

Abstract
Background
DNA microarrays can be used to quickly and sensitively identify several different pathogens in one step. Our previously developed DNA microarray, based on the detection of variable regions in the 16S rDNA gene (rrs), which are specific for each selected bacterial genus, allowed the concurrent detection of Borrelia spp., Anaplasma spp., Francisella spp., Rickettsia spp. and Coxiella spp.
Methods
In this study, we developed a comprehensive detection system consisting of a second generation DNA microarray and quantitative PCRs. New oligonucleotide capture probes specific for Borrelia burgdorferi s.l. genospecies and Candidatus Neoehrlichia mikurensis were included. This new DNA microarray system required substantial changes in solution composition, hybridization conditions and post-hybridization washes.
Results
This second generation chip displayed high specificity and sensitivity. The specificity of the capture probes was tested by hybridizing the DNA microarrays with Cy5-labeled, PCR-generated amplicons encoding the rrs genes of both target and non-target bacteria. The detection limit was determined to be 103 genome copies, which corresponds to 1–2 pg of DNA. A given sample was evaluated as positive if its mean fluorescence was at least 10% of the mean fluorescence of a positive control. Those samples with fluorescence close to the threshold were further analyzed using quantitative PCRs, developed to identify Francisella spp., Rickettsia spp. and Coxiella spp. Like the DNA microarray, the qPCRs were based on the genus specific variable regions of the rrs gene. No unspecific cross-reactions were detected. The detection limit for Francisella spp. was determined to be only 1 genome copy, for Coxiella spp. 10 copies, and for Rickettsia spp., 100 copies.
Conclusions
Our detection system offers a rapid method for the comprehensive identification of tick-borne bacteria, which is applicable to clinical samples. It can also be used to identify both pathogenic and endosymbiontic bacteria in ticks for eco-epidemiological studies, tick laboratory colony testing, and many other applications.
Keywords: Tick-borne bacteria, DNA microarray, Quantitative PCR
Background
Tick transmitted diseases are a serious and permanent public health problem. In Europe, the most frequent and most epidemiologically important vector is the hard tick Ixodes ricinus. It transmits viral, bacterial and protozoan agents to humans and animals. The most common and important tick-transmitted disease in the northern hemisphere, Lyme borreliosis, is caused by spirochetes from the Borrelia burgdorferi sensu lato (s.l.) complex. It currently includes 19 different genospecies [1]. The considerable genotypic and phenotypic heterogeneity of the B. burgdorferi s.l. complex has been linked to differences in pathogenicity, clinical symptoms and ecology [2-4]. The Borrelia genus also includes a second group of spirochetes, called the relapsing fever group. The spirochetes of this group are transmitted mainly by soft ticks, but can also utilize some hard ticks as vectors [5].
Anaplasmoses are also common tick-borne, zoonotic bacterial diseases. The causative agents are intracellular gram-negative bacteria that belong to the family Anaplasmataceae [6]. The genus Anaplasma consists of Anaplasma marginale, Anaplasma ovis, Anaplasma bovis and Anaplasma platys. While they are primary of veterinary significance, A. phagocytophilum can cause granulocytic anaplasmosis in humans as well as horses and dogs and tick-borne fever in ruminants [7]. A relatively new member of the family Anaplasmataceae is Candidatus Neoehrlichia mikurensis [8]. It infects endothelial cells and most infection symptoms depend on the physical status of the patient. The illness predominantly develops in immunocompromised patients [9-12].
Less common bacteria that may be transmitted by Ixodes ricinus amongst other tick species include Francisella spp. and Coxiella spp. Coxiella burnetii is the causative agent of Q-fever, which can be either an acute or chronic disease. Francisella tularensis causes tularemia, a febrile disease with myalgia and headache and when left untreated, it can cause a high mortality rate [13]. Most cases of disease caused by both C. burnetii and F. tularensis result from non-vector transmission.
Another European tick-borne obligate intracellular parasite, which is also globally distributed, is Rickettsia spp. The genus Rickettsia contains many species which form several biogroups, including the typhus fever group, the spotted fever group and the group causing tick-borne lymphadenopathy or Dermacentor spp. - borne necrosis - erythema - lymphadenopathy (TIBOLA or DEBONEL) [14]. Many other Rickettsia species have been recently identified, but are not yet well described, including the human pathogens R. helvetica and R. aeschlimannii[15,16].
