCC 9261: AOXM00000000; S. Virchow str. ATCC 51955: AOXL00000000; S. Worthington str. ATCC 9607: AOXK00000000.
Results
Whole Genome Sequencing and SNP Discovery
Our total data set of 156 S. enterica genomes included 102 newly sequenced draft genomes and 49 published genomes (13 complete and 41 drafts available at NCBI). The final taxon sampling was composed of five outgroup and 151 ingroup S. enterica subsp. enterica strains, spanning 78 serovars (supplementary data S1, Supplementary Material online). Three SNP matrices were determined for this data set: A core matrix containing 6,827 SNPs, a 95% majority matrix (kmer present in ≥95% of isolates) containing 119,750 SNPs, and a total matrix containing 653,038 SNPs. The inclusion of draft genomes allowed us to use standard NGS technology to collect genome-scale data, although the data missing from these genomes meant that our core SNP matrix was too conservative to allow us to perform downstream phylogenetic analyses because such analyses require that SNPs be present in each of the genomes used. The opposite held true for the total SNP matrix, which included all SNPs present for every genome, including SNPs derived from mobile elements (phages and plasmids), SNPs present in lineage-specific genes, and SNPs called due to sequence error. The 95% majority SNP matrix included SNPs that were present in 95% of the taxa or 148 of the 156 genomes. They accounted for inherent missing data in the draft genomes while omitting mobile regions and low-quality SNPs that could potentially add noise or obscure phylogeny (supplementary data S2, Supplementary Material online).
Phylogeny and Diversification
Phylogenetic analysis of the 95% majority matrix resulted in an ML tree with strong support for the monophyly of subspecies S. enterica subsp. enterica (fig. 1). Within the S. enterica subsp. enterica subspecies, we uncovered a deep split that delineates two sister lineages, Clade A and Clade B, which confirms what others have previously reported (Falush et al. 2006; den Bakker, Switt, Govoni, et al. 2011). In addition to very strong bootstrap support for both clades, we also uncovered 24 unique SNPs for Clade A and six unique SNPs for Clade B.
Fig. 1.—

Phylogenetic tree based on the maximum-likelihood method implemented in RAxML. Bold black branches represent 90–100% bootstrap support. Bold gray branches represent 70–90% bootstrap support. Numbers associated with branches are SNPs unique to that lineage. For the purposes of this figure, the long outgroup branches were shortened; however, the original tree is available for download at TreeBase.org. Three antigen characters are mapped onto this phylogeny: O group, Phase 1 (H) flagellar antigen, and Phase 2 (H) flagellar antigen.
Figure 1 shows both clades and their sublineages. Within Clade B, S. Infantis (B1 in fig. 1) is an early diverging lineage and the sister group to the remaining serovars in Clade B. Several strongly supported lineages emerge from this group, some of which were suggested in the den Bakker et al.’s Figure 1 phylogeny (den Bakker, Switt, Govoni, et al. 2011). We also found two strongly supported lineages: B2 (S. Miami + S. Javiana) and B3 (S. Poona + S. Rubislaw + S. Abaetetuba) that diverge before the well-supported Clade B4 lineage that contains the most serovar diversity. The majority of serovars (18 out of 20) in Clade B are monophyletic. Two serovars do not appear to be natural groups: S. Abaetetuba is nested within a paraphyletic S. Rubislaw, and S. Give appears to be polyphyletic with two strains that arose independently. Across the monophyletic serovars, there are very strong bootstrap values along with numerous unique SNPs defining each of them (listed above the branches in fig. 1). Salmonella Montevideo revealed the most diversity with three divergent lineages.
Two major lineages comprise Clade A: A1 and A2 (fig. 1). Within lineage A1, there are 17 serovars, of which four serovars are polyphyletic: S. Agona, S. Senftenberg, S. Kentucky, and S. Paratyphi B. Clade A2 contains 45 serovars, demonstrating the most diversity within the S. enterica subsp. enterica. Salmonella Typhi, S. paraphyphi A, and S. Mississippi group into a well-supported lineage we are calling “Section Typhi” (fig. 1). Salmonella Paratyphi C is sister to S. Choleraesuis with very strong support, and three other independent lineages of S. Paratyphi B are scattered throughout group A2. Although there is only moderate support for most of the deep nodes in A2, there are several very strongly supported sublineages. For example, a group we call “Section Enteritidis” is composed of the serovars S. Enteritidis, S. Gallinarum, S. Pullorum, S. Dublin, and S. Berta, which have a well-documented relationship (Vernikos et al. 2007; Achtman et al. 2012; Allard et al. 2013). “Section Typhimurium” contains the S. Typhimurium + S. 4,[5],12:I- complex, along with S. Saintpaul, part of S. Paratyphi B, S. Heidelberg, and S. Virchow. What we refer to as “Section Newport II” contains a diverse set of S. Newports, one S. Muenchen, and one S. Litchfield. The remaining lineages in A2 were well-supported but contained fewer taxa. Although most of the serovars appear to be monophyletic, several were not. Our analysis revealed multiple independent lineages of S. Newport, two of S. Bareilly, two of S. Saintpaul, and two of S. Muenchen.
