
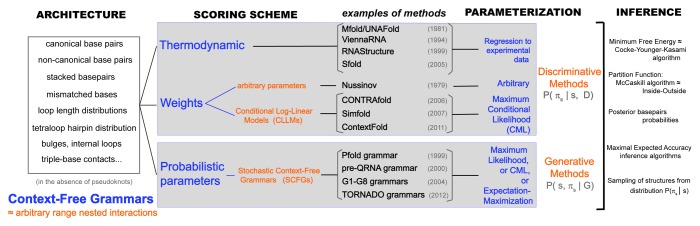
Figure 1.Unified description of different methods for single-sequence RNA secondary structure prediction. The menu of elements that define a method are: architecture, scoring scheme, parameterization and inference method. The architecture consists of the list of features which, in turn, determine the number of parameters of the model. The different architectures one can devise for a nested RNA secondary structure all fall into the category of a Context-Free Grammar (CFG). Any architecture can be implemented using either thermodynamic, weights or probabilistic parameters. Both weight and probabilistic schemes can be trained on data (statistical). There are statistical weight schemes such as CLLMs. Statistical probabilistic schemes for RNA folding are usually stochastic CFGs (SCFGs). Notice that SCFGs are a subset of CFGs. SCFGs describe models with a probabilistic scheme, while the concept CFG applies to all scoring schemes. The assignment of values for the parameters (parameterization) depends on the scoring scheme used. Thermodynamic models take values as kcal/mol free-energy estimations from experimental data. Conditional Log-Linear models use methods that require numerical optimization (CML and also online training). Probabilistic models are usually trained by maximum likelihood methods, which simply require obtaining frequencies of occurrences in the training set [and the addition of at least Laplace (+1) priors]. Once an architecture, scoring scheme and parameterization are in place (that is, a “model”), one can use different algorithms to infer plausible secondary structures. Unlike training, which is specific for the different scoring schemes, the folding algorithms (usually dynamic programming algorithms) are essentially identical for all parameterizations (although oftentimes they have different names). A side note; the term “probabilistic” often leads to confusion. In the end, all scoring schemes (probabilistic or not) can give us insight into the probabilistic distribution of structures (πs) for a given sequence (s) (the so-called Boltzmann ensemble in a thermodynamic scheme). For instance, one can calculate the distribution’s partition function (via the McCaskill or inside algorithms) or rigorously sample structures from that distribution. However, what is normally referred to as a “probabilistic” model is one in which the parameters of the model are themselves probabilities. Probabilistic models are “generative” models, which means that in addition to the Boltzmann ensemble per sequence, they also provide insight into the joint distribution for the ensemble of sequences and structures. With a probabilistic method, one can quite naturally generate sequences together with their structures according to the model.