

Abstract
Dicationic 2,6-diphenylpyrazines, aza-analogues and prodrugs were synthesized; evaluated for DNA affinity, activity against Trypanosoma brucei rhodesiense (T. b. r.) and Plasmodium falciparum (P. f.) in vitro, efficacy in T. b. r. STIB900 acute and T. b. brucei GVR35 CNS mouse models. Most diamidines gave poly(dA-dT)2 ΔTm values greater than pentamidine, IC50 values: T. b. r. (4.8 to 37 nM) and P. f. (10 to 52 nM). Most diamidines and prodrugs gave cures for STIB900 model (11, 19a and 24b 4/4 cures); 12 3/4 cures for GVR35 model. Metabolic stability half-life values for O-methylamidoxime prodrugs did not correlate with STIB900 results.
1. Introduction
Tropical protozoan diseases, such as malaria and human African trypanosomiasis (HAT), affect millions of people in large parts of the world.1 Malaria caused by Plasmodium falciparum (P. f.) leads to 665,000 deaths each year.2 HAT (or sleeping sickness), another devastating disease, is caused by Trypanosoma brucei rhodesience (T. b. r.) and Trypanosoma brucei gambiense (T. b. g.), which is eventually fatal without treatment. There are below 10,000 reported cases and 30,000 estimated cases of HAT a year mostly in sub-Saharan Africa.3, 4 HAT has two stages: an early stage in which the parasites reside and proliferate in the hemolymphatic system and a second stage in which the parasites cross the blood-brain barrier (BBB) and infect the central nervous system (CNS). 5 The drugs currently in use against both malaria and HAT are far from satisfactory; most drugs suffer from poor oral bioavailability and severe side effects. For example, the second stage HAT drug melarsoprol causes a reactive encephalopathy which leads to death in 5% of treated patients.6 Furthermore, for both diseases the cases of treatment failure because of drug resistance have increased in recent years. For second stage HAT, there are only two drugs melarsoprol and eflornithine available for monotherapy and NECT a combination therapy with niturtimox and eflornithine.7 Therefore, there is an urgent need for development of more effective, orally available and less toxic drugs against these tropical protozoan diseases.
Although numerous DNA binding aromatic diamidines exhibit potent antiprotozoal activity against these tropical diseases, pentamidine 1 (Fig. 1) is the only one which has seen significant clinical use in humans.8 Pentamidine has been used to treat first stage T. b. g. HAT, antimony-resistant leishmaniasis, and Pneumocystis jiroveci pneumonia. Because the amidine groups are protonated at physiological pH, pentamidine has low oral availability and requires parenteral administration, which makes its clinical use difficult in remote regions. Furamidine 2a (Fig. 1), a diphenyl furan diamidine analogue, is the active metabolite of the di-O-methylamidoxime prodrug pafuramidine 2c (Fig. 1) which reached phase III clinical trials against first-stage HAT and P. jiroveci pneumonia, and phase II clinical trials against malaria.1, 5, 8, 9 Due to hepatic and renal toxicity of pafuramidine in humans observed in an additional safety study paralleling the phase III trials, the further development of pafuramidine has been terminated. Introduction of a nitrogen atom into one or both of the terminal phenyl rings of furamidine resulted in aza-analogues of furamidine 3a and 4a (Fig. 1), which exihibited more potent in vivo activities against HAT than pentamidine and furamidine.10, 11 The O-methylamidoxime prodrugs of azafuramidines 3c and 4c (Fig. 1) were found to be quite effective in the second stage HAT GVR35 CNS mouse model 5 and 3c was effective in a vervet monkey model of second stage HAT.12 As yet, only very few compounds have shown good activity in the GVR35 CNS mouse model. Since the aza-analogue 4c is better tolerated than 3c in toxicity studies in vervet monkeys,5 it is under further evaluation as a possible preclinical candidate for treatment of the second stage HAT. Unexpectedly, the parent diamidine 4a also showed potent activity in the CNS mouse model on intraperitoneal injection; therefore, 4a may have potential to become a treatment option for second stage HAT.5
Figure 1.

Aromatic diamidine antiprotozoal agents.
Due to the promising results from the furamidine series, a large number of furamidine related diamidines have been synthesized.8, 13 Recently, we have described a series of linear terphenyl diamidine 5 (Fig. 1) and their aza-analogues (e.g. 6), which showed significant DNA minor groove binding affinity and low nanomolar antiprotozoal activity against T. b. r. and P. f.14–16 The in vivo efficacy for three of those aza-analogues in the T. b. r. STIB900 acute mouse model is much superior to that of furamidine, and comparable to the azafuramidines. Unfortunately, the di-amidoximes and di-O-methylamidoxime prodrugs of the terphenyl dicationic analogues showed poor bioconversion and were not effective on oral administration.14 In this study we have explored a series of novel curved dicationic 2,6-diarylpyrazines, their aza-analogues and their prodrugs which are isomeric with their linear 2,5-pyrazines.14–16 Herein, we describe the synthesis, DNA binding affinity, in vitro activities against T. b. r. and P. f. and in vivo activities in the T. b. r. STIB900 acute mouse model and T. b. brucei GVR35 CNS mouse model for these curved 2,6-diarylpyrazines.
2. Chemistry
The synthesis of the parent dicationic 2,6-diphenylpyrazine 10 begins with Suzuki coupling of 2,6-dichloropyrazine (7) with 4-cyanophenylboronic acid (8) to yield the diphenylpyrazine di-nitrile 9 (Scheme 1).14–16 The di-nitrile 9 was converted to the diamidine 10 by the action of lithium trimethylsilylamide [LiN(TMS)2] in THF. The di-amidoxime prodrug 11 was obtained by reaction of the di-nitrile 9 with hydroxylamine and followed by O-methylation with dimethylsulfate in the presences of lithium hydroxide to yield the corresponding di-O-methylamidoxime prodrug 12. 10
Scheme 1. Reagents and conditions.

(i) Pd(PPh3)4, Na2CO3, toluene, 80 °C; (ii) (a) LiN(TMS)2, THF; (b) HCl(gas), EtOH; (iii) NH2OH-HCl, KOt-Bu, DMSO; (iv) LiOH, (CH3)2SO4, DMF.
Employing the related Stille coupling process starting with 2,6-di(tri-n-butylstannyl)pyrazine the symmetrical di-nitriles 14a and 14b were made in one step (Scheme 2).17 The di-nitriles 14a–b were converted to the diamidines 15a–b using LiN[TMS]2 as previously mentioned.
Scheme 2. Reagents and conditions.
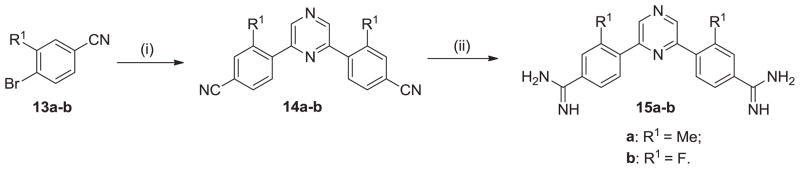
(i) 2,6-bis(tri-n-butylstannyl)pyrazine, Pd(PPh3)4, xylene, 120 °C; (ii) (a) LiN(TMS)2, THF; (b) HCl(gas), EtOH.
The dissymmetric mono-aza analogues 19a–b were made as outlined in Scheme 3. The pyridyl rings are introduced in the first step by performing Stille coupling between 2-chloro-6-(tri-n-butylstannyl)pyrazine and the appropriate bromocyanopyridines 16a–b.17 Subsequently, a Suzuki reaction between the 6-(pyridyl)-2-chloropyrazines 17a–b and 4-cyanophenylboronic acid yields the dissymmetric di-nitriles 18a–b. The di-nitriles were converted into the corresponding diamidines 19a–b, the amidoxines 20a,b and the O-methylamidoximes 21a–b as discussed previously for Scheme 1.
Scheme 3.

Regents and conditions: (i) 2-chloro-6-(tri-n-butylstannyl)pyrazine, Pd(PPh3)4, xylene, 120 °C; (ii) Pd(PPh3)4, Na2CO3, toluene, 80 °C; (iii) (a) LiN(TMS)2, THF; (b) HCl(gas), EtOH; (iv) NH2OH-HCl, KOt-Bu, DMSO; (v) LiOH, (CH3)2SO4, DMF.
The synthesis of the symmetrical di-aza analogues is presented in Scheme 4. It this case the needed di-nitriles 22a–b are made directly by Stille coupling of the bromocyanopridines 16a–b with 2,6-di(tri-n-butylstannyl)pyrazine. The di-nitriles were converted into the corresponding diamidines 23a–b, the amidoxines 24a–b and the O-methylamidoximes 25a–b as discussed previously for Scheme 1.
Scheme 4. Reagents and conditions.

(i) 2,6-bis(tri-n-butylstannyl)pyrazine, Pd(PPh3)4, xylene, 120 °C; (ii) (a) LiN(TMS)2, THF; (b) HCl(gas), EtOH; (iii) NH2OH-HCl, KOt-Bu, DMSO; (iv) LiOH, (CH3)2SO4, DMF.
3. Biology
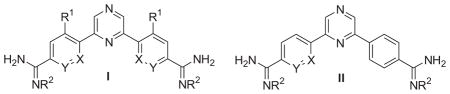
The results for the evaluation of the dicationic 2,6-diarylpyrazine analogues and their prodrugs against T. b. r. and P. f. and their DNA binding affinities are shown in Table 1. For comparative purposes, the analogous data for pentamidine (1), furamidine (2a), azafuramidines 3a, 4a and 2,5-diphenyl pyrazine diamidine (6) are also included in Table 1. 10, 16
Table 1.
DNA affinities and antiprotozoan activity for 2,6-diarylpyrazine diamidines.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Code | Structure type | X | Y | R1 | R2 | ΔTm (°C) a |
T. b. r. IC50 (nM) b |
P. f. IC50 (nM) b |
Cytotoxicity IC50 (nM) c |
| 1 pentam idne | / | / | / | / | / | 12.6 | 2.2 | 46.4 | 2,100 |
| 2a | / | / | / | / | / | 25 | 4.5 | 15.5 | 6,400 |
| 3a | / | / | / | / | / | 19.3 | 6.5 | 6.5 | 77,900 |
| 4a | / | / | / | / | / | 15.5 | 21 | 83 | 83,000 |
| 6 | / | / | / | / | / | 8.0 | 18 | 0.4 | 42,500 |
| 10 | I | CH | CH | H | H | 15.1 | 5.8 | 10 | 80,800 |
| 11 | I | CH | CH | H | OH | / | 7,950 | 870 | 2,900 |
| 12 | I | CH | CH | H | OMe | / | 18,900 | 410 | 58,700 |
| 15a | I | CH | CH | Me | H | 5.1 | 462 | 323 | 117,000 |
| 15b | I | CH | CH | F | H | 8.2 | 27 | 27 | 29,800 |
| 19a | II | N | CH | / | H | 13.1 | 14 | 52 | 117,000 |
| 20a | II | N | CH | / | OH | / | 7,780 | 8,400 | 24,600 |
| 21a | II | N | CH | / | OMe | / | 25,300 | 5,600 | >180,000 |
| 19b | II | CH | N | / | H | 15.1 | 6.0 | 10 | 34,100 |
| 20b | II | CH | N | / | OH | / | 10,800 | 453 | 5,000 |
| 21b | II | CH | N | / | OMe | / | 194,000 | 453 | >185,000 |
| 23a | I | N | CH | H | H | 11.2 | 37 | 31 | 139,000 |
| 24a | I | N | CH | H | OH | / | 90,600 | 7,010 | >196,000 |
| 25a | I | N | CH | H | OMe | / | 119,600 | 7,390 | 20,100 |
| 23b | I | CH | N | H | H | 12.1 | 4.8 | 85 | 24,400 |
| 24b | I | CH | N | H | OH | / | 95,400 | 785 | >190,000 |
| 25b | I | CH | N | H | OMe | / | 7,870 | 328 | >212,000 |
The T. b. r. (Trypanosoma brucei rhodesiense) strain was STIB900, and the P. f. (Plasmodium falciparum) strain was K1. The IC50 values are the mean of two independent assays. Individual values differed by less than 50% of the mean see ref 17.
The interaction of diamidines with nuclear and kinetoplast DNA has been shown to be an important part of their mode of antiparasitic action.8f The ΔTm values of these dicationic 2,6-diphenylpyrazines and their aza-analogues range from high values of 15.1 °C to low ones of 5.1 °C, as shown in Table 1. The parent diamidine 10 showed a ΔTm value of 15.1 °C, which is lower than that of furamidine (2a) (ΔTm = 25 °C) and higher than that of pentamidine (1) (ΔTm = 12.6 °C). In comparison to 2a the ΔTm value of 10 is consistent with its increased hydrophilic property as a result of the additional two nitrogen atoms in the central pyrazine ring of 10. This result may further suggest that the hydrophobic component is important for minor groove DNA binding. The ΔTm value (15.1°C) for 10 is higher than that of the linear 2,5-isomer 6 (ΔTm = 8.0°C). One possible explanation for this result may be that compound 10 presents an approximately crescent shape which more closely fits the curvature of the DNA minor groove, similar to furamidine; and the linear isomer 6 needs incorporation of a water molecule in the complex to simulate the curved structure of DNA minor groove.20 The compounds 15a and 15b, methyl and fluorine substituted analogues, showed much lower ΔTm values, which may be due to their twisted shape. Introduction of a nitrogen atom meta to the amidine group in one or both of the phenyl rings in the parent compound 10 leads to decreased DNA binding affinity: compound 19a with one nitrogen resulted in a 2 °C decrease in ΔTm value and compound 23a with two nitrogen atoms gave a ΔTm value with a 3.9 °C decrease. However, their corresponding ortho-isomers 19b and 23b showed a smaller influence on the DNA binding affinity: compound 19b, with one nitrogen atom, showed the same ΔTm value as that of the parent compound 10 and compound 23b, with two nitrogen atoms, showed a ΔTm value reduced by 3.0 °C. These results are consistent with the effect of nitrogen substitution relationships found in the previous study of the aza-analogues of furamdine (2a).10 It is also noteworthy that the compounds which exhibit the higher ΔTm values showed the higher antitrypanosomal activity whereas weaker binding compounds show lower activity (see below) which is consistent with previous observations that while a threshold level of binding appears essential for activity a direct correlation between DNA affinity and in vitro activity is neither expected nor found.8f A similar trend was not found for the antiplasmodial activity.
These 2,6-diarylpyrazine diamidines exhibited significant in vitro antitrypanosomal and antiplasmodial activity at the low nanomolar level. The parent dicationic 2,6-diphenylpyrazine 10 showed an IC50 value of 6 nM against T. b. r., comparable to that of pentamidine and furamidine. The antiplasmodial activity of 10 (IC50 = 10 nM) is approximately five-fold more active than pentamidine and slightly less active than furamidine. The antiprotozoal activity of 10 is similar to that of the azafuramidine 3a. Compared to the isomer 2,5-dicationic diphenylpyrazine 6, there are significant differences: the 2,6-isomer 10 was three times more active than 2,5-isomer 6 against T. b. r.; conversely, 10 was 25-fold less potent than 6 against P. f. The 2,6-isomer 10 lost the good selectivity for P. f. versus T. b. r. for which the 2,5-isomer 6 showed 45-fold selectivity for P. f., compared to T. b. r. These differences may be due, in part, to the fact that the 2,6-diphenyl pyrazine diamidine 10 is an approximately crescent shaped molecule which more closely fits the curvature of the DNA minor groove, much similar to furamidine;8, 19 however, its dicationic isomer 2,5-diphenylpyrazine 6 is a linear molecule which presumably requires the incorporation of a water molecule into the recognition complex to simulate the curved structure of DNA minor groove.20 The introduction of a methyl group or fluorine atom into the meta-position to the amidine group on both of the phenyl rings yielded compounds 15a and 15b which showed more than a sixty- and four-fold loss of their antiprotozoal activities against T. b. r. and P. f., respectively. Compound 19a and 23a, in which a nitrogen atom has been placed meta to the amidine group in one or both of the phenyl rings, showed a two- and six-fold decrease in potency compared to the parent diamidine 10 against T. b. r. and a ten- and six-fold decreased potency against P. f. The two analogues 19b and 23b, in which the nitrogen atoms are ortho to the amidine, exhibited equivalent potency against T. b. r. and a similar or eight-fold decrease in activity against P. f., compared to the parent compound 10. It is noted that the compounds 19b and 23b in which the nitrogen atoms are ortho to the amidine exhibited higher activity against T. b. r. than the corresponding meta-isomers 19a and 23a. However, a similar trend was not found for P. f. activity. Ten potential di-amidoxime and di-O-methylamidoxime prodrugs of the dicationic 2,6-diphenylpyrazine and aza-analogues were prepared. As expected, these amidoximes and O-methylamidoxime prodrugs showed low antiprotozoal activity when tested in vitro due to the absence of metabolizing enzymes.10
Given the promising in vitro T. b. r. activity of these new diamidines, except the methyl analogue 15a which showed only moderate antitrypanosomial activity, we have evaluated them and their prodrugs in the stringent STIB900 acute mouse model for T. b. r. which mimics first stage disease.5, 9, 18 Since the diamidines exhibit quite high pKa values (10–11) and therefore unlikely to cross the intestinal barrier they were administered intraperitoneally ; the prodrugs were designed to enhance oral bioavailability and hence were given orally. The results are shown in Table 2 (diamidines) and Table 3 (prodrugs).
Table 2.
In vitro and in vivo anti-trypanosomal activity of 2,6-diarylpyrazine diamidines in the STIB900 mouse model.a
| Code | T. b. r. IC50(nM) |
Dosage (ip, mg/kg) b | Cures c | Survival (days) d |
|---|---|---|---|---|
| 1 | 2.2 | 20 | 2/4 | >57.5 |
| 5 | 1/4 | >38 | ||
| 2a | 4.5 | 20 | 3/4 | >57.75 |
| 5 | 1/4 | >46 | ||
| 3a | 6.5 | 20 | 4/4 | >60 |
| 5 | 3/4 | >54.5 | ||
| 4a | 21 | 20 | 4/4 | >60 |
| 5 | 3/4 | >49.5 | ||
| 6 | 18 | 5 | 0/4 | 36.5 |
| 10 | 5.8 | 5 | 3/4 | >53.5 |
| 15a | 462 | / | / | / |
| 15b | 27 | 5 | 0/4 | >41 |
| 19a | 14 | 5 | 4/4 | >60 |
| 19b | 6 | 5 | 2/4 | >60 |
| 23a | 37 | 5 | 2/4 | >53.75 |
| 23b | 4.8 | 5 | 1/4 | >36.5 |
Table 3.
In vivo anti-trypanosomal activity of 2,6-diarylpyrazine diamidine prodrugs in the STIB900 mouse model.a
| Code | Dosage (po, mg/kg)c | Cures d | Survival (days)e |
|---|---|---|---|
| 2b(2a) | 100 | 0/4 | 50 |
| 2c(2a) | 25 | 4/4 | >60 |
| 10 | 4/4 | >60 | |
| 3c(3a) | 25 | 4/4 | >60 |
| 4c(4a) | 25 | 2/4 | >42 |
| 11(10) | 25 | 4/4 | >60 |
| 10 | 0/4 | 22.5 | |
| 12(10) | 25 | 3/4 | >53.5 |
| 10 | 3/4 | >56.5 | |
| 20a(19a) | 25 | 2/4 | >45.75 |
| 21a(19a) | 25 | 3/4 | >57.5 |
| 10 | 1/4 | >49 | |
| 20b(19b) | 25 | 2/4 | >50 |
| 21b(19b) | 25 | 2/4 | >50 |
| 24a(23a) | 25 | 0/4 | 23.75 |
| 25a(23a) | 25 | 2/4 | >38.75 |
| 24b(23b) | 25 | 4/4 | >60 |
| 10 | 1/4 | >33.75 | |
| 25b(23b) | 25 | 1/4 | >43.25 |
For comparative purposes Table 2 and Table 3 also contain in vivo data for the dicationic analogues pentamidine (1), furamidine (2a), azafuramidines 3a and 4a, the dicationic 2,5-diphenylpyrazine 6 and the prodrugs 2b, 2c, 3c and 4c in the same mouse model. 10, 14, 16, 23 On intraperitoneal dosing at 5 mg/kg all of the tested dications show a significant increase in survival time for the treated animals compared to untreated controls. The parent compound 10 gave 3/4 cures at a dose of 5 mg/kg, which is superior to that of pentamidine (1) and furamidine (2a) (1/4 cure), and is as effective as the azafuramidines 3a and 4a (3/4 cure). The linear 2,5-isomer 6 was less effective and gave no cure but did show an increase in mean survival time. The fluorine substituted analogue 15b gave also no cures in the in vivo model which is consistent with its lower in vitro activity against T. b. r. The four aza-analogues 19a, 19b, 23a and 23b exhibited identical or better results to that for furamidine at a dose of 5 mg/kg. The best result obtained was for compound 19a, which showed 4/4 cures at a dose of 5 mg/kg. It is noteworthy that although compounds 19a and 23a, in which the nitrogen atom is meta to the amidine group, exhibited a significant loss of in vitro potency against T. b. r. compared to their isomeric compounds 19b and 23b, in which the nitrogen atom is ortho to the amidine, the in vivo efficacy of 19a (4/4 cure) and 23a (2/4 cure) was superior to the ortho isomers 19b (2/4 cure) and 23b (1/4 cure). This result may be due to pharmacokinetic differences between the ortho and meta isomers and/or the differential involvement of transporters. In general, the results for the aza-pyrazine analogues are consistent with those observed for the aza-furamidine system.10
Although previous studies of di-amidoxime and di-O-methylamidoxime prodrugs of the linear terphenyl diamidine analogues showed poor bioconversion and were not curative on oral administration,14 all the di-amidoxime and di-O-methylamidoxime prodrugs of the dicationic 2,6-diphenylpyrazine and aza-analogues showed activity when they were administered orally to the mice during this study. In vitro metabolic stability studies using mouse liver microsomes showed that di-O-methylamidoxime prodrugs were biotransformed at different rates, with 12, 21b, and 25b showing shorter half-life than 21a and 25a (Table 4). However, this difference did not translate into activity in the STIB900 mouse model, as the latter generally gave more cures, with the exception of 12 (Table 3). This disconnect is likely due to a recent finding that intrahepatic binding and efflux of diamidines formed in the hepatocytes, rather than enzymatic biotransformation of prodrugs, determined the disposition of active diamidine metabolites.22 It should also be noted that there was a marked interspecies difference in the metabolic stability of di-O-methylamidoxime prodrugs, as liver microsomes derived from humans metabolized the prodrugs much faster than those from mice (Table 4). This could be due to species differences in the enzyme activity and expression level of CYP4F/cyp4f enzymes, which were shown to be responsible for catalyzing the primary O-demethylation of the di-O-methylamidoxime pafuramidine in the human liver and intestinal microsomes.23
Table 4.
In vitro metabolic stability of 2,6-diarylpyrazine diamidine prodrugs
| Code | Mouse t1/2 (min)a | Human t1/2 (min)a |
|---|---|---|
| 2b | 29 ± 10# | NDb |
| 2c | 150 ± 10c | 6.8 ± 2c |
| 11 | 1.9 ± 0.1# | ND |
| 12 | 26 ± 6 | 8.6 ± 0.9 |
| 20a | 30 ± 4# | ND |
| 21a | 210 ± 100 | 7.6 ± 0.3 |
| 20b | 36 ± 5# | ND |
| 21b | 35 ± 6 | 6.5 ±1.2 |
| 24a | 20 ± 0.4# | ND |
| 25a | 200 ± 70 | 70 ± 23 |
| 24b | 51 ± 1.4# | ND |
| 25b | 59 ± 10 | 14 ± 6 |
Bis-amidoxime prodrugs were incubated with mouse liver S9 fraction, instead of liver microsomes.
Mean ± standard deviations of triplicate determinations.
ND, not determined.
Substrate concentration for 2c was 3 μM and its t1/2 was shown as mean and range of duplicate determinations.
Very interestingly, treatment with the amidoxime produgs 11 and 24b resulted in cures of all mice at the oral dose of 25 mg/kg, superior to the corresponding O-methylamidoxime prodrugs 12 (3/4 cure) and 25b (1/4 cure) at the same dosage. Both of the amidoximes 11 and 24b are more potent than the amidoxime prodrug 2b of furamidine which gave no cure at an oral dose of 100 mg/kg (Table 3).24 These results do not parallel those observed in the furamidine series, which showed that the O-methylamidoximes are more effective than the amidoximes.10 The O-methylamidoxime prodrug 12 of the parent compound 10 gave 3/4 cures at the oral dose of 10 mg/kg. To evaluate the bioconversion of the di-amidoxime prodrugs, mouse liver S9 fractions were used as they contain cytochrome b5 and cytochrome b5 reductase, which are likely required to reduce amidoxime to amidine.25 All di-amidoxime prodrugs examined in this study were efficiently metabolized (half-lives ranged from 2 to 51 min; Table 4), supporting their potential as prodrugs to generate active diamidine metabolites in vivo, but failed to explain the superiority of oral 24b over other di-amidoximes (except 11) in the STIB900 mouse model. As discussed above, this observation underscores the role of intrahepatic binding and efflux from hepatocytes, rather than bioconversion, in determining the disposition and activity of active diamidine metabolites in vivo.
Given the potent in vivo activity found in the T. b. r. STIB900 acute mouse model 11 and 12 were selected for study in the T. b. brucei GVR35 mouse model for second stage disease.5 In sharp contrast to the results found in the STIB900 model, 11 was not effective at a dosage of 100 mg/kg for five days in the GVR35 model providing no cures and only a modest increase (69 days) in survival time (Table 5). This shows that compound 11 is only able to remove trypanosomes from the hemolymphatic compartment. However, at the same dosage 12 gave 2/5 cures. To achieve cures in the CNS mouse model drugs must cross the blood brain barrier and reach trypanocidal levels in the CSF and CNS. These additional barriers often result in CNS drug levels lower than that in blood. Hence, to attempt to compensate for this circumstance where possible we further test with increased doses. When the dosing of 12 was extended to 10 days 3/4 cures were noted showing that this compound is penetrating into the brain in sufficient concentration to cure CNS infection. The activity of 12 in the GVR35 CNS model, with 2/5 mice cured at dosage of 100 mg/kg for five days compares favorably with that for pafuramidine 2c which showed 3/5 cured at the same dosage with the five day regimen. The efficacy of 12 is somewhat reduced from that of the O-methylamidoxime prodrug 4c for treatment of second stage HAT which gave 5/5 cures at the same dosage (Table 5).5 Nevertheless, prodrug 12 is one of only a very few compounds which have shown good activity in this CNS model. The compounds 24b and 25a were also selected for study in the T. b. brucei GVR35 CNS mouse model. Both compounds were not curative at the oral dosage of 100 mg/kg for five days but they extended the survival time of mice similarly to control mice treated with diminazine (at 40mg/kg ip single dose) which is a diamidine curing only first stage disease. The results for the amidoxime and O-methylamidoxime prodrugs of this series of dications provides stimulation for further evaluation of their efficacy and toxicity.
Table 5.
In vivo anti-trypanosomal activity of 2,6-diarylpyrazine diamidine prodrugs in the GVR35 CNS mouse model.a
| Codeb | Dosage (po, mg/kg)/no. of days administered | No. of mice cured c | MSDd |
|---|---|---|---|
| 2ce(2a) | 100/5 | 3/5 | >167.8 |
| 3ce(3a) | 100/5 | 5/5 | >180 |
| 4ce(4a) | 100/5 | 5/5 | >180 |
|
| |||
| 11(10) | 100/5 | 0/5 | 69 |
| 12(10) | 100/5 | 2/5 | >173.8 |
| 100/10 | 3/4 | >180.0 | |
| 24b(23b) | 100/5 | 0/5 | 67.2 |
| 25a(23a) | 100/5 | 0/5 | 95.2 |
See Ref. 5 for details of GVR35 CNS mouse model.
Code for parent of prodrug in parenthesis.
Cure defined as survival for more than 180 days after infection without showing a parasitemia relapse.
MSD (mean survival days) was determined for mice with and without parasitemia relapse.
Data from Ref. 5
4. Conclusions
A series of dicationic 2,6-diphenylpyrazines and aza analogues have been prepared which exhibited DNA binding which is consistent with a role in their mode of action, showed potent in vitro activity against both T. b. r. and P. f., and gave promising results on intraperitoneal administration in the stringent T. b. r. STIB900 mouse model. The diamidines 10 and 19a exhibited in vivo efficacy (3/4 or 4/4 cures at 5 mg/kg dosage, ip) in the STIB900 model, superior to that of furamidine (2a), and comparable to or better than the azafuramidines 3a and 4a. Eight of the ten prodrugs of the dicatonic 2,6-diphenylpyrazine and aza-analogues showed good oral activity, giving cures in the STIB900 acute mouse model. The potent O-methylamidoxime prodrug 12 also showed good in vivo oral efficacy in the GVR35 second stage mouse model. This series of dicationic 2,6-diphenylpyrazine analogues and their prodrugs merit further evaluation for treatment of both stages of HAT.
5. Experimental Section
5.1 Biology
5.1.1 Efficacy Studies
The in vitro assays13 with T. b. r. STIB 900 and P. f. K1 strain as well as the efficacy studies in an acute mouse model for T. b. r. STIB 9005 were carried out as previously reported. The studies in the T. b. brucei GVR35 mouse model for second stage disease were performed as previously described.5 All protocols and procedures for the mouse models used in the current study were reviewed and approved by the local veterinary authorities of Canton Basel-Stadt, Switzerland. The data was generated at the time the determination of survival was still accepted by the authorities.
5.1.2 Tm Measurements
Thermal melting experiments were conducted with a Cary 300 spectrophotometer. Cuvettes for the experiment were mounted in a thermal block and the solution temperatures monitored by a thermistor in the reference cuvette. Temperatures were maintained under computer control and increased at 0.5 °C/min. The experiments were conducted in 1 cm path length quartz curvettes in CAC 10 buffer (cacodylic acid 10mM, EDTA 1mM, NaCl 100mM with NaOH added to give pH = 7.0). The concentrations of DNA were determined by measuring its absorbance at 260 nm. A ratio of 0.3 moles compound per mole of DNA was used for the complex and DNA alone was used as a control.14 ΔTm values were determined by the peak in first derivative curves (dA/dT).26
5.1.3 In vitro metabolic stability assays
The procedures used were similar to a reported method.23a Substrate stock solutions were prepared in DMSO. DMSO content was kept at 0.5% (v/v) in final incubations. Incubation mixtures (final volume 0.25 ml) consisted of 10 μM substrate and 0.5 mg/ml pooled liver microsomes from human or mouse (XenoTech, Lenexa, KS) for O-methylamidoxime prodrugs, or liver S9 fraction from mouse for amidoxime prodrugs. Reactions were carried out in 100 mM phosphate buffer (pH 7.4) containing 3.3 mM MgCl2. Mouse liver S9 fractions were prepared from male Swiss Webster mice (25–30 g) as previously reported. 27 Briefly, four volumes of 0.25 M sucrose containing 0.1 M KCl and 1 mM EDTA was added to mouse liver and homogenized on ice with a sonic dismembrator (Fisher Scientific, Fair Lawn, NJ). The homogenate was centrifuged at 9,000×g for 20 min at 4°C and then the supernatant fraction (S9 fraction) was collected and aliquoted before storing at −78°C. After a 5-min pre-equilibration period at 37°C, the metabolic stability reactions (in triplicate) were initiated by adding the cofactor (1 mM β-NADPH for microsomal incubations or a cocktail of 1 mM β-NADPH, 1 mM NADH, and 3.3 mM UDPGA for incubations with S9 fractions) and kept at 37°C. Aliquots (100 μl) of the reaction mixtures were removed at 0, 15, 30, and 60 min and individually mixed with 100 μl of ice-cold acetonitrile. The mixtures were vortex-mixed, and precipitated protein was removed by centrifugation at 1,400×g for 15 min. The supernatant fractions were analyzed immediately by HPLC/UV. 23a In vitro half-lives were obtained using the one-phase exponential decay model with plateau set at zero (GraphPad Prism® 5.0, San Diego, CA).
5.2 Chemistry
5.2.1 General materials and methods
Melting points were determined on a Mel-Temp 3.0 melting point apparatus, and are uncorrected. TLC analysis was carried out on silica gel 60 F254 precoated aluminum sheets using UV light for detection. 1H and 13C NMR spectra were recorded on a Varian Unity Plus 300 MHz or Bruker 400 MHz spectrometer using indicated solvents. Mass spectra was obtained from the Georgia State University Mass Spectrometry Laboratory, Atlanta, GA. Elemental analysis were performed by Atlantic Microlab Inc., Norcross, GA, and are within ±0.4 of the theoretical values. The compounds reported as salts frequently analyzed correctly for fractional moles of water and/or other solvents; in each case 1H NMR spectra was consistent with the analysis. All chemicals and solvents were purchased from Aldrich Chemical Co., VWR International, or Combi-Blocks, Inc.
5.2.2 2,6-Di-(4′-cyanophenyl)pyrazine (9)
To a stirred solution of 2,6-dichloropyrazine (7, 2.0 g, 13.4 mmol) in toluene (56 mL) under nitrogen atmosphere at 80 °C was added 27 mL of 2 M aqueous solution of Na2CO3 followed by 4-cyanophenylboronic acid 8 (4.34 g, 29.5 mmol) in 30 mL of methanol. After 30 minutes, tetrakis(triphenylphosphine) palladium (1.34 g, 1.16 mmol) was added to the reaction mixture. The reaction mixture was stirred overnight at 80 °C. After cooling to room temperature, water was added to the mixture. The solution was filtered, and the precipitate was washed with water and MeOH. The crude product was purified by recrystallization to afford the title compound 9 (2.92 g, 77% yield); mp 298.5–299 °C. 1H NMR (DMSO-d6): δ 8.04 (d, J = 8.1 Hz, 4H), 8.47 (d, J = 8.1 Hz, 4H), 9.41 (s, 2H). 13C NMR (DMSO-d6): δ 148.8, 142.1, 139.9, 133.0, 127.7, 118.6, 112.6. MS: m/z 282 (M+). Anal. Calc. for C18H10N4: C, 76.58; H, 3.57; N, 19.85. Found: C, 76.33; H, 3.55; N, 19.76.
5.2.3 2,6-Di-(4′-amidinophenyl)pyrazine hydrochloride (10)
The above dinitrile 9 (0.14 g, 0.48 mmol), suspended in freshly distilled THF (4 mL), was treated with lithium trimethylsilylamide (1 M solution in THF, 2.5 mL, 2.5 mmol), and the reaction was allowed to stir overnight. The reaction mixture was then cooled to 0 °C and to which was added HCl saturated ethanol (3 mL), whereupon a precipitate started forming. The mixture was allowed to stir overnight, after which it was diluted with ether, and the precipitate was filtered. The diamidine was purified by neutralization with 1 N NaOH followed by filtration of the resultant solid and washing with water. Finally, the free base was stirred with ethanolic HCl overnight and diluted with ether, and the solid formed was filtered and dried to give the diamidine salt 10 (0.14 g, 76% yield); mp >300 °C. 1H NMR (DMSO-d6): δ 8.05 (d, J = 7.6 Hz, 4H), 8.53 (d, J = 7.6 Hz, 4H), 9.31 (s, 4H), 9.48 (s, 2H), 9.57 (s, 4H). 13C NMR (DMSO-d6): δ 165.1, 149.1, 141.6, 140.5, 129.0, 128.9, 127.2. Anal. Calc. for C18H16N6-2.0HCl-2.3H2O: C, 50.19; H, 5.29; N, 19.51. Found: C, 50.31; H, 4.92; N, 19.18.
5.2.4 2,6-Di-(4′-N-hydroxyamidinophenyl)pyrazine hydrochloride (11)
A mixture of hydroxylamine hydrochloride (4.9 g, 70.8 mmol) in anhydrous DMSO (60 mL) was cooled to 5 °C under nitrogen. Potassium tert-butoxide (7.95 g, 70.8 mmol) was added in portions and the mixture was stirred for 30 minutes. To this mixture was added the above dinitrile 9 (1.0 g, 3.54 mmol) and the mixture was stirred overnight. The reaction mixture was poured slowly into a beaker with ice water and stirred for 15 minutes. The white precipitate was filtered and washed with water to afford the free base of 11. The free base was stirred with ethanolic HCl overnight and diluted with ether, and the precipitate which formed was collected by filtration to give the title compound HCl salt 11 in 87% yield; mp >300 °C. 1H NMR (DMSO-d6): δ 3.86 (s, 6H), 4.64 (s, 2H), 7.96 (d, J = 8.4 Hz, 4H), 8.51 (d, J = 8.4 Hz, 4H), 9.25 (s, 4H), 9.43 (s, 2H), 11.42 (s, 2H), 13.13 (s, 2H). 13C NMR (DMSO-d6): δ 158.8, 149.1, 141.8, 140.0, 128.9, 127.3, 126.7. Anal. Calc. for C18H16N6O2-2.2HCl-2.0H2O: C, 46.53; H, 4.82; N, 18.09. Found: C, 46.59; H, 4.45; N, 17.80.
5.2.5 2,6-Di-(4′-N-methoxyamidinophenyl)pyrazine hydrochloride (12)
A solution of lithium hydroxide monohydrate (0.24 g, 5.74 mmol) in water (4 mL) was added dropwise to the mixture of the free base of the above di-amidoxime 11 (0.43 g, 1.44 mmol) in DMF (26 mL) at room temperature. The reaction mixture was stirred for 30 minutes at room temperature. Dimethylsulfate (0.45 g, 3.59 mmol) was added to the reaction mixture and the mixture was stirred at room temperature overnight. The reaction mixture was poured slowly into a beaker with ice water and stirred for 15 minutes. The precipitate was filtered and washed with water to afford the free base of 12. The free base was stirred with ethanolic HCl overnight and diluted with ether, and the precipitate which formed was collected by filtration to give the title compound 12 in 58% yield; mp 244–246 °C. 1H NMR (DMSO-d6): δ 3.49 (s, 8H), 3.83 (s, 6H), 7.90 (d, J = 8.4 Hz, 4H), 8.38 (d, J = 8.4 Hz, 4H), 9.33 (s, 2H). 13C NMR (DMSO-d6): δ 156.7, 149.2, 141.6, 139.3, 128.5, 128.2, 127.1, 63.1. Anal. Calc. for C20H20N6O2-2.0HCl-1.5H2O: C, 50.43; H, 5.29; N, 17.64. Found: C, 50.28; H, 5.17; N, 17.46.
5.2.6 2,6-Di-(4′-amidino-2′-methylphenyl)pyrazine hydrochloride (15a)
A solution of 2,6-di(tri-n-butylstannyl)pyrazine (77% purity)17 (4.04 g, 4.68 mmol), 4-bromo-3-methylbenzonitrile 13a (2.02 g, 10.30 mmol), tetrakis(triphenylphosphine) palladium (0.56 g, 0.56 mmol) in degassed xylene (80 mL) was heated at 120 °C under nitrogen atmosphere for 24 h. After cooling to room temperature, the mixture was filtered and the precipitate was washed with xylene and ether. The crude product was purified by recrystallization (DMF) to afford compound 14a in 62% yield [mp 239–141 °C (dec). 1H NMR (DMSO-d6): δ 2.43 (s, 6H), 7.74 (d, J = 8.1 Hz, 2H), 7.83 (d, J = 8.1 Hz, 2H), 7.89 (s, 2H), 8.95 (s, 2H). 13C NMR (DMSO-d6): δ 151.8, 143.5, 140.8, 137.8, 134.4, 131.0, 129.9, 118.5, 111.8, 19.8] and it was used directly in the next step without further characterization.
The same procedure described for the preparation of 10 was used starting with the above dinitrile 14a; 52% yield; mp 250–252 °C (dec). 1H NMR (DMSO-d6): δ 2.46 (s, 6H), 7.80 (d, J = 8.1 Hz, 2H), 7.84 (d, J = 8.1 Hz, 2H), 7.89 (s, 2H), 8.98 (s, 2H), 9.32 (s, 4H), 9.52 (s, 4H). 13C NMR (DMSO-d6): δ 165.2, 152.1, 143.4, 141.2, 137.1, 130.6, 130.6, 128.6, 125.9, 20.2. Anal. Calc. for C20H20N6-2.5HCl-1.0H2O: C, 52.96; H, 5.44; N, 18.53. Found: C, 52.96; H, 5.31; N, 18.26.
5.2.7 2,6-Di-(4′-amidino-2′-fluorophenyl)pyrazine hydrochloride (15b)
The same procedure described above for the preparation of 14a was used starting with 4-bromo-3-fluorobenzonitrile 13b; to give 14b 62% yield [mp >300 °C. 1H NMR (DMSO-d6): δ 8.12–8.16 (m, 2H), 8.37 (d, J = 8.4 Hz, 2H), 8.49 (d, J = 10.4 Hz, 2H), 9.48 (s, 2H). HRMS: m/z 319.0800 (M+1) (calculated for C18H9N4F2, 319.0795)] and it was used directly in the next step without further characterization.
The same procedure described for the preparation of 10 was used starting with the above dinitrile 14b; 60% yield; mp >300 °C. 1H NMR (DMSO-d6): δ 7.89–7.92 (m, 2H), 8.37 (d, J = 8.4 Hz, 2H), 8.44 (d, J = 12.4 Hz, 2H), 9.46 (s, 4H), 9.49 (s, 2H), 9.63 (s, 4H). 13C NMR (DMSO-d6): δ 161.9, 159.2 (d, J = 248.2 Hz), 147.9 (d, J = 2.3 Hz), 142.0, 141.6 (d, J = 8.6 Hz), 130.7, 122.8 (d, J = 2.9 Hz), 118.3 (d, J = 13.7 Hz), 114.4 (d, J = 23.5 Hz). HRMS: m/z 353.1311 (M+1) (calculated for C18H15N6F2, 353.1326). Anal. Calc. for C18H14F2N6-2.0HCl-1.3H2O: C, 48.18; H, 4.18; N, 18.73. Found: C, 48.37; H, 4.11; N, 18.67.
5.2.8 2-Chloro-6-(5′-cyanopyridin-2′-yl)pyrazine (17a)
A solution of 2-chloro-6-(tri-n-butylstannyl)pyrazine17 (3.62 g, 9.0 mmol), 2-bromo-5-cyanopyridine 16a (1.67 g, 9.0 mmol), tetrakis(triphenylphosphine) palladium (0.52 g, 0.45 mmol) in degassed xylene (20 mL) was heated at 120 °C under nitrogen atmosphere overnight. After cooling to room temperature, the mixture was filtered. Most of the solvent was removed under reduced pressure; the precipitate which formed was filtered and washed with xylene and ether. The crude product was purified by column chromatography on silica gel (eluent hexane/ethyl acetate (5/1)) to give the title compound 17a in 67% yield; mp 148–150 °C. 1H NMR (DMSO-d6): δ 8.41 (d, J = 8.4 Hz, 1H), 8.51 (dd, J = 2.0, 8.4 Hz, 1H), 8.97 (s, 1H), 9.20 (d, J = 2.0 Hz, 1H), 9.51 (s, 1H). 13C NMR (DMSO-d6): δ 154.9, 152.6, 148.5, 147.9, 145.7, 141.7, 141.0, 121.2, 116.7, 110.1. Anal. Calc. for C10H5ClN4: C, 55.44; H, 2.33; N, 25.86. Found: C, 55.55; H, 2.28; N, 25.83.
5.2.9 2-(4′-cyanophenyl)-6-(5′-cyanopyridin-2′-yl)pyrazine (18a)
The same procedure described for 2,6-di-(4′-cyanophenyl)pyrazine 9 was used by employing 2-chloro-6-(5′-cyanopyridin-2′-yl)pyrazine 17a and 4-cyanophenylboronic acid 8 to furnish the compound 18a in 71% yield; mp 293–295 °C. 1H NMR (DMSO-d6): ): δ 8.04 (d, J = 8.4 Hz, 2H), 8.50 (d, J = 8.4 Hz, 2H), 8.53 (dd, J = 2.0, 8.4 Hz, 1H), 8.71 (d, J = 8.4 Hz, 1H), 9.19 (d, J = 2.0 Hz, 1H), 9.48 (s, 1H), 9.57 (s, 1H). HRMS: m/z 284.0930 (M+1) (calculated for C17H10N5, 284.0936). Anal. Calc. for C17H9N5: C, 72.08; H, 3.20; N, 24.75. Found: C, 71.85; H, 3.17; N, 24.64.
5.2.10 2-(4′-amidinophenyl)-6-(5′-amidinopyridin-2′-yl)pyrazine hydrochloride (19a)
The same procedure described for the preparation of 10 was used starting with the above dinitrile 18a; 90% yield; mp 296–298 °C (dec). 1H NMR (DMSO-d6): δ 8.05 (d, J = 8.4 Hz, 2H), 8.46 (dd, J = 2.0, 8.4 Hz, 1H), 8.58 (d, J = 8.4 Hz, 2H), 8.77 (d, J = 8.4 Hz, 1H), 9.17 (d, J = 2.0 Hz, 1H), 9.27 (s, 2H), 9.40 (s, 2H), 9.56 (s, 2H), 9.56 (s, 1H), 9.62 (s, 1H), 9.73 (s, 2H). 13C NMR (DMSO-d6): δ 165.1, 163.7, 157.2, 149.0, 148.9, 148.2, 143.5, 142.1, 140.1, 137.9, 129.2, 128.9, 127.4, 125.2, 120.9. Anal. Calc. for C17H15N7-3.0HCl-1.7H2O: C, 44.64; H, 4.72; N, 21.44. Found: C, 44.67; H, 4.52; N, 21.44.
5.2.11 2-(4′-N-hydroxyamidinophenyl)-6-(5′-N-hydroxyamidinopyridin-2′-yl)pyrazine hydrochloride (20a)
The same procedure described for the preparation of 11 was used starting with the above dinitrile 18a; yield 89%; mp 283–285 °C (dec). 1H NMR (DMSO-d6): δ 3.43 (s, 8H), 7.95 (d, J = 8.4 Hz, 2H), 8.36 (dd, J = 2.0, 8.4 Hz, 1H), 8.52 (d, J = 8.4 Hz, 2H), 8.68 (d, J = 8.4 Hz, 1H), 9.08 (d, J = 2.0 Hz, 1H), 9.47 (s, 1H), 9.57 (s, 1H). 13C NMR (DMSO-d6): δ 158.8, 156.9, 156.8, 149.0, 148.7, 148.2, 143.4, 142.0, 139.8, 137.9, 128.9, 127.5, 126.8, 122.9, 121.1. Anal. Calc. for C17H15N7O2-3.0HCl-0.8H2O: C, 43.16; H, 4.18; N, 20.72. Found: C, 43.15; H, 4.22; N, 20.62.
5.2.12 2-(4′-N-methoxyamidinophenyl)-6-(5′-N-methoxyamidinopyridin-2′-yl)pyrazine hydrochloride (21a)
The same procedure described for the preparation of 12 was used starting with the above compound 20a; 29% yield; mp 213–215 °C (dec). 1H NMR (DMSO-d6): δ 3.43 (s, 8H), 3.83 (s, 3H), 3.85 (s, 3H), 7.92 (d, J = 8.4 Hz, 2H), 8.29 (dd, J = 2.0, 8.4 Hz, 1H), 8.39 (d, J = 8.4Hz, 2H), 8.57 (d, J = 8.4 Hz, 1H), 9.03 (d, J = 2.0 Hz, 1H), 9.38 (s, 1H), 9.52 (s, 1H). 13C NMR (DMSO-d6): δ 157.5, 155.1, 152.7, 149.0, 148.4, 147.7, 143.0, 141.8, 139.5, 136.6, 128.7, 127.4, 127.3, 126.1, 121.0, 63.4, 62.3. Anal. Calc. for C19H19N7O2-3.0HCl-0.7H2O: C, 45.70; H, 4.72; N, 19.63. Found: C, 45.91; H, 4.67; N, 19.29.
5.2.13 2-Chloro-6-(2′-cyanopyridin-5′-yl)pyrazine (17b)
The same procedure described for 2-chloro-6-(5′-cyanopyridin-2′-yl)pyrazine 17a was used by employing 2-chloro-6-(tri-n-butylstannyl)pyrazine 17 and 2-bromo-5-cyanopyridine 16b to furnish the title compound 17b in 52% yield; mp 149–151 °C. 1H NMR (DMSO-d6): δ 8.24 (d, J = 8.4 Hz, 1H), 8.71 (dd, J = 2.0, 8.4 Hz, 1H), 8.91 (s, 1H), 9.45 (d, J = 2.0 Hz, 1H), 9.46 (s, 1H). 13C NMR (DMSO-d6): δ 149.4, 148.1, 147.7, 144.7, 141.3, 136.0, 133.5, 133.4, 129.2, 117.3. Anal. Calc. for C10H5ClN4: C, 55.44; H, 2.33; N, 25.86. Found: C, 55.67; H, 2.30; N, 25.60.
5.2.14 2-(4′-amidinophenyl)-6-(2′-amidinopyridin-5′-yl)pyrazine hydrochloride (19b)
The same procedure described for 2,6-di-(4′-cyanophenyl)pyrazine 9 was used by employing 2-chloro-6-(2′-cyanopyridin-5′-yl)pyrazine 17b and 4-cyanophenylboronic acid 8 to furnish the compound 18b in 88% yield [mp 271–273 °C. 1H NMR (DMSO-d6): δ 8.06 (d, J = 8.4 Hz, 2H), 8.26 (d, J = 8.4 Hz, 1H), 8.51 (d, J = 8.4 Hz, 2H), 8.90 (dd, J = 2.0, 8.4 Hz, 1H), 9.48 (s, 1H), 9.50 (s, 1H), 9.63 (d, J = 2.0Hz, 1H). 13C NMR (DMSO-d6): δ 149.5, 149.0, 147.0, 142.6, 142.4, 139.6, 135.9, 134.6, 133.3, 133.0, 129.2, 127.8, 118.6, 117.4, 112.7] and it was used directly in the next step without further characterization.
The same procedure described for the preparation of 10 was used starting with the above dinitrile 18b; yield 87%; mp >300 °C. 1H NMR (DMSO-d6): δ 8.04 (d, J = 8.8 Hz, 2H), 8.52 (d, J = 8.4 Hz, 1H), 8.55 (d, J = 8.8 Hz, 2H), 9.02 (dd, J = 2.0, 8.4 Hz, 1H), 9.21 (s, 2H), 9.46 (s, 2H), 9.51 (s, 2H), 9.51 (s, 1H), 9.54 (s, 1H), 9.65 (d, J = 2.0 Hz, 1H), 9.70 (s, 2H). 13C NMR (DMSO-d6): δ 165.1, 161.6, 149.3, 148.1, 147.2, 144.7, 142.6, 142.4, 140.2, 136.4, 135.3, 129.3, 128.9, 127.4, 123.6. Anal. Calc. for C17H15N7-2.0HCl-1.55H2O: C, 48.83; H, 4.84; N, 23.44. Found: C, 49.15; H, 4.78; N, 23.07.
5.2.15 2-(4′-N-hydroxyamidinophenyl)-6-(2′-N-hydroxyamidinopyridin-5′-yl)pyrazine hydrochloride (20b)
The same procedure described for the preparation of 11 was used starting with the above dinitrile 18b; yield 87%; mp 291–293 °C (dec). 1H NMR (DMSO-d6): δ 3.44 (s, 4H), 7.95 (d, J = 8.4 Hz, 2H), 8.24 (d, J = 8.4 Hz, 1H), 8.53 (d, J = 8.8 Hz, 2H), 8.87 (dd, J = 2.0, 8.4 Hz, 1H), 9.21 (s, 2H), 9.45 (s, 1H), 9.49 (s, 1H), 9.56 (d, J = 2.0 Hz, 1H), 11.01 (s, 1H), 11.20 (s, 1H). 13C NMR (DMSO-d6): δ 158.7, 154.7, 149.2, 147.9, 147.4, 145.0, 142.3, 142.1, 139.7, 136.0, 134.4, 128.9, 127.4, 126.8, 123.0. Anal. Calc. for C17H15N7O2-2.0HCl-1.4H2O: C, 45.63; H, 4.46; N, 21.91. Found: C, 45.99; H, 4.40; N, 21.56.
5.2.16 2-(4′-N-methoxyamidinophenyl)-6-(2′-N-methoxyamidinopyridin-5′-yl)pyrazine hydrochloride (21b)
The same procedure described for the preparation of 12 was used starting with the above compound 20b; yield 37%; mp 160–162 °C (dec). 1H NMR (DMSO-d6): δ 3.86 (s, 3H), 3.88 (s, 3H), 3.89 (s, 8H), 7.96 (d, J = 8.0 Hz, 2H), 8.10 (d, J = 8.4 Hz, 1H), 8.46 (d, J = 8.0 Hz, 2H), 8.87 (dd, J = 2.0, 8.4 Hz, 1H), 9.41 (s, 1H), 9.43 (s, 1H), 9.47 (d, J = 2.0 Hz, 1H). 13C NMR (DMSO-d6): δ 157.3, 150.7, 149.3, 148.3, 147.9, 147.2, 141.8, 141.7, 139.5, 135.5, 132.8, 128.7, 127.6, 127.2, 121.1, 63.4, 62.0. Anal. Calc. for C19H19N7O2-2.0HCl-2.0H2O: C, 46.92; H, 5.18; N, 20.16. Found: C, 46.98; H, 5.09; N, 19.93.
5.2.17 2,6-Di-(5′-cyanopyridin-2′-yl)pyrazine (22a)
The same procedure described for 2,6-di-(4′-cyano-2′-methylphenyl)pyrazine 14a was used by employing 2,6-di(tri-n-butylstannyl)pyrazine and 2-bromo-5-cyanopyridine 16a to furnish the title compound 22a in 58% yield; mp 264–266 °C (dec).1H NMR (DMSO-d6): δ 8.69 (dd, J = 2.1, 8.4 Hz, 2H), 8.80 (d, J = 8.4 Hz, 2H), 9.23 (s, 2H), 9.67 (d, J = 2.1 Hz, 2H). 13C NMR (DMSO-d6): δ 155.9, 152.6, 147.8, 143.8, 141.6, 121.4, 116.9, 110.0. Anal. Calc. for C16H8N6: C, 67.60; H, 2.84; N, 29.56. Found: C, 67.41; H, 2.73; N, 29.31.
5.2.18 2,6-Di-(5′-amidinopyridin-2′-yl)pyrazine hydrochloride (23a)
The same procedure described for the preparation of 10 was used starting with the above dinitrile 22a; 50% yield; mp 277–279 °C (dec). 1H NMR (DMSO-d6): δ 8.48 (dd, J = 2.4, 8.4 Hz, 2H), 8.86 (d, J = 8.1 Hz, 2H), 9.18 (d, J = 2.4 Hz, 2H), 9.41 (s, 4H), 9.72 (s, 2H), 9.74 (s, 4H). 13C NMR (DMSO-d6): δ 163.7, 156.9, 149.0, 148.1, 143.6, 138.0, 125.2, 121.1. Anal. Calc. for C16H14N8-2.0HCl-2.5H2O: C, 44.05; H, 4.85; N, 25.68. Found: C, 44.08; H, 4.67; N, 25.39.
5.2.19 2,6-Di-(5′-N-hydroxyamidinopyridin-2′-yl)pyrazine hydrochloride (24a)
The same procedure described for the preparation of 11 was used starting with the above dinitrile 22a in 97% yield; mp 274–276 °C (dec).1H NMR (DMSO-d6-D2O): δ 3.52 (s, 6H), 8.32 (dd, J = 2.0, 8.4 Hz, 2H), 8.74 (d, J = 8.4 Hz, 2H), 9.03 (d, J = 2.0 Hz, 2H), 9.65 (s, 2H), 11.08 (s, 2H). 13C NMR (DMSO-d6): δ 156.4, 156.2, 148.6, 148.1, 143.4, 137.6, 123.5, 121.2. Anal. Calc. for C16H14N8O2-2.0HCl-2.1H2O: C, 41.68; H, 4.42; N, 24.30. Found: C, 41.71; H, 4.30; N, 24.30.
5.2.20 2,6-Di-(5′-N-methoxyamidinopyridin-2′-yl)pyrazine hydrochloride (25a)
The same procedure described for the preparation of 12 was used starting with the above compound 24a; yield 58%; mp 216–218 °C (dec). 1H NMR (DMSO-d6): δ 3.82 (s, 6H), 4.00 (s, 6H), 6.70 (s, 2H), 8.27 (dd, J = 1.8, 8.4 Hz, 2H), 8.64 (d, J = 8.4 Hz, 2H), 9.04 (d, J = 1.8 Hz, 2H), 9.61 (s, 2H). 13C NMR (DMSO-d6): δ 154.5, 151.4, 148.4, 147.4, 142.8, 135.9, 127.2, 121.1, 61.9. Anal. Calc. for C18H18N8O2-3.0HCl-2.3H2O: C, 40.85; H, 4.88; N, 21.17. Found: C, 41.01; H, 4.89; N, 21.07.
5.2.21 2,6-Di-(2′-cyanopyridin-5′-yl)pyrazine (22b)
The same procedure described for 2,6-di-(4′-cyano-2′-methylphenyl)pyrazine 14a was used by employing 2,6-di(tri-n-butylstannyl)pyrazine and 5-bromo-2-cyanopyridine 16b to furnish the title compound 22b; 70% yield; mp >300 °C. 1H NMR (DMSO-d6): δ 8.27 (d, J = 8.0 Hz, 2H), 8.94 (dd, J = 2.0, 8.0 Hz, 2H), 9.54 (s, 2H), 9.66 (d, J = 2.0 Hz, 2H). 13C NMR (DMSO-d6): δ149.6, 147.2, 143.0, 136.1, 134.4, 133.4, 129.2, 117.4. Anal. Calc. for C16H8N6: C, 67.60; H, 2.84; N, 29.56. Found: C, 67.34; H, 2.73; N, 29.28.
5.2.22 2,6-Di-(2′-amidinopyridin-5′-yl)pyrazine hydrochloride (23b)
The same procedure described for the preparation of 10 was used starting with the above dinitrile 22b; 83% yield; mp 249–251 °C (dec). 1H NMR (DMSO-d6): δ 8.58 (d, J = 8.1 Hz, 2H), 9.06 (dd, J = 1.2, 8.1 Hz, 2H), 9.59 (s, 4H), 9.61 (s, 2H), 9.68 (d, J = 1.2 Hz, 2H), 9.78 (s, 4H). 13C NMR (DMSO-d6): δ 151.3, 147.9, 147.6, 147.3, 142.0, 135.6, 133.0, 121.5. Anal. Calc. for C16H14N8-2.0HCl-1.4H2O: C, 46.14; H, 4.55; N, 26.91. Found: C, 46.42; H, 4.52; N, 26.52.
5.2.23 2,6-Di-(2′-N-hydroxyamidinopyridin-5′-yl)pyrazine hydrochloride (24b)
The same procedure described for the preparation of 11 was used starting with the above dinitrile 22b; 69% yield; mp 280–282 °C (dec). 1H NMR (DMSO-d6): δ 8.40 (dd, J = 2.1, 8.4 Hz, 2H), 8.80 (d, J = 8.4 Hz, 2H), 8.82 (s, 2H), 9.10 (d, J = 2.1 Hz, 2H), 9.68 (s, 2H), 11.28 (s, 2H). 13C NMR (DMSO-d6): δ 155.0, 148.0, 147.6, 144.8, 142.7, 136.2, 134.4, 123.2. Anal. Calc. for C16H14N8O2-3.0HCl-0.75H2O: C, 40.61; H, 3.94; N, 23.68. Found: C, 40.81; H, 4.18; N, 23.35.
5.2.24 2,6-Di-(2′-N-methoxyamidinopyridin-5′-yl)pyrazine hydrochloride (25b)
The same procedure described for the preparation of 12 was used starting with the above compound 24b; 50% yield; mp 89–91 °C. 1H NMR (DMSO-d6): δ 3.85 (s, 6H), 8.07 (d, J = 8.4 Hz, 2H), 8.72 (dd, J = 2.4, 8.4 Hz, 2H), 9.43 (s, 2H), 9.46 (d, J = 2.4 Hz, 2H). 13C NMR (DMSO-d6): δ 150.1, 149.0, 148.3, 146.9, 141.5, 135.0, 131.8, 119.9, 61.2. Anal. Calc. for C18H18N8O2-2.0HCl-0.7H2O: C, 46.60; H, 4.65; N, 24.15. Found: C, 46.80; H, 4.79; N, 23.95.
Acknowledgments
This work was supported by The Bill and Melinda Gates Foundation through a subcontract with the Consortium of Parasitic Drug Development (CPDD) (RB, WDW, DWB) and by NIH grant AI064200 (WDW, DWB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organisation. WHO | Malaria. 2012 Fact sheet N°94. http://www.who.int/mediacentre/factsheets/fs094/en/
- 3.Simarro PP, Jannin J, Cattand P. Plos Med. 2008;68:e55. doi: 10.1371/journal.pmed.0050055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organisation. WHO | African trypanosomiasis (sleeping sickness) 2012 Fact sheet N°259. http://www.who.int/mediacentre/factsheets/fs259/en/
- 5.Wenzler T, Boykin DW, Ismail MA, Hall JE, Tidwell RR, Brun R. Antimicrob Agents Chemother. 2009;53:4185. doi: 10.1128/AAC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy PGE. Ann Neurol. 2008;64:116. doi: 10.1002/ana.21429. [DOI] [PubMed] [Google Scholar]
- 7.(a) Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Baudin E, Buard V, Kazadi-Kyanza S, Ilunga M, Mutangala W, Pohlig GG, Schmid C, Karunakara U, Torreele E, Kande V. Lancet. 2009;374:56. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]; (b) Simarro PP, Franco J, Diarra A, Postigo JAR, Jannin J. Parasitology. 2012;139:842. doi: 10.1017/S0031182012000169. [DOI] [PubMed] [Google Scholar]
- 8.(a) Tidwell RR, Boykin DW. Dicationic DNA Minor Groove Binders as Antimicrobial agents. In: Demeunynck M, Bailly C, Wilson WD, editors. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes. Vol. 2. Wiley-VCH; New York: 2003. pp. 414–460. [Google Scholar]; (b) Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, Boykin DW. Curr Med Chem-Anti-Cancer agents. 2005;5:389. doi: 10.2174/1568011054222319. [DOI] [PubMed] [Google Scholar]; (c) Soeiro MNC, de Souza EM, Stephens CE, Boykin DW. Expert Opin Invest Drugs. 2005;14:957. doi: 10.1517/13543784.14.8.957. [DOI] [PubMed] [Google Scholar]; (d) Dardonville C. Expert Ther Pat. 2005;15:1241. [Google Scholar]; (e) Werbovetz KA. Curr Opin Invest Drugs. 2006;7:147. [PubMed] [Google Scholar]; (f) Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Biochimie. 2008;90:999. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thuita JK, Karanja SM, Wenzler T, Mdachi RE, Ngotho JM, Kagira JM, Tidwell R, Brun R. Acta Tropica. 2008;108:6. doi: 10.1016/j.actatropica.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Ismail MA, Brun R, Easterbrook JD, Tanious FA, Wilson WD, Boykin DW. J Med Chem. 2003;46:4761. doi: 10.1021/jm0302602. [DOI] [PubMed] [Google Scholar]
- 11.Ansede JH, Voyksner RD, Ismail MA, Boykin DW, Tidwell RR, Hall JE. Xenobiotica. 2005;35:211. doi: 10.1080/00498250500087671. [DOI] [PubMed] [Google Scholar]
- 12.Thuita JK, Wang MZ, Kagira JM, Denton CL, Paine MF, Mdachi RE, Murilla GA, Ching S, Boykin DW, Tidwell RR, Hall JE, Brun R. PLoS Negl Trop Dis. 2012;6:e1734. doi: 10.1371/journal.pntd.0001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Berger O, Kanti A, van Ba CT, Vial H, Ward SA, Biagini GA, Gray PG, O’Neill PM. ChemMedChem. 2011;6:2094. doi: 10.1002/cmdc.201100265. [DOI] [PubMed] [Google Scholar]; (b) Chackal-Catoen S, Miao Y, Wilson WD, Wenzler T, Brun R, Boykin DW. Bioorg Med Chem. 2006;14:7434. doi: 10.1016/j.bmc.2006.07.024. [DOI] [PubMed] [Google Scholar]; (c) Patrick DA, Bakunov SA, Bakunova SM, Kumar EVKS, Lombardy RJ, Jones SK, Bridge SA, Zhirnov O, Hall JE, Wenzler T, Brun R, Tidwell RR. J Med Chem. 2007;50:2468. doi: 10.1021/jm0612867. [DOI] [PubMed] [Google Scholar]; (c) Ismail MA, Arafa RK, Wenzler T, Brun R, Tanious FA, Wilson WD, Boykin DW. Bioorg Med Chem. 2008;16:683. doi: 10.1016/j.bmc.2007.10.042. [DOI] [PubMed] [Google Scholar]; (e) Bakunov SA, Bakunova SM, Wenzler T, Ghebru M, Werbovetz KA, Brun R, Tidwell RR. J Med Chem. 2010;53:254. doi: 10.1021/jm901178d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ismail MA, Arafa RK, Brun R, Wenzler T, Miao Y, Wilson DW, Generaux C, Bridges A, Hall JE, Boykin DW. J Med Chem. 2006;49:5324. doi: 10.1021/jm060470p. [DOI] [PubMed] [Google Scholar]
- 14.Hu L, Arafa RK, Ismail MA, Wenzler T, Brun R, Munde M, Wilson WD, Nzimiro S, Samyesudhas S, Werbovetz KA, Boykin DW. Bioorg Med Chem Lett. 2008;18:247. doi: 10.1016/j.bmcl.2007.10.091. [DOI] [PubMed] [Google Scholar]
- 15.Hu L, Arafa RK, Ismail MA, Patel A, Munde M, Wilson WD, Wenzler T, Brun R, Boykin DW. Bioorg Med Chem. 2009;17:6651. doi: 10.1016/j.bmc.2009.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darabantu M, Boully L, Turck A, Ple N. Tetrahedron. 2005;61:2897. [Google Scholar]
- 17.Bakunova SM, Bakunov SA, Patrick DA, Kumar EVKS, Ohemeng KA, Bridges AS, Wenzler T, Barszcz T, Kilgore Jones S, Werbovetz KA, Brun R, Tidwell RR. J Med Chem. 2009;52:2016. doi: 10.1021/jm801547t. [DOI] [PubMed] [Google Scholar]
- 18.Goodsell D, Dickerson RE. J Med Chem. 1986;29:727. doi: 10.1021/jm00155a023. [DOI] [PubMed] [Google Scholar]
- 19.(a) Nguyen B, Lee MP, Hamelberg D, Bailly C, Brun R, Neidle S, Wilson WD. J Am Chem Soc. 2002;124:13680. doi: 10.1021/ja027953c. [DOI] [PubMed] [Google Scholar]; (b) Nguyen B, Hamelberg D, Bailly C, Colson J, Stenek J, Brun R, Neidle S, Wilson WD. Biophys J. 2004;86:1028. doi: 10.1016/s0006-3495(04)74178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Miao Y, Lee MPH, Parkinson GN, Batista-Parra A, Ismail MA, Neidle S, Boykin DW, Wilson DW. Biochemistry. 2005;44:14701. doi: 10.1021/bi051791q. [DOI] [PubMed] [Google Scholar]
- 20.Boykin DW, Kumar A, Xiao G, Wilson WD, Bender BC, McCurdy DR, Hall JE, Tidwell RR. J Med Chem. 1998;41:124. doi: 10.1021/jm970570i. [DOI] [PubMed] [Google Scholar]
- 21.Yan GZ, Brouwer KLM, Pollack GM, Wang MZ, Tidwell RR, Hall JE, Paine MF. J Pharmacol Exper Therap. 2011;337:503. doi: 10.1124/jpet.110.177220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Wang MZ, Saulter JY, Usuki E, Cheung YL, Hall M, Bridges AS, Loewen G, Parkinson OT, Stephens CE, Allen JL, Zeldin DC, Boykin DW, Tidwell RR, Parkinson A, Paine MF, Hall JE. Drug Metab Disp. 2006;34:1985. doi: 10.1124/dmd.106.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang MZ, Wu JQ, Bridges AS, Zeldin DC, Kornbluth S, Tidwell RR, Hall JE, Paine MF. Drug Metab Disp. 2007;35:2067. doi: 10.1124/dmd.107.016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansede JH, Anbazhagan M, Brun R, Easterbrook JD, Hall JE, Boykin DW. J Med Chem. 2004;47:4335. doi: 10.1021/jm030604o. [DOI] [PubMed] [Google Scholar]
- 24.Saulter JY, Kurian JR, Trepanier LA, Tidwell RR, Bridges AS, Boykin DW, Stephens CE, Anbazhagan M, Hall JE. Drug Metab Disp. 2005;33:1886. doi: 10.1124/dmd.105.005017. [DOI] [PubMed] [Google Scholar]
- 25.Wilson WD, Tanious FA, Fernandez-Saiz M, Rigl CT. Methods in Mol Biol., Drug–DNA Interaction Protocols. 1997;90:219. doi: 10.1385/0-89603-447-X:219. [DOI] [PubMed] [Google Scholar]
- 26.Prochaska HJ, Talalay P, Sies H. J Biol Chem. 1987;262:1931. [PubMed] [Google Scholar]