

Abstract
Enteropeptidase (enterokinase [E.C.3.4.21.9]) is a serine protease of the intestinal brush border in the proximal small intestine. It activates the pancreatic proenzyme trypsinogen, which, in turn, releases active digestive enzymes from their inactive pancreatic precursors. Congenital enteropeptidase deficiency is a rare recessively inherited disorder leading, in affected infants, to severe failure to thrive. The genomic structure of the proenteropeptidase gene (25 exons, total gene size 88 kb) was characterized in order to perform DNA sequencing in three clinically and biochemically proved patients with congenital enteropeptidase deficiency who were from two families. We found compound heterozygosity for nonsense mutations (S712X/R857X) in two affected siblings and found compound heterozygosity for a nonsense mutation (Q261X) and a frameshift mutation (FsQ902) in the third patient. In accordance with the biochemical findings, all four defective alleles identified are predicted null alleles leading to a gene product not containing the active site of the enzyme. These data provide first evidence that proenteropeptidase-gene mutations are the primary cause of congenital enteropeptidase deficiency.
Introduction
Enteropeptidase (synonym: enterokinase [E.C.3.4.21.9]) was discovered, by N. P. Schepovalnikow in 1899 (Walther 1900), as a factor that is contained in the duodenum and that is capable of activating pancreatic juice to digest fibrin. Enteropeptidase acts as a sequence-specific protease (Maroux et al. 1971) activating trypsinogen by cleaving off an inhibitory portion. Trypsin, in turn, releases chymotrypsin, carboxypeptidases, elastases, and also lipases from their inactive pancreatic precursors. Intestinal activation of pancreatic precursors is the physiologic mechanism preventing the damage that proteases would cause if they were active within the pancreatic-duct system. Consequently, protein digestion is expected to be largely dependent on enteropeptidase activity. Enteropeptidase is exclusively expressed in the brush border of the proximal small intestine (Eggermont et al. 1971). Recently, enteropeptidase has been reported to be activated from an inactive precursor (proenteropeptidase) by duodenase, a newly discovered serine protease expressed in the duodenum (Zamolodchikova et al. 2000).
Hadorn et al. (1969) first identified a case of congenital enteropeptidase deficiency (MIM 226200) in an infant with failure to thrive, chronic diarrhea, low serum protein, and generalized edema. Trypsin activity in a duodenal-juice sample was low but was restored to normal when isolated enteropeptidase was added. Several similar cases were described subsequently (Polonovski et al. 1970, Haworth et al. 1971; Pardou et al. 1975; Follett and Macdonald 1976; Lebenthal et al. 1976; Ghishan et al. 1983; Green et al. 1984; Marshall et al. 1989). These reports consider two families with affected siblings (Tarlow et al. 1970; Haworth et al. 1975), including one family with affected individuals of either gender, a finding suggesting autosomal recessive inheritance (Haworth et al. 1975). On the basis of the isolation of a partial bovine enteropeptidase cDNA (LaVallie et al. 1993), Kitamoto et al. (1994, 1995) cloned the cDNAs containing the complete coding regions of bovine and human proenteropeptidase and mapped the human gene to chromosome 21q21, by FISH. According to the deduced amino acid sequence, enteropeptidase is a serine protease. The active two-chain enteropeptidase is derived from a single-chain precursor (Kitamoto et al. 1994).
The characterization of the genomic organization of the proenteropeptidase gene has enabled us to perform mutation analysis of genomic DNA from the original patients in whom enteropeptidase deficiency was diagnosed >25 years ago. Here we provide evidence that proenteropeptidase-gene mutations are indeed the molecular cause of congenital enteropeptidase deficiency in these patients.
Material and Methods
Genomic-Library Screening and Determination of Gene Structure
A human genomic DNA PAC library (Ioannou et al. 1994) was screened with an α[32P]-dATP–labeled proenteropeptidase cDNA probe (Screening Service of the Resource Center of the German Human Genome Project, Heidelberg/Berlin; see the Resource Center/Primary Database web site). Two overlapping clones representing the entire proenteropeptidase gene (clones LLNLP704L19841Q3 and LLNLP704L19841Q3) were identified, and their identity was confirmed by PCR and FISH analyses (Lichter et al. 1990). Exon-exon PCR products were generated from PAC DNA templates and were sequenced from either end by the rhodamine fluorescent dideoxy-dye–terminator method (Perkin-Elmer), to clarify the gene structure.
Samples from Patients
Blood samples were donated in accordance with consent principles. In family 1 (Haworth et al. 1975), two affected siblings of either gender had been diagnosed on the basis of biochemical testing. Both of these patients and their parents were available for DNA analysis. In family 2, a single case had been reported (Haworth et al. 1971). In this man, the coincidence of congenital enteropeptidase deficiency and celiac disease had been reported only recently (Moroz et al. 2001). His parents and two daughters were available for DNA analysis. In both families, residual enteropeptidase activity in affected individuals was below the limit of detection. All patients were untreated and free of symptoms in adulthood.
Mutation Analysis in Patients with Congenital Enteropeptidase Deficiency
Oligonucleotide primers corresponding to exon-flanking intronic sequences (table 1) were used to amplify and directly sequence all 25 coding exons from blood genomic DNA, by an ABI 377 DNA sequencer (Perkin-Elmer). Polymorphisms were originally detected in patient samples. Population studies of these polymorphisms were performed on genomic DNA from unrelated healthy German blood donors.
Table 1.
Sequences of Oligonucleotide Primers Annealing to Exon-Flanking Intronic DNA, Used for Amplification and Sequencing of Coding Exons of the Proenteropeptidase Gene
|
Oligonucleotide Primer(5′→3′) |
||
| Fragment | Forward | Reverse |
| Exon 1 | CAGTTCTTAAATTAGCAAGCC | CTGACACTAAAGTGTGTACATTC |
| Exon 2 | CAGAGCTAACACATCAGGC | TCACAGTGAGAAAATGGTG |
| Exon 3 | ACTCTAAGCAGAACATAAAGATTG | GCCACTTTGCTTAATCCTC |
| Exon 4 | GAATTGAACACAAATCAGTGG | GTAAATGTGTCACCTCAGG |
| Exon 5 | AATGACATACTTCTAAATGGACAC | TAGCCCTAAATATGTTGTTTACTG |
| Exon 6 | TAAATATTCCTATGGCAGTTGAAG | AGTGAGGGAGGATAAAACAGAAG |
| Exon 7 | AATGTTTTGCAGCCAATTTGAATC | AAAGCAATAAGACGTTGCATCAG |
| Exon 8 | GATATCATATTATGTGGTGTTCAC | AAAATCAAAGGGAAAACATACCAC |
| Exon 9 | ATAGATCAACTGACAAACTGATAG | TAAGTTCTAAGAAGAGAAAGATGC |
| Exon 10 | GAAGTATAAGCTGTATAAGGTTAG | GACTTTTGCATTTAATGCTGCTC |
| Exons 11–13 | TCTTTCCTGCTTACAAAGTCTAC | GTGCCCAGCTAATTTGTGTTTG |
| Exon 14 | AGCATTTACCCTAACATGACTC | ATTTATAATCTACTTTGCTGCGTC |
| Exon 15 | TGAAATTCTCAAGAGAGTGAAGC | CGTTTACAACCTTTACAATATTCAG |
| Exon 16 | GAGATGTGGGGTACATTTC | ATTCTGTACACCTTTGCTG |
| Exon 17 | GTACTCCAGAATCATCCATTTG | TCTTTCTTTGACACTGTAGAGTC |
| Exon 18 | TATAGTCACAGTGCTGTGC | GTAATAAAAAGGTTTGCAAATCTC |
| Exon 19 | GTCCATAGCATTAAGGAAC | CAAATCTGAGTGGGTTCAAC |
| Exons 20 and 21 | CATATATGAGACATTAAGATGTCC | AATGTATCTCTATTTTCATACATGAC |
| Exon 22 | TTCTGAAATGTCATGATGAAGATC | GCTTTTATGAATTAATGC |
| Exon 23 | CTAAAGGGCCACCAGTGGTAGC | CATTCACAGGATATTATGACAG |
| Exon 24 | AGTTCATTCAAGGATGCATGTTG | ATGAAATAACTATAACACCAGTGC |
| Exon 25 | TGAGAATATATACAGATGACTTGC | CCATGCTTTCTAGAGTAGAATGG |
| Exon 12 forward | CTCTGAAAAGGACTTATTCTAATG | |
| Exon 13 forward | CAAGAAGAATCAAGTCAGAG | |
| Exon 21 forward | GAGTTTTATGTCACCTGAAG | |
Results
Structural Organization of the Proenteropeptidase Gene
The human proenteropeptidase gene consists of 25 exons (24 introns) and spans ∼88 kb of genomic DNA sequence. Exon-flanking intronic sequence of a length sufficient to allow exon amplification was obtained for all coding exons. After deposition of our data in the GenBank database (GenBank accession numbers Y19124–Y19143), the proenteropeptidase-gene sequence (GenBank accession number AL078474) was independently released as part of the chromosome 21 sequencing project. This database entry confirmed our findings.
Mutation Analysis in Patients with Congenital Enteropeptidase Deficiency
In family 1, segregation analysis using a newly identified polymorphic CA repeat in intron 6 showed that the two affected siblings had inherited identical alleles. Sequencing of genomic DNA containing the entire coding region revealed two heterozygous nonsense mutations (2135C→G [S712X] in exon 18 and 2569C→T [R857X] in exon 22; fig. 1). Segregation analysis of the defective alleles (fig. 2) demonstrated that both patients had, in fact, inherited one defective allele from each parent, thus yielding proof of compound heterozygosity. In family 2, a heterozygous nonsense mutation was identified in exon 8 (781C→T [Q261X]; fig. 1). In exon 23, we found a heterozygous deletion of 2 bp (del 2707–8GT) leading to a frameshift mutation (FsQ902; fig. 1). This mutation results in a premature termination of translation at amino acid 930, following 28 false amino acid residues at the carboxy terminus. Segregation analysis including parents and progeny of the index patient clearly demonstrated compound heterozygosity (fig. 2).
Figure 1.
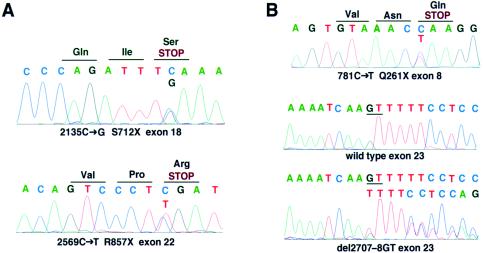
Mutations in the proenteropeptidase gene. A, Analysis of index patient in family 1. Nonsense mutations were identified in exons 18 and 22. B, Analysis of index patient in family 2, which led to identification of a nonsense mutation in exon 8 and of a frameshift mutation in exon 23.
Figure 2.
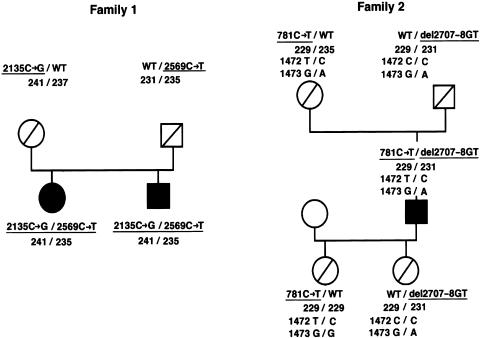
Segregation analysis of families with congenital enteropeptidase deficiency. The sequence alterations of the pathogenic mutation are underlined; the three-digit numerals 241, 237, 231, 229, and 235 denote the fragment lengths of the intron 6 polymorphic CA repeat. In both families, defective alleles are inherited from heterozygous parents. Note that the 1472C→T (A491V) mutation (or rare polymorphism) in family 2 is part of an allele also affected by a nonsense mutation. Position 1473 is dimorphic (G/A) and informative in family 2 (i.e., the 781C→T mutation segregates with the T allele) but not in family 1.
Identification and Characterization of Polymorphisms
Sequencing of exon-intron boundaries led to the identification of a polymorphic site, a CA repeat at the 5′ end of intron 6, that was characterized further. The analysis of DNA samples from 81 normal individuals (162 chromosomes) revealed the existence of 10 different alleles. The number of CA repeats ranged from 18 to 27. The observed heterozygosity index was 0.82 (calculated, 0.84), indicating Hardy-Weinberg equilibrium.
A frequent single-nucleotide dimorphism (1473G→A [A491A]) was detected in exon 13. Allele frequencies and distributions are given in table 2. Additionally, we identified a sequence alteration (1472C→T [A491V]) within exon 13 of family 2 but not in any of 96 normal chromosomes analyzed; it was linked to a nonsense mutation (Q261X; fig. 2), such that it could not be determined whether it confers functional compromise. This finding can be interpreted either as a defective allele having acquired another mutation or as a rare polymorphism not appearing in 96 normal chromosomes.
Table 2.
Distribution of Genotypes of a Frequent Single-Nucleotide Dimorphism (1473G→A) in Exon 13 of the Proenteropeptidase Gene in Healthy Unrelated Individuals from Southern Germany
Data are based on allele frequencies of 0.73 and 0.27 for the G and A alleles, respectively, and indicate Hardy-Weinberg equilibrium.
χ2 = 0.05; P=.73.
Discussion
Congenital enteropeptidase deficiency is characterized by severe protein malabsorption during early infancy. To date, the diagnosis has been made solely on the basis of the inability of a duodenal-juice sample to activate trypsinogen and in vitro restoration by the addition of purified enteropeptidase (Hadorn et al. 1969). The human proenteropeptidase cDNA has been cloned, and the gene has been mapped (Kitamoto et al. 1995). Mutation analyses of the proenteropeptidase gene, to trace the biochemical defect back to a genetic defect, have not previously been performed. To amplify and sequence genomic DNA from affected individuals, we clarified the exon-intron structure of the proenteropeptidase gene. Interestingly, the exon organization reflects the modular-domain organization of the proenteropeptidase-gene product as observed by Kitamoto et al. (1994, 1995) (fig. 3). With the exception of the serine protease domain, functional analyses of enteropeptidase domains have not yet been performed. It is remarkable that primary-structure “modules” other than the serine protease domain are shared by some serine proteases, such as C1r (fig. 3).
Figure 3.
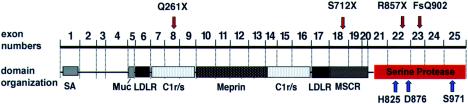
Position of mutations (red arrows), in relation to proenteropeptidase exon organization, domains, and amino acid residues forming the active site of the serine protease domain (H825, D876, and S971 [blue arrows]). All four mutations identified are null mutations predicting the absence of a correctly formed active site. The previously described modular structure of proenteropeptidase domains, based on primary-structure comparison, correlates with exon boundaries. SA = signal/anchor sequence; LDLR = LDL receptor–like domain; Muc = mucin-domain; Meprin = meprin-like domain; C1r/s = complement component C1r-like domain; MSCR = macrophage scavenger receptor–like domain.
Sequencing the entire coding region, we identified compound heterozygosity for mutations in the affected individuals of both families investigated. Newly identified polymorphisms have been instrumental in segregation analyses. Since the active serine protease domain is positioned at the carboxy terminus (Kitamoto et al. 1994), terminations of translation amino terminally to this domain—such as in the cases of Q261X in exon 8 and S712X in exon 18—must be predicted to result in abolishment of enzymatic function. The two other mutations lead to truncation within the serine protease domain. The three-dimensional structure of the serine protease domain of enteropeptidase (Protein Data Bank accession number 1ekb) showing the expected catalytic triad of serine proteases is conserved (Lu et al. 1999) and consists of H825, D876, and S971 in human enteropeptidase (Kitamoto et al. 1995). Residue S971 is the active-site nucleophile and is required for substrate cleavage, as depicted in figure 2a in the report by Lu et al. (1999). This active-site residue is predicted to be missing in proteins encoded by alleles with the mutations R857X (exon 22) and del2707–2708GT (FsQ902, exon 23). Although not studied in direct-expression experiments, both the nature of the mutations found and their location in relation to the active site of the enzyme allow no prediction other than that these alleles must be null alleles. This is in accordance with the biochemical phenotype described earlier in these patients. Our data lead to the conclusion that proenteropeptidase-gene defects are the molecular cause of congenital enteropeptidase deficiency.
In the index patient in family 2, evidence of celiac disease has been reported only very recently (Moroz et al. 2001). In light of this, it is important that we have found disruptive mutations in the proenteropeptidase gene. We thereby have confirmed that enteropeptidase deficiency is not secondary to morphological changes observed in celiac disease.
All patients investigated had been diagnosed as infants, >25 years ago. In adulthood, they have had apparently normal lives, with absence of gastrointestinal symptoms and with normal body weight, even when pancreatic-enzyme substitution has been discontinued. With this knowledge, we had expected to identify missense mutations that might allow for the possibility that some residual enteropeptidase activity had not been detected in the original investigations; however, since all mutations found predict abolishment of enzymatic function, we conclude that protein digestion depends on the action of enteropeptidase during infancy only. Activation of trypsinogen by enteropeptidase-independent mechanisms must be predicted to occur. In fact, it has previously been shown that trypsinogen does have an inherent activity by which it can self-activate, albeit at a very slow rate (Kay and Kassell 1971). Furthermore, it is known, from earlier studies, that trypsin, once released from its precursor, promotes further activation of trypsinogen, in a positive-feedback fashion (Davie and Neurath 1955). This explains the observation that active trypsin, chymotrypsin, and carboxypeptidase A are detected in the feces of patients with enteropeptidase deficiency (Hadorn et al. 1969). We speculate that the self-activation of trypsinogen and the activation by trypsin independently of enteropeptidase activity may suffice for protein digestion in the human adult but not in the human infant, given that the infant's relative demand for protein digestion is much higher. Alternative, unknown mechanisms of enteropeptidase-independent trypsinogen activation may also exist.
Congenital enteropeptidase deficiency is most likely underdiagnosed. In cases of failure to thrive during early infancy, accurate therapy, either by the use of hydrolyzed infant formula or by the substitution of pancreatic enzymes, is often initiated prior to a precise diagnosis, because of the lack of a convenient test. DNA analysis may improve this situation.
Congenital enteropeptidase deficiency may be genetically heterogeneous. It is conceivable that duodenase mutations resulting in defective activation of proenteropeptidase may lead to disease similar to enteropeptidase deficiency. As more patients are genetically analyzed in the future, it will be interesting to see whether genetic defects in the proenteropeptidase gene can be identified in every case; if not, such patients would be candidates for duodenase-gene mutations.
Acknowledgments
We thank Dr. Peter Lichtner for FISH analyses and Dr. Stefan Kammerer for helpful discussions. J.E.S. is an Investigator of the Howard Hughes Medical Institute, and these studies were supported in part by Howard Hughes Medical Institute grant DK50053 and by Digestive Disease Research Core Center (DDRCC) grant P30 DK525474 from the National Institutes of Health. A.H. would like to acknowledge the molecular-biology training received in the laboratories of Drs. T. E. Weaver and J. A. Whitsett at Children’s Hospital Medical Center, Cincinnati. This article contains work submitted as a thesis by C.B., in partial fulfilment of the requirements for the medical degree at Ludwig-Maximilians-University, Munich.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for sequence data [accession numbers Y19124–Y19143 and AF246125])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for congenital enteropeptidase deficiency [MIM 226200])
- Protein Data Bank, http://www.rcsb.org/pdb/ (for the three-dimensional structure of the serine protease domain of bovine enteropeptidase [accession number 1ekb])
- Resource Center/Primary Database, http://www.rzpd.de/ (for the α[32P]-dATP–labeled proenteropeptidase cDNA probe [Screening Service of the Resource Center of the German Human Genome Project, Heidelberg/Berlin])
References
- Davie EW, Neurath H (1955) Identification of a peptide released during autocatalytic activation of trypsinogen. J Biol Chem 212:515–529 [PubMed] [Google Scholar]
- Eggermont E, Molla AM, Tytgat G, Rutgeerts L (1971) Distribution of enterokinase activity in the human intestine. Acta Gastroenterol Belg 34:655–662 [PubMed] [Google Scholar]
- Follett GF, Macdonald TH (1976) Intestinal enterokinase deficiency. Acta Paediatr Scand 65:653–655 [DOI] [PubMed] [Google Scholar]
- Ghishan FK, Lee PC, Lebenthal E, Johnson P, Bradley CA, Greene HL (1983) Isolated congenital enterokinase deficiency. Recent findings and review of the literature. Gastroenterology 85:727–731 [PubMed] [Google Scholar]
- Green JR, Bender SW, Posselt HG, Lentze MJ (1984) Primary intestinal enteropeptidase deficiency. J Pediatr Gastroenterol Nutr 3:630–633 [DOI] [PubMed] [Google Scholar]
- Hadorn B, Tarlow MJ, Lloyd JK, Wolff OH (1969) Intestinal enterokinase deficiency. Lancet 1:812–813 [DOI] [PubMed] [Google Scholar]
- Haworth JC, Gourley B, Hadorn B, Sumida C (1971) Malabsorption and growth failure due to intestinal enterokinase deficiency. J Pediatr 78:481–490 [DOI] [PubMed] [Google Scholar]
- Haworth JC, Hadorn B, Gourley B, Prasad A, Troesch V (1975) Intestinal enterokinase deficiency: occurrence in two sibs and age dependency of clinical expression. Arch Dis Child 50:277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou PA, Amemiya CT, Garnes J, Kroisel PM, Shizuya H, Chen C, Batzer MA, de Jong PJ (1994) A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nat Genet 6:84–89 [DOI] [PubMed] [Google Scholar]
- Kay J, Kassell B (1971) The autoactivation of trypsinogen. J Biol Chem 246:6661–6665 [PubMed] [Google Scholar]
- Kitamoto Y, Veile RA, Donis-Keller H, Sadler JE (1995) cDNA sequence and chromosomal localization of human enterokinase, the proteolytic activator of trypsinogen. Biochemistry 34:4562–4568 [DOI] [PubMed] [Google Scholar]
- Kitamoto Y, Yuan X, Wu Q, McCourt DW, Sadler JE (1994) Enterokinase, the initiator of intestinal digestion, is a mosaic protease composed of a distinctive assortment of domains. Proc Natl Acad Sci USA 91:7588–7592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVallie ER, Rehemtulla A, Racie LA, DiBlasio EA, Ferenz C, Grant KL, Light A, McCoy JM (1993) Cloning and functional expression of a cDNA encoding the catalytic subunit of bovine enterokinase. J Biol Chem 268:23311–23317 [PubMed] [Google Scholar]
- Lebenthal E, Antonowicz I, Shwachman H (1976) Enterokinase and trypsin activities in pancreatic insufficiency and diseases of the small intestine. Gastroenterology 70:508–512 [PubMed] [Google Scholar]
- Lichter P, Tang CJ, Call K, Hermanson G, Evans GA, Housman D, Ward DC (1990) High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 247:64–69 [DOI] [PubMed] [Google Scholar]
- Lu D, Futterer K, Korolev S, Zheng X, Tan K, Waksman G, Sadler JE (1999) Crystal structure of enteropeptidase light chain complexed with an analog of the trypsinogen activation peptide. J Mol Biol 292:361–373 [DOI] [PubMed] [Google Scholar]
- Maroux S, Baratti J, Desnuelle P (1971) Purification and specificity of porcine enterokinase. J Biol Chem 246:5031–5039 [PubMed] [Google Scholar]
- Marshall G, Mitchell JD, Tobias V, Messina IM (1989) Arrhythmogenic right ventricular dysplasia in a child with congenital enteropeptidase deficiency and hypogammaglobulinaemia. Aust Paediatr J 25:106–108 [DOI] [PubMed] [Google Scholar]
- Moroz SP, Hadorn B, Rossi TM, Haworth JC (2001) Celiac disease in a patient with a congenital deficiency of intestinal enteropeptidase. Am J Gastroenterol 96:2251–2254 [DOI] [PubMed] [Google Scholar]
- Pardou A, Cadranel S, Rodesch P, Eggermont E, Loeb H (1975) Déficience en entérokinase: a propos d'une observation. Pédiatrie 30:544 [PubMed] [Google Scholar]
- Polonovski C, Laplane R, Alison F, Navarro J (1970) Pseudodéficit entrypsinogène par déficit en entérokinase: étude clinique. Arch Fr Pediatr 27:677–688 [PubMed] [Google Scholar]
- Tarlow MJ, Hadorn B, Arthurton MW, Lloyd JK (1970) Intestinal enterokinase deficiency: a newly-recognized disorder of protein digestion. Arch Dis Child 45:651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A (1900) Die Physiologie des Darmsaftes (in “Verdauung” chap). In: von Nencki M, Andreasch R (eds) Jahres-Bericht über die Fortschritte der Thier-Chemie. Vol 29. Verlag von JF Bergmann, Wiesbaden, Germany [Google Scholar]
- Zamolodchikova TS, Sokolova EA, Lu D, Sadler JE (2000) Activation of recombinant proenteropeptidase by duodenase. FEBS Lett 466:295–299 [DOI] [PubMed] [Google Scholar]