Clade A2 also exhibits high pathogenicity. Six serovars in this lineage are on the Center for Disease Control’s top ten list for foodborne diseases: S. Enteritidis (the most prevalent foodborne pathogen in the world), S. Typhimurium, I4, [5],12:i:-, S. Newport, S. Muenchen, and S. Heidelberg (http://www.cdc.gov/foodnet/data/trends/tables/table5.html, last accessed June 3, 2013). Also present in A2 are the typhoidal Salmonella: S. Typhi and S. Paratyphi A, B, and C.
We also looked at broader patterns of diversification across the ML phylogeny (fig. 1). A lineage-through-time plot showed evidence for two episodes of elevated serovar diversification (fig. 2). Early in the evolutionary history of S. enterica, there appears to have been a relatively long period of gradually increasing diversity followed by a plateau representing a constant rate of diversification. A second much more punctual increase in the diversification rate appears to have occurred within the recent past. This pattern is congruent with our phylogeny, which showed high levels of diversification early in the divergence process followed by relatively long branches with additional short branches indicative of diversification at the tips (fig. 1).
Fig. 2.—
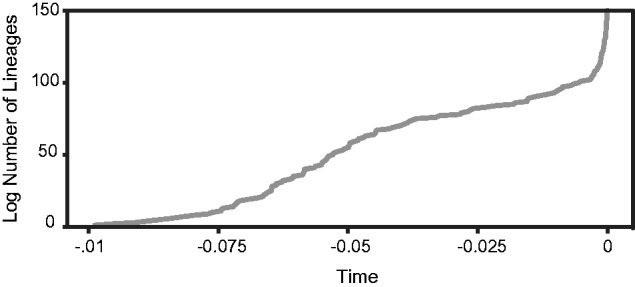
Lineage-through-time plots illustrating fluctuations in diversification rate throughout the evolutionary history of S. enterica.
The degree of genealogical exclusivity exhibited by the 35 serovars for which we had multiple samples was generally quite high (table 1). Specifically, 22 serovars had genealogical sorting index (GSI) values of 1, indicating that all samples from these serovars formed a monophyletic group. Thirteen other serovars had GSI values <1, and only one serovar (S. Give) showed a phylogenetic distribution that was not significantly different from random. Among serovars that were not monophyletic (i.e., GSI < 1), isolates were clustered relatively close to one another in a way that statistically supported describing them as genealogically exclusive groups. This was true even for the polyphyletic S. Newport samples, which formed two clades.
Table 1.
Serovar-Level Characters and Statistics
| Salmonella enterica subsp. enterica serovar | Number of Strains | GSI | Antigen Characters |
||
|---|---|---|---|---|---|
| O Group | H Phase 1 | Phase 2 | |||
| 4,[5],12:i:- | 2 | 0.245 | 4 | i | — |
| Abaetetuba | 1 | NA | 11 | k | 1,5 |
| Abony | 1 | NA | 4 | b | e,n,x |
| Adelaide | 1 | NA | 35 | f,g | — |
| Agona | 4 | 0.258 | 4 | f,g,s | [1,2] |
| Alachua | 1 | NA | 35 | z4,z23 | — |
| Albany | 1 | NA | 8 | z4,z24 | — |
| Anatum | 2 | 1.000 | 3,10 | e,h | 1,6 |
| Baildon | 1 | NA | 9,46 | a | e,n,x |
| Bareilly | 6 | 0.364 | 7 | y | 1,5 |
| Berta | 1 | NA | 9 | [f],g,[t] | — |
| Braenderup | 2 | 1.000 | 4 | e,h | e,n,z15 |
| Bredeney | 1 | NA | 4 | l,v | 1,7 |
| Cerro | 1 | NA | 18 | z4,z23 | [1,5] |
| Chester | 1 | NA | 4 | e,h | e,n,x |
| Choleraesuis | 3 | 1.000 | 7 | c | 1,5 |
| Cubana | 1 | NA | 13 | z29 | — |
| Derby | 1 | NA | 4 | f,g | [1,2] |
| Dublin | 2 | 1.000 | 9 | g,p | — |
| Eastbourne | 1 | NA | 9 | e,h | 1,5 |
| Enteritidis | 3 | 1.000 | 9 | g,m | — |
| Gallinarum | 2 | 0.497 | 9 | — | — |
| Gaminara | 2 | 1.000 | 16 | d | 1,7 |
| Give | 2 | 0.105 | 3,10 | l,v | 1,7 |
| Hadar | 2 | 1.000 | 8 | z10 | e,n,x |
| Hartford | 1 | NA | 7 | y | e,n,x |
| Havana | 1 | NA | 13 | f,g,[s] | — |
| Heidelberg | 3 | 1.000 | 4 | r | 1,2 |
| Hvittingfoss | 1 | NA | 16 | b | e,n,x |
| Indiana | 1 | NA | 4 | z | 1,7 |
| Infantis | 1 | NA | 7 | r | 1,5 |
| Inverness | 2 | 1.000 | 38 | k | 1,6 |
| Javiana | 3 | 1.000 | 9 | l,z28 | 1,5 |
| Johannesburg | 1 | NA | 40 | b | e,n,x |
| Kentucky | 4 | 0.235 | 8 | i | z6 |
| Litchfield | 1 | NA | 8 | l,v | 1,2 |
| London | 1 | NA | 3,10 | l,v | 1,6 |
| Manhattan | 1 | NA | 8 | d | 1,5 |
| Mbandaka | 1 | NA | 7 | z10 | e,n,z15 |
| Meleagridis | 1 | NA | 3,10 | e,h | l,w |
| Miami | 1 | NA | 9 | a | 1,5 |
| Minnesota | 2 | 1.000 | 21 | b | e,n,x |
| Mississippi | 1 | NA | 13 | b | 1,5 |
| Montevideo | 5 | 1.000 | 7 | g,m,[p],s | [1,2,7] |
| Muenchen | 3 | 0.392 | 8 | d | 1,2 |
| Muenster | 2 | 1.000 | 3,10 | e,h | 1,5 |
| Nchanga | 2 | 1.000 | 3,10 | l,v | 1,2 |
| Newport | 7 | 0.480 | 8 | e,h | 1,2 |
| Norwich | 1 | NA | 7 | e,h | 1,6 |
| Ohio | 1 | NA | 7 | b | l,w |
| Oranienburg | 2 | 1.000 | 7 | m,t | [z57] |
| Panama | 1 | NA | 28 | l,v | 1,5 |
| Paratyphi A | 3 | 1.000 | 2 | a | [1,5] |
| Paratyphi B | 9 | 0.270 | 4 | b | 1,2 |
| Paratyphi C | 1 | NA | 6,7 | c | 1,5 |
| Pomona | 1 | NA | 28 | y | 1,7 |
| Poona | 1 | NA | 13 | z | 1,6 |
| Pullorum | 2 | 1.000 | 9 | — | — |
| Rissen | 1 | NA | 7 | f,g | — |
| Rubislaw | 2 | 0.497 | 11 | r | e,n,x |
| Saintpaul | 4 | 0.216 | 4 | e,h | 1,2 |
| Schwarzengrund | 2 | 1.000 | 4 | d | 1,7 |
| Senftenberg | 6 | 0.397 | 1,3,19 | g,[s],t | — |
| Sloterdijk | 1 | NA | 4 | z35 | z6 |
| Soerenga | 1 | NA | 30 | i | l,w |
| Stanley | 1 | NA | 4 | d | 1,2 |
| Stanleyville | 1 | NA | 4 | z4,z23 | [1,2] |
| Tallahassee | 1 | NA | 8 | z4,z32 | — |
| Tennessee | 3 | 1.000 | 7 | z29 | [1,2,7] |
| Thompson | 1 | NA | 7 | k | 1,5 |
| Typhi | 2 | 1.000 | 9 | d | — |
| Typhimurium | 4 | 0.745 | 4 | i | 1,2 |
| Uganda | 1 | NA | 3,10 | l,z13 | 1,5 |
| Urbana | 2 | 1.000 | 30 | b | e,n,x |
| Virchow | 2 | 1.000 | 7 | r | 1,2 |
| Wandsworth | 1 | NA | 39 | b | 1,2 |
| Weltevreden | 1 | NA | 3,10 | r | z6 |
| Worthington | 1 | NA | 13 | z | l,w |
Note.—GSI, genealogical sorting index. NA, not applicable.
Genetic Clusters
Using a model-based Bayesian clustering method, we found that samples generally clustered in a pattern matching the phylogenetic analyses (fig. 3). Focusing on broader groups, at k = 2 (i.e., the number of clusters to which samples could be assigned), our results provide additional support for Clades A and B; but we also found that there is a degree of admixture or similarity in SNP profiles among some samples that break out into a third relatively distinct cluster at k = 3. At k = 5, there is another cluster composed only of S. Paratyphi A isolates (fig. 3).
Fig. 3.—

Bayesian clustering results for values of k = 2–5 based on the matrix containing SNPs present in at least 95% of the samples (outgroups were excluded). Different colors represent different clusters and the bars represent different individuals. The extent to which different colors comprise a bar is indicative of the degree of admixture. Samples are in the same order as they are in the ML phylogeny (fig. 1), which is shown for comparison.
SNP Annotation
Each of the 119,750 SNPs derived by a reference-free method was mapped to the reference genome, S. Typhimurium LT2 (GenBank: AE006468), for annotation and deposited in NCBI’s dbSNP database accessible with the following Submitter SNP (ss) accession numbers: 749616252–749736198. Detailed annotation was extracted for Clades A and B SNPs, which were the major clades of interest (table 2). There are 24 SNPs unique to the Clade B lineage. Four of these fell within intergenic regions, two are nonsynonymous and 18 are synonymous substitutions within protein coding regions. Both of the nonsynonymous substitutions are in transcriptional activators or genes that activate transcription. The Clade A lineage, although large, showed only six unique SNPs. One occurred within an unannotated region, one was nonsynonymous and the remaining four were synonymous substitutions.
Table 2.
SNP Annotations for Clade A and Clade B as Determined by kSNP
| kSNP Locus ID | Annot. | aa Change | Codon Change | Pos. in CDS | AE006468 Location | Gene | Locus Tag | Strand | Product Name |
|---|---|---|---|---|---|---|---|---|---|
| Clade B SNPs | |||||||||
| 115230 | Coding | A_A | GCC_GCG | 345 | 2417643 | STM2310 | − | Isochorismate synthase | |
| 139194 | Coding | P_P | CCC_CCG | 90 | 729566 | gltI, menF | STM0665 | − | Glutamate/aspartate transporter |
| 198788 | Coding | S_S | AGC_AGT | 126 | 2402046 | yfaZ | STM2294 | − | Putative inner membrane protein |
| 22659 | Coding | V_V | GTC_GTT | 125 | 1784052 | sapA | STM1692 | + | ABC superfamily peptide transport protein |
| 245765 | Coding | V_V | GTC_GTT | 206 | 1276770 | plsX | STM1192 | + | Putative fatty acid/phospholipid synthesis protein |
| 2656 | Coding | T_T | ACC_ACT | 57 | 17316 | STM0017 | − | Putative protein | |
| 266398 | Coding | A_A | GCC_GCT | 30 | 1518497 | STM1441 | − | Putative inner membrane protein | |
| 267325 | Coding | L_L | CTG_TTG | 370 | 552354 | fsr | STM0493 | − | Putative MFS family of transport protein |
| 34745 | Intergenic | 80406 | + | Upstream of STM0068, transcriptional regulator | |||||
| 365928 | Intergenic | 1332201 | + | Upstream of STM1246, reduced macrophage survival protein | |||||
| 370045 | Coding | S_S | TCA_TCG | 460 | 2727192 | lepA | STM2583 | − | GTP-binding elongation factor |
| 389879 | Coding | I_I | ATC_ATT | 97 | 729545 | gltI | STM0665 | − | Glutamate/aspartate transporter |
| 407961 | Coding | A_T | ACG_GCG | 26 | 2929670 | mig-14 | STM2782 | + | Putative transcription activator |
| 421386 | Coding | P_P | CCC_CCT | 43 | 2022982 | yecG | STM1927 | + | Putative universal stress protein |
| 444415 | Coding | V_V | GTA_GTT | 109 | 4820845 | yhhI | STM4566 | − | Putative cytoplasmic protein |
| 457785 | Intergenic | 3515395 | + | Upstream of STM3348, serine endoprotease | |||||
| 461849 | Coding | T_T | ACC_ACT | 26 | 2438962 | nuoB | STM2327 | − | NADH dehydrogenase I chain B |
| 559334 | Coding | G_G | GGG_GGT | 294 | 1524368 | tyrS | STM1449 | + | Tyrosine tRNA synthetase |
| 572551 | Coding | T_T_T | ACA_ACC_ACT | 650 | 2618849 | rihC | STM2503 | − | Putative diguanylate cyclase |
| 609237 | Coding | P_P | CCA_CCG | 13 | 60202 | mig-14 | STM0051 | + | Putative purine nucleoside hydrolase |
| 631239 | Coding | E_K | AAA_GAA | 56 | 2929760 | STM2782 | + | Putative transcription activator | |
| 632030 | Coding | A_A_A | GCA_GCC_GCT | 502 | 132233 | leuA | STM0113 | − | 2-isopropylmalate synthase |
| 647650 | Intergenic | 1528293 | − | Upstream of STM1542, putative POT family peptide transport protein | |||||
| 78823 | Coding | L_L | TTA_TTG | 41 | 1832990 | cls | STM1739 | + | Cardiolipin synthase |
| Clade A SNPs | |||||||||
| 135358 | Intergenic | 2449145 | + | Upsteam of STM2338, phosphotransacetylase | |||||
| 267325 | Coding | L_L | CTG_TTG | 370 | 552354 | fsr | STM0493 | − | Putative MFS family of transport protein |
| 271444 | Coding | Y_Y | TAC_TAT | 279 | 59782 | STM0050 | + | Putative nitrite reductase | |
| 370045 | Coding | S_S | TCA_TCG | 460 | 2727192 | lepA | STM2583 | − | GTP-binding elongation factor |
| 510549 | Coding | D_N | AAC_GAC | 144 | 2401994 | yfaZ | STM2294 | − | Putative inner membrane protein |
| 572551 | Coding | T_T_T | ACA_ACC_ACT | 650 | 2618849 | STM2503 | − | Putative diguanylate cyclase | |
Note.—SNPs that produce nonsynonymous amino acid substitutions are highlighted in Bold.
Detailed annotation was also summarized for the well-supported “sections” highlighted in figure 1. “Section Typhi” had 51 unique SNPs, “section Enteritidis” had 34 SNPs, “section Typhimurium” had nine SNPs, and “section Newport II” had six SNPs (supplementary data S3, Supplementary Material online). In summary, 11 SNPs occurred in intergenic regions, twenty-one are nonsynonymous and the rest are synonymous substitutions within protein coding regions.
Clustered Regularly Interspaced Short Palindromic Repeats
CRISPR loci 1 and 2 were determined and aligned for 126 Salmonella strains. The alignment revealed mixed homology across serovars (supplementary data S4, Supplementary Material online) with an increase of shared spacers toward the ancestral end of the CRISPR array, nearest to the trailer. For example, spacer 1 in CRISPR 2 is shared across 41 genomes representing multiple serovars. Spacers are only added to the leader (5′) end of the CRISPR array, which allows us to assume unidirectional deletion determinations.
Both spacer alignments are hypotheses, and each revealed the degradation of many internal spacers. This is more obvious when multiple strains are sampled within a single serovar, as is the case within S. Typhimurium for both CRISPR 1 and 2. Each CRISPR sequence was mapped onto the SNP phylogeny and pruned for missing taxa (fig. 4). The first four spacers are shown along with the total spacer number in each array, with their length represented as a bar chart. Spacer numbers vary within and between serovars and across the CRISPR loci. The median number of spacers in CRISPR 1 and 2 are 13 and 14, respectively, which also accounts for S. Mbandaka in CRISPR 2, which is an outlier with an unusually large array (113 spacers).
Fig. 4.—

ML phylogeny from figure 1, pruned for strains for which we have CRISPR data (126 in-house collected draft genomes plus published complete genomes). The four most ancestral spacers were extracted from the CRISPR alignment in supplementary data S4, Supplementary Material online and mapped onto the tree. Spacers with the same coloring represent the exact same underlying sequence. Different coloring represents different underlying sequence. Blue bars represent CRISPR length, which was determined from the number of unaligned spacers for each CRISPR locus. Spacer deletions are represented by a black square with an x.
Phylogenetic Signal
Antigen character states for each of the 78 serovars were derived from Kaufman and White’s antigenic formulas (Grimont and Weill 2007) and mapped onto the ML phylogeny (fig. 1). We considered O group factors and two flagellar (H) antigens: H Phase 1 and H Phase 2. As shown in figure 1, the selected antigen character states appear in clusters on the phylogeny, but most are not monophyletic (i.e., O group 4 appears to have arisen at least four separate times). Their GSI scores only reveal a subset of character states that show significant genealogical exclusivity (or monophyly) (table 3). Despite these patterns, all three characters showed significant phylogenetic signals (P = 4.9E−09, 2.51E−09, and 1.39E−15, respectively) when compared with a star phylogeny (table 4).
Table 3.
GSI Values for the Antigen Character States
| Antigens | Num Taxa | GSI | P-value |
|---|---|---|---|
| O group | |||
| 8 | 11 | 0.27 | 0.001 |
| 3,10 | 9 | 0.14 | 0.490 |
| 16 | 2 | 0.05 | 0.893 |
| 7 | 13 | 0.26 | 0.003 |
| 4 | 20 | 0.16 | 0.461 |
| 9 | 9 | 0.24 | 0.010 |
| 35 | 2 | 0.13 | 0.165 |
| 13 | 4 | 0.15 | 0.158 |
| 1,3,19 | 2 | 0.11 | 0.226 |
| 30 | 2 | 0.06 | 0.774 |
| 28 | 2 | 0.24 | 0.050 |
| 11 | 2 | 1.00 | 0.008 |
| Flagellar antigen (H) Phase 1 | |||
| D | 7 | 0.17 | 0.130 |
| e,h | 11 | 0.14 | 0.544 |
| l,v | 7 | 0.16 | 0.168 |
| b | 10 | 0.15 | 0.361 |
| y | 4 | 0.10 | 0.546 |
| z10 | 2 | 0.10 | 0.284 |
| a | 3 | 0.10 | 0.395 |
| i | 5 | 0.15 | 0.173 |
| r | 5 | 0.17 | 0.078 |
| – | 2 | 0.49 | 0.017 |
| z4,z23 | 3 | 0.10 | 0.474 |
| k | 2 | 0.07 | 0.607 |
| c | 2 | 1.00 | 0.007 |
| f,g | 3 | 0.13 | 0.169 |
| z | 3 | 0.11 | 0.297 |
| z29 | 2 | 0.24 | 0.052 |
| g,s,t | 2 | 0.11 | 0.226 |
| f,g,s | 3 | 0.20 | 0.038 |
| Flagellar antigen (H) Phase 2 | |||
| 1,2 | 20 | 0.24 | 0.011 |
| 1,5 | 15 | 0.27 | 0.002 |
| 1,6 | 5 | 0.16 | 0.122 |
| e,n,x | 10 | 0.21 | 0.043 |
| e,n,z15 | 2 | 0.10 | 0.288 |
| – | 16 | 0.28 | 0.001 |
| l,w | 4 | 0.16 | 0.106 |
| 1,7 | 7 | 0.26 | 0.003 |
| 1,2,7 | 2 | 0.06 | 0.766 |
| z6 | 4 | 0.25 | 0.010 |
P <0.05 are in bold.
Table 4.
MultiState Lambda Test for Phylogenetic Signal
| Character | Ultrametric Tree |
Star Phylogeny (Null) |
Lambda Test |
||||
|---|---|---|---|---|---|---|---|
| Trait1.lnl | Trait1.q | Trait1.treeParam | Trait1.lnl.1 | Trait1.q.1 | Likelihood Ratio | P from Chi-Squared Test | |
| O antigens | −226.91 | −1.74 | 0.90 | −244.18 | 11.34 | 34.53 | 4.19E-09 |
| H antigen, phase 1 | −269.36 | −3.41 | 0.98 | −287.13 | −16.63 | 35.53 | 2.51E-09 |
| H antigen, phase 2 | −185.02 | −1.44 | 0.90 | −216.91 | −14.40 | 63.78 | 1.39E-15 |
Discussion
Using these 156 draft and complete Salmonella genomes enabled us to present the most highly resolved phylogeny for S. enterica subsp. enterica to date. This will be a valuable tool for understanding the phylogenetic and population genetic diversity among Salmonella food pathogens and will promote quicker and more accurate tracebacks when future foodborne illness outbreaks occur.
Despite the limitations stated previously of inferring phylogeny from an SNP matrix, this approach has yielded far more variable characters for inferring evolutionary history than any previous study. Although many of the relationships reconstructed in this study are consistent with previous reports, our increased data collection and taxon sampling provide a better context for interpreting the evolutionary history in this group. For example, previous studies showed the S. Typhi group as sister to the remaining Clade A (den Bakker, Switt, Govoni, et al. 2011; Fricke et al. 2011; Leekitcharoenphon et al. 2012; Desai et al. 2013). By contrast, our study clearly places the Typhi group as a derived lineage nested within Clade A. The four named lineages in this study (fig. 1) all showed previous phylogenetic support or had additional antigen characters that supported the relationship. “Section Typhi’s” antigen characters were variable, but these relationships were previously seen by eight studies (McClelland et al. 2004; Vernikos et al. 2007; den Bakker, Switt, Govoni, et al. 2011; Fricke et al. 2011; Jacobsen et al. 2011; Brankatschk et al. 2012; Yue et al. 2012). “Section Typhimurium” serovars are characterized by an O:4 group, and most have H: 1,2 Phase 2 flagellar antigens as well as previous phylogenetic support (den Bakker, Switt, Govoni, et al. 2011; Fey et al. 2012; Leekitcharoenphon et al. 2012). Along with previous molecular support (Vernikos et al. 2007; Didelot et al. 2011; Brankatschk et al. 2012; Leekitcharoenphon et al. 2012), “Section Enteritidis” serovars are characterized by having mostly O:9 groups and no Phase 2 flagellar antigens. Although no previous study uncovered the “Section Newport II” relationships (Newport + Muenchen + Litchfield), the lineage is very strongly supported in our ML tree and is also congruent with antigen characters: all have O:8 groups and 1,2 phase-2 flagellar antigens.
There has also been strong support coalescing around the sister relationship between S. Paratyphi C and S. Choleraesuis (Kingsley and Bäumler 2000; Liu et al. 2009; Soyer et al. 2009; Didelot et al. 2011; Fricke et al. 2011; Jacobsen et al. 2011; Trujillo et al. 2011; Achtman et al. 2012; Brankatschk et al. 2012). Because this is such a long, strongly supported branch (455 unique SNPs on our ML tree), it will be interesting to watch as our taxon sampling grows to include all ∼1,500 S. enterica subsp. enterica serovars. Taxon sampling, nucleotide character type, and phylogenic methods all influence inferred phylogeny, as shown by several earlier studies that reveal different relationships within our named sections (Bäumler et al. 1998; Kingsley and Bäumler 2000; Zou et al. 2013). Multilocus sequencing typing methods (Achtman et al. 2012) are mostly consistent with our results but suffer from zero resolution at the deeper nodes.
Our GSI analyses generally support the current taxonomic alignments; however, future research should investigate the nonmonophyletic serovars to explain the incongruity between experimentally based taxonomic alignments and what has been predicted by the genealogical species concept (Baum and Shaw 1995). Early molecular literature showed evidence of multiple independent origins for S. Paratyphi B (Barker et al. 1988), which we also recover (fig. 1 shows four independent S. Paratyphi B lineages). As mentioned in the Introduction, we also recovered a polyphyletic S. Newport II and S. Newport III, which was expected based on earlier investigations (Sangal et al. 2010; Achtman et al. 2012; Cao et al. 2013) (fig. 1). Little is known regarding multiple origins for the other serovars we found to be polyphyletic.
Overall, the three antigen characters (O group and two flagellar [H] antigens) revealed significant phylogenetic signals (table 4), but only a few antigen states had a significant GSI value, indicating that the character distribution for most of the antigens cannot be distinguished from random. Despite this, several characters did reveal a significant GSI (table 3), suggesting some degree of shared ancestry with other antigen characters. A few of the most common O groups are highlighted on the ML tree (fig. 1). Although it is possible that the deeper branching is incorrect, it is highly improbable that all of the O:4 strains shared the same most recent common ancestor, and, in fact, this is reflected in its GSI value (P = 0.46).
Without approaching a more complete taxon sampling for all ∼1,500 serovars within S. enterica subsp. enterica, any hypotheses regarding patterns of antigen evolution will be difficult to fully test. However, our study does suggest that the genes responsible for the O groups and phase 1 flagellar antigen traits are not evolving in a linear fashion, which, in turn, suggests that HGT, convergent evolution, or another mechanism may play an important role in these evolutionary patterns. Phase 2 antigens appear to have more shared ancestry, and this is reflected by the higher proportion of significant GSI values (table 3).
Analyzing character states for the CRISPR regions is much more complex than analyzing for discrete antigen characters. Between any given pair of Salmonella serovars, the CRISPR locus can be entirely replaced, leaving no homologous spacers to compare. Fabre et al. (2012) analyzed the utility of the CRISPR locus to “type” Salmonella strains or to determine serovar-level taxonomy. Our analysis provides an evolutionary framework to view the phylogenetic patterns of CRISPR diversity (fig. 4). Spacers can be deleted anywhere in the locus (spacer decay noted with “x” boxes), but they are only added to the 5′ end, leaving the 3′-most spacer as the most ancestral. When we look at all the CRISPR locus alignments (supplementary data S4, Supplementary Material online), we find evidence of decay scattered through each CRISPR locus. For example, S. Stanleyville, S. Gallinarum, and S. Pullorum appear to have lost the first four spacers in CRISPR2. If there is no evidence of homology, the 3′ spacer assumes position number 1.
Visualizing the first four spacers highlights the most informative ancestral pattern, whereas the length bar alludes to diversity even within serovars (e.g., length variation with S. Montevideo strains). There is also a striking reduction of CRISPR length in both locus 1 and locus 2 across several Clade A2 lineages. Overall, the diversity revealed by the uniqueness of the first four spacers closely resembles taxonomic diversity: There are 77 unique CRISPR 1 categories and 80 CRISPR 2 categories. If the CRISPR region is evolving in a linear fashion, we should expect closely related serovars to share more of the ancestral spacers. Although this pattern emerges in some parts of the tree (e.g., “Section Enteritidis”), it is more common to see shared ancestral spacers spanning the ML tree. For example, the same four ancestral CRISPR1 spacers are shared across three unrelated lineages: Three strains of S. Seftenberg (str. 604314, 316235162, and 423984-2), two S. Anatums, and one S. Virchow. So, although the CRISPR 1 and 2 loci might contain the appropriate level of variation for typing, and perhaps subtyping, within diverse serovars, the stability of the loci within and between serovars needs further investigation before its utility as an identification tool can be established.
Conclusions
We present the largest, most-resolved phylogeny of S. enterica subsp. enterica to date. Past typing methods gave us a very coarse view of strain diversity. Antigen screening, PFGE, multi-locus sequence typing (MLST), and a newly proposed CRISPR typing will all produce some level of ID; however, these methods are not currently able to place a new unknown strain into an evolutionary context, and that lack of context prevents more robust track-and-trace of contamination sources. Our research contributes the largest genome-scale phylogenetic framework to date toward a fine-scale reference phylogeny for all 1,500 S. enterica subsp. enterica serovars.
Based on our analysis, antigen characters and CRISPR loci reveal nonphylogenetic patterns. Although these patterns raise interesting evolutionary questions, they call into question the utility of relying on these characters for identification. For this reason, whole genome sequencing, SNP discovery, and phylogenetic analysis are quickly emerging as the standard for disease tracking.
Supplementary Material
Supplementary data S1–S4 and supplementary table S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Shea Gardner at Lawrence Livermore National Laboratory for the development of kSNP v. 2. They would also like to sincerely thank several people at FDA-CFAN: Charles Wang, Cong Li, and Andrea Ottesen for generating draft Salmonella genomes; David W. Weingaertner for assistance with fig. 4; Lili Velez and Barbara Berman for manuscript editing. Wesley Morovic at DuPont contributed helpful CRISPR sequence analyses, and Philippe Horvath provided the CRISPR DB II macro. And finally, the manuscript was greatly improved by critical comments from the reviewers. No human subjects or animals were used in this study. All authors have read the manuscript and agreed to its contents, subject matter, and author line order. These data are novel and have not been previously published elsewhere. Disclosure forms provided by the authors will be available with the full text of this article. This work was supported by the Center for Food Safety and Applied Nutrition at the US Food and Drug Administration.
Literature Cited
- Achtman M, et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012;8:e1002776. doi: 10.1371/journal.ppat.1002776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard MW, et al. On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE Pattern JEGX01.0004. PLoS One. 2013;8:e55254. doi: 10.1371/journal.pone.0055254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker RM, et al. Types of Salmonella Paratyphi B and their phylogenetic significance. J Med Microbiol. 1988;26:285–293. doi: 10.1099/00222615-26-4-285. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Horvath P. CRISPR: new horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3:143–162. doi: 10.1146/annurev-food-022811-101134. [DOI] [PubMed] [Google Scholar]
- Baum D, Shaw KL. Genealogical perspectives on the species problem. In: Hoch PC, Stephenson AC, editors. Experimental and molecular approaches to plant biosystematics. St. Louis (MO): Missouri Botanical Garden; 1995. [Google Scholar]
- Bäumler AJ, Tsolis RM, Ficht TA, Adams LG. Evolution of host adaptation in Salmonella enterica. Infect Immun. 1998;66:4579–4587. doi: 10.1128/iai.66.10.4579-4587.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- Boisvert S, Laviolette F, Corbeil J. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. 2010;17:1519–1533. doi: 10.1089/cmb.2009.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd EF, Wang FS, Whittam TS, Selander RK. Molecular genetic relationships of the Salmonellae. Appl Environ Microbiol. 1996;62:804–808. doi: 10.1128/aem.62.3.804-808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankatschk K, Blom J, Goesmann A, Smits THM, Duffy B. Comparative genomic analysis of Salmonella enterica subsp. enterica serovar Weltevreden foodborne strains with other serovars. Int J Food Microbiol. 2012;155:247–256. doi: 10.1016/j.ijfoodmicro.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Brown E, Kotewicz M, Cebula T. Detection of recombination among Salmonella enterica strains using the incongruence length difference test. Mol Phylogenet Evol. 2002;24:102–120. doi: 10.1016/s1055-7903(02)00222-1. [DOI] [PubMed] [Google Scholar]
- Cao G, et al. Phylogenetics and differentiation of Salmonella newport lineages by whole genome sequencing. PLoS One. 2013;8:e55687. doi: 10.1371/journal.pone.0055687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings MP, Neel MC, Shaw KL. A genealogical approach to quantifying lineage divergence. Evolution. 2008;62(9):2411–2422. doi: 10.1111/j.1558-5646.2008.00442.x. [DOI] [PubMed] [Google Scholar]
- den Bakker HC, Switt AI, Govoni G, et al. Genome sequencing reveals diversification of virulence factor content and possible host adaptation in distinct subpopulations of Salmonella enterica. BMC Genomics. 2011;12:425. doi: 10.1186/1471-2164-12-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PT, et al. Evolutionary genomics of Salmonella enterica subspecies. MBio. 2013;4(2):c00579-12. doi: 10.1128/mBio.00579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, et al. Recombination and population structure in Salmonella enterica. PLoS Genet. 2011;7:e1002191. doi: 10.1371/journal.pgen.1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre L, et al. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One. 2012;7:e36995. doi: 10.1371/journal.pone.0036995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, et al. Mismatch induced speciation in Salmonella: model and data. Philos Trans R Soc Lond B Biol Sci. 2006;361:2045–2053. doi: 10.1098/rstb.2006.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7:574–578. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey PD, et al. Assessment of whole genome mapping in a well-defined outbreak of Salmonella enterica serotype Saintpaul. J Clin Microbiol. 2012;50:3063–3065. doi: 10.1128/JCM.01320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WF, et al. Comparative genomics of 28 Salmonella enterica isolates. evidence for CRISPR-mediated adaptive sublineage evolution. J Bacteriol. 2011;193:3556–3568. doi: 10.1128/JB.00297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SN, Slezak T. Scalable SNP analyses of 100+ bacterial or viral genomes. J Forensic Res. 2010;1:107. [Google Scholar]
- Grimont PAD, Weill F-X. Antigenic formulae of the Salmonella serovars. 2007 WHO Collaborating Centre for Reference and Research on Salmonella, Paris (France): World Health Organization. [Google Scholar]
- Harmon LJ, Weir JT, Brock CD, Glor RE. GEIGER: investigating evolutionary radiations. Bioinformatics. 2008;24:129–131. doi: 10.1093/bioinformatics/btm538. [DOI] [PubMed] [Google Scholar]
- Horvath P, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi A, Iyoda S, Ohnishi M, EHEC Study Group Molecular characterization reveals three distinct clonal groups among clinical shiga toxin-producing Escherichia coli strains of serogroup O103. J Clin Microbiol. 2012;50:2894–2900. doi: 10.1128/JCM.00789-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen A, Hendriksen RS, Aaresturp FM, Ussery DW, Friis C. The Salmonella enterica pan-genome. Microbiol Ecol. 2011;62:487–504. doi: 10.1007/s00248-011-9880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, et al. Phylogenetic analysis of non-O157 Shiga toxin-producing Escherichia coli strains by whole-genome sequencing. J Clin Microbiol. 2012;50:4123–4127. doi: 10.1128/JCM.02262-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley RA, Bäumler AJ. Host adaptation and the emergence of infectious disease: the Salmonella paradigm. Mol Microbiol. 2000;36:1006–1014. doi: 10.1046/j.1365-2958.2000.01907.x. [DOI] [PubMed] [Google Scholar]
- Leekitcharoenphon P, Lukjancenko O, Friis C, Aarestrup FM, Ussery DW. Genomic variation in Salmonella enterica core genes for epidemiological typing. BMC Genomics. 2012;13:88. doi: 10.1186/1471-2164-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W-Q, et al. Salmonella Paratyphi C: genetic divergence from Salmonella Choleraesuis and pathogenic convergence with Salmonella Typhi. PLoS One. 2009;4:e4510. doi: 10.1371/journal.pone.0004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, et al. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet. 2004;36:1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- McQuiston JR, et al. Molecular phylogeny of the Salmonellae: relationships among Salmonella species and subspecies determined from four housekeeping genes and evidence of lateral gene transfer events. J Bacteriol. 2008;190:7060–7067. doi: 10.1128/JB.01552-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- R Core Development Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2012. [Google Scholar]
- Sangal V, et al. Evolution and population structure of Salmonella enterica serovar Newport. J Bacteriol. 2010;192:6465–6476. doi: 10.1128/JB.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan E, et al. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitkin ES, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyer Y, Orsi RH, Rodriguez-Rivera LD, Sun Q, Wiedmann M. Genome wide evolutionary analyses reveal serotype specific patterns of positive selection in selected Salmonella serotypes. BMC Evol Biol. 2009;9:264. doi: 10.1186/1471-2148-9-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Trujillo S, Keys CE, Brown EW. Evaluation of the taxonomic utility of six-enzyme pulsed-field gel electrophoresis in reconstructing Salmonella subspecies phylogeny. Infect Genet Evol. 2011;11:92–102. doi: 10.1016/j.meegid.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Vernikos GS, Thomson NR, Parkhill J. Genetic flux over time in the Salmonella lineage. Genome Biol. 2007;8:R100. doi: 10.1186/gb-2007-8-6-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue M, et al. Diversification of the Salmonella fimbriae: a model of macro- and microevolution. PLoS One. 2012;7:e38596. doi: 10.1371/journal.pone.0038596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, et al. Meta-analysis of pulsed-field gel electrophoresis fingerprints based on a constructed Salmonella database. PLoS One. 2013;8:e59224. doi: 10.1371/journal.pone.0059224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.