Considering all the serious diseases that humans can potentially be exposed to after a tick-bite, an unambiguous diagnostic tool is essential for identifying them. The most reliable modern diagnostic tools employ serological tests, including ELISA (enzyme linked immunoabsorbent assay), Western blot, indirect immunofluorescence assay (IFA), a microagglutination test, and in the case of rickettsial infection, the Weil-Felix test [17]. Unfortunately, these methods are only indirect and do not allow illnesses to be diagnosed in the early stages of infection. Another major limitation of serology is cross-reactivity [18], application of the non-standardized antigen preparations and discrepancies in test procedures among laboratories can lead to different test results. Furthermore, identification of Candidatus N. mikurensis using serology is presently not possible and A. phagocytophilum and E. chaffeensis antigens do not interact with Candidatus N. mikurensis antibodies [19]. The primary approach for detecting Candidatus N. mikurensis therefore relies on PCR-based methods.
Molecular biology approaches offer the advantages of directly detecting these pathogens during early infection along with better taxonomic classification. The most common techniques employ conventional, nested, or quantitative PCR (qPCR) targeted to a genus or species specific gene, such as 16S rDNA gene (rrs), gltA, omp, ospA or ospC[20-23]. Another method, commonly used for identifying B. burgdorferi s.l., targets the 5S-23S rDNA (rrfA-rrlB) intergenic spacer followed by genotyping using RFLP or SSCP [24,25]. These tests target the rDNA genes because they are minimally affected by horizontal gene transfer. Typically, these genes have hypervariable regions, specific for each bacterial genus, which are flanked by conserved regions [26].
The more recent, microarray-based techniques are high-throughput large-scale screening systems for the simultaneous identification of several target amplicons. DNA microarrays are used in many fields of research, including transcription profile analysis and DNA-DNA or protein–protein interactions. Microarrays have been developed for the identification of microorganisms in soil extracts [27], for the detection of multiple pathogens [28-30] and for differentiating between different Borrelia genospecies [31]. These techniques employ DNA or RNA as a template for the preparation of a target product which is suitable for passive hybridization with complementary DNA fragments or oligonucleotides bound to the surface of a slide. The stringency and hybridization efficiency is regulated by solution composition and temperature.
An alternative to the DNA microarray is an electronic microarray - biosensor, which can be prepared using standard complementary metal oxide semiconductor (CMOS) technology. This “smart” biosensor uses an electric field to regulate the stringency, transport and active hybridization of nucleic acids [32,33]. An electronic microarray based on the genus-specific variability of the rrs gene has already been developed for the detection of marine bacterial species [34].
In this study, we report the development of a detection system combining a second generation DNA microarray with qPCR for the detection of pathogens in vectors or in clinical samples. A second generation DNA microarray is basically an epoxy glass slide with bound capture oligonucleotides, which code for the hypervariable regions of the rrs gene, specific for each bacterial genus. The target DNA is amplified, Cy5-labeled using nested PCR and passively hybridized with capture probes on the microarray. We also developed qPCRs employing the genus-specific, hypervariable regions of rrs for Coxiella spp., Francisella spp. and Rickettsia spp. to confirm the DNA microarray results.
Methods
Bacterial isolates and genomic DNA preparation
A DNA microarray was designed to detect bacteria from Borrelia spp., Anaplasma spp., Francisella spp., Rickettsia spp., Coxiella spp. and Candidatus N. mikurensis. The DNA of A. phagocytophilum, R. africae, R. slovaca, F. tularensis subsp. holarctica and C. burnetii Nine Mile phase II were from laboratory stocks [28]. The DNA from different Borrelia species and Candidatus N. mikurensis was isolated from questing ticks collected in Slovakia using the Qiagen DNeasy Blood and Tissue kit (Qiagen, Hilden Germany). Positive samples were identified using previously described PCR methods and sequenced [19,25]. Borrelial DNA was also isolated from cultures kindly supplied by Dr. Ian Livey (Baxter, Orth, Austria). DNA samples from non-targeted bacteria used as negative hybridization controls [28] were also taken from laboratory stocks.
Sequence selection of capture probes
The sequences of the DNA microarray capture probes Bv, Be, Bg1 used for the detection of Borrelia spp, C1 and Cv for detection of Coxiella spp., Av and A3 for detection of Anaplasma spp., F1v, F2v, Fa and F2 for detection of Francisella spp. and R1, Rv and Re for detection of Rickettsia spp. were previously published in Blaškovič and Barák [28]. New probes for detecting the DNA of Borrelia spp. and Candidatus N. mikurensis were designed (Table 1). The sequences for the new capture probes were chosen based on the hypervariable regions of the 16S rDNA genes (rrs). These were identified by an alignment of 16S rDNA (rrs) sequences from GenBank and the Ribosomal Database Project II (RDPII) [35]. The sequences of the new capture probes were tested based on melting temperature and secondary structure prediction by Integrated DNA Technologies’ OligoAnalyzer 3.1 online software [36]. The hybridization specificity of the designed probes was also analyzed using a Blast search [37].
Table 1.
Nucleotide sequences of PCR primers and probes
| Oligonucleotide | Sequence (5′-3′) | Target bacteria | Source |
|---|---|---|---|
|
DNA microarray amplification |
|
|
|
| 16S27f |
GAGAGTTTGATCCTGGCTCAG |
Almost all eubacteria |
Modified oligo fD1[38] |
| 16S1495r |
CTACGGCTACCTTGTTACGA |
Almost all eubacteria |
Modified oligo fD1[38] |
|
qPCR amplification |
|
|
|
| CbqPCR F |
GGGAAACTCGGGCTAATACC |
Coxiella spp. |
This study |
| CbqPCR R |
CACGAGGTCCGAAGATCC |
Coxiella spp. |
This study |
| CbqPCR P |
FAM-CCCGCTTTGCTCCAAAGAGATTATG-TAMRA |
Coxiella spp. |
This study |
| RcqPCR F |
GCTTAACCTCGGAATTGCTT |
Rickettsia spp. |
This study |
| RcqPCR R |
CGTCAGTTGTAGCCCAGATG |
Rickettsia spp. |
This study |
| RcqPCR P |
HEX-CCTTCGCCACCGGTGTTCCT-TAMRA |
Rickettsia spp. |
This study |
| FrqPCR F |
ATTAAAGGTGGCCTTTGTGC |
Francisella spp. |
This study |
| FrqPCR F2 |
ATTAAAGGTGGCTTTCGGGC |
Francisella spp. |
This study |
| FrqPCR R |
ACCAACTAGCTAATCCAACGC |
Francisella spp. |
This study |
| FrqPCR P |
Cy5-AGGCTCATCCATCTGCGGCA-BHQ2 |
Francisella spp. |
This study |
|
Capture probes |
|
|
|
| A3 |
CGGCTATCTGGTCCGGTACTGAC |
Anaplasma spp. |
[28] |
| Av |
GCTGAATGTGGGGATTTTTTATCTCTGT |
Anaplasma spp. |
[28] |
| Be |
AAGGGTGGAATCTGTTGATATCAGG |
Borrelia spp. |
[28] |
| Bg1 |
CTGGTGTAAGGGTGGAATCTGTTGA |
Borrelia spp. |
[28] |
| Bg2 |
TCAGAAAGAATACCGGAGGCGAAGG |
Borrelia spp. |
This study |
| Bsp1 |
GGAATAAGCTTTGTAGGAAATGGCAAAGTGATGACG |
Borrelia spp. |
This study |
| Bv |
ACTTGGTGTTAACTAAAAGTTAGTACCGA |
Borrelia spp. |
[28] |
| Bv2 |
TATCAGGAAGAATACCGGAGGCGAA |
Borrelia spp. |
This study |
| C1 |
AATATCCTTGGGCGTTGACGTTACC |
Coxiella spp. |
[28] |
| Cv |
ACTAGCTGTTGGGAAGTTCACTTCTTAGT |
Coxiella spp. |
[28] |
| F1v |
ACTAGCTGTTGGAGTCGGTGTAAAGG |
Francisella spp. |
[28] |
| F2 |
TAGAGGAATGGGGAATTTCTGGTGT |
Francisella spp. |
[28] |
| F2v |
ACTAGCTGTTGGATTCGGTGTAAAGG |
Francisella spp. |
[28] |
| Fa |
AATAGCCTTGGGGGAGGACGTTAC |
Francisella spp. |
[28] |
| NM |
CTATTTAAACTAGAGATCGAGAGAGGATAGTGG |
C. Neoehrlichia mikurensis |
This study |
| R1 |
TAGAGTRTAGTAGGGGATGATGGAA |
Rickettsia spp. |
[28] |
| Rv |
GCTAGATATCGGAAGATTCTCTTTCGG |
Rickettsia spp. |
[28] |
| Re | GTGGTCGCGGATCGCAGAGA | Rickettsia spp. | [28] |
For qPCR, three genus-specific oligonucleotides and dual-labeled probe sets were designed. The first set bound exclusively to the Coxiella spp. 16S rDNA gene (rrs), the second set was specific for the Rickettsia spp. 16S rDNA gene (rrs), and the last set was designed to bind the Francisella spp. 16S rDNA gene (rrs). The unique region of the Coxiella spp. 16S rDNA gene (rrs) was identified by aligning the 16S rDNA (rrs) sequences and comparing them to the previously published primers and probes for the 16S rDNA gene (rrs) of Coxiella burnetii[22]. This region was used to design oligonucleotides and dual-labeled probes using GenScript (GenScript USA Inc., Piscataway, NJ, USA). The same strategy was used to generate oligonucleotides and dual-labeled probes for the Francisella spp. and Rickettsia spp. [39,40]. Like the DNA microarray capture probes, the qPCR probes and oligonucleotides were validated based on melting temperature, predicted secondary structure folding and hybridization specificity as described above.
PCR amplification
The sequences of all oligonucleotides and the probes used in this study are listed in Table 1.
PCR amplification for DNA microarray
The 16S rDNA (rrs) target genes of the targeted bacteria were amplified by nested PCR. The first cycle used 3 μl of genomic DNA, 1× high yield buffer complete with 2 mM MgCl2 (Jena Bioscience, Germany), 200 μM of each dNTP, 1 μM of 16S27f (forward) and 16S1495r (reverse) primers (Table 1) and 1U Taq Pol (Jena Bioscience, Germany). The primers 16S27f and 16S1495r are slightly modified fD1 and rP2 general eubacterial primers [38]. The second cycle was used to incorporate the fluorescent labeled Cy5-dUTP into the PCR product of the first amplification. The incorporation was performed as recommended by the manufacturer. Thus, the total 20 μl reaction volume contained 1× high yield buffer complete with 2 mM MgCl2 (Jena Bioscience, Germany), 100 μM of dATP, dCTP, dGTP, 50 μM of dTTP, 50 μM Cy5-dUTP (Jena Bioscience, Germany), 0.5 μM 16S27f and 16S1495r primers (Table 1) and 1U Taq Pol (Jena Bioscience, Germany). 1 μl of the PCR product from the first cycle was used as the template for the second cycle. The cycling conditions in both PCRs were the same. The initial denaturation was performed for 2 minutes at 94°C followed by 30 cycles of 94°C for 30 seconds, 52°C for 30 seconds, 72°C for 1 minute and 30 seconds. The program ended with final elongation at 72°C for 5 minutes.
PCR amplification for quantitative PCRs (qPCRs)
TaqMan probes for qPCRs were synthetized by Microsynth AG, Austria. The CbPr probe was covalently bound at the 5′end with a FAM fluorophore and at the 3′end with a TAMRA quencher; the RLOqPCRPr probe was covalently bound at the 5′end with a HEX fluorophore and at the 3′end with a TAMRA; and the FrqPCRPr probe was covalently bound with Cy5 at the 5′end and BHQ-2 at the 3′end. The cycling conditions for all qPCRs were the same. The initial denaturation was performed for 2 minutes at 95°C, followed by 40 cycles at 95°C for 25 seconds and 50°C for 1 minute. The reaction mixture consisted of 300 nM of forward and reverse primes, 200 nM dual-labeled probes, 1×TaqMan Master Mix (Bioron, Germany), 4 mM MgCl2 and 5 μl of template DNA.
DNA microarray preparation and scanning
Epoxy coated slides were used for DNA microarray prefabrication. The procedure was performed as recommended by the manufacturer (Corning Incorporated, USA). Briefly, the microarray capture probes were diluted in a printing solution, consisting of 150 mM sodium phosphate, pH 8.5 and 0.01% SDS, and printed onto slides in a final spotting concentration of 30 nM. The epoxy slides were spotted at room temperature in 55–70% relative humidity and stored overnight at room temperature. All capture probes were spotted onto slides in triplicate. These prefabricated slides were blocked in a prehybridization solution (5× SSC, 0,1% SDS and 0.1 mg/ml BSA) at 42°C for 1 hour, washed 3 times in 0.1× SSC for 5 minutes and once more in purified water for 30 seconds. After washing, the slides were dried by centrifugation at 1 600 × g for 2 minutes. The Cy5-labelled target PCR products from the nested PCRs were diluted in a hybridization solution consisting of 5× SSC, 10% formamide, 0.1% SDS and 0.1 mg/ml of sonicated salmon sperm DNA, denaturated for 5 minutes in boiling water, shortly spun down, and cooled to room temperature. The target PCR products were pipetted onto microarray slides and covered with cover slips. The hybridization was performed at 42°C for 12–16 hours. After hybridization, the microarray slides were immersed in 2× SSC and 0.1% SDS at 42°C to gently release the cover slips from the slides and washed again for 5 minutes in 2× SSC and 0.1% SDS at 42°C. The final washing consisted of two washes in 1× SSC for 2 minutes at room temperature and two washes in 0.1× SSC for 1 minute at room temperature. The slides were dried by centrigufation at 1 600 × g for 2 minutes and scanned at 635 nm on a MARs Micro Array Scanner (DITABIS - Digital Biomedical Imaging Systems AG, Germany) with SpotScout Pro at 50 μm resolution. The fluorescence intensity for a given spot is represented by the mean feature pixel intensity at 635 nm minus the median background intensity at this wavelength (F635 Mean – B635). Since all slides were spotted in triplets, the reported measurements are the mean values of three measurements. An “M probe” consisting of mixed PCR fragments from the first PCRs was used as a positive control (details were previously published in Blaškovič and Barák [28]. M probe fragments were spotted onto the microarray plates in three places as capture probes. This was done in triplicate. In addition to their use as a positive control, the M probes also aided in grid location during the scanning and made it possible to performing the final negative sample evaluation. The lower border of a saturated spot was defined as 25% of the saturated spot area. All fluorescence intensity values were normalized with respect to the M probe and are reported as percentages of the M probe intensity (defined to be 100%).
Determination of the limit of detection (LOD)
The concentration of genomic DNA isolated from the targeted tick-borne bacteria was quantified using a GeneQuant spectrophotometer (LABFISH, Germany) and the number of copies per μl was calculated using an on-line DNA copy number calculator [41]. The DNA was diluted to a starting concentration of 105 copies/μl for Coxiella spp. and Rickettsia spp. and 104 copies/μl for Francisella spp. This starting solution was then serially diluted 10-fold to prepare a series of solutions from 105 or 104 copies of genomic DNA (gDNA) per μl down to 1 copy/μl (that is, there were six dilutions: 105, 104, 103, 102, 101, and 100). To determine the microarray LOD, 1 μl of these diluted DNAs were used as templates for the amplifications of the rrs gene and the products of these first PCRs were Cy5-labeled using nested PCRs as described above. The labeled amplicons were then hybridized with the prefabricated microarray and scanned and evaluated.
A very similar approach was used to determine the detection limit of qPCR. The same DNA dilutions were used as the templates and genus specific oligonucleotides and probes were used to amplify the rrs genes.
The highest target DNA dilution which still returned a positive result was determined the detection limit of the DNA microarray or qPCR. Three independent experiments were run for each dilution series. Each qPCR experiment consisted of one of the target gDNA dilutions and a mix of non-target gDNAs as negative control. To determine the detectable copy number, an absolute quantification method was employed. The mean quantification cycle (Cq) was converted to a log starting quantity using a linear equation derived from the standard curves.
Results
Development of a DNA microarray for the detection of tick-borne pathogens
A DNA microarray is an efficient and simple tool for detecting a wide spectrum of tick-borne pathogens in a single step. A first generation DNA microarray for detecting tick-borne bacteria was developed previously by Blaškovič and Barák [28]. In that study, the amplification of target DNA by symmetric/asymmetric PCR appeared to be quite complicated. The authors were not able to obtain a single stranded amplicon using asymmetric PCR when DNA from B. burgdorferi, C. burnetii and R. africae was used as a template. They therefore recommended using symmetric PCR for the amplification of all targets involved in the study. The limit of detection (LOD) was also not determined for this assay.
The first steps for upgrading this DNA microarray were to improve the amplification of the 16S rDNA (rrs) gene to enable detection of all bacteria present in the analyzed samples and to increase the efficiency of Cy5-dUTP incorporation into the PCR products. A nested PCR was designed whose first cycle was used to amplify the target gene in high yield. In the second cycle, Cy5-dUTP was incorporated into the PCR amplicon. Crucial for the success of this PCR was the selection of a Taq-polymerase, which was able to both efficiently incorporate modified nucleotides into PCR fragments and to amplify the gene in high yield. Several types of Taq-polymerases, including DyNAzyme EXT DNA Polymerase, Taq DNA Polymerase, (Thermo Fisher Scientific, USA), Taq DNA Polymerase with Standard Taq (Mg-free) Buffer (New England Biolabs, USA), and many cycling conditions were tested (not shown). The best Taq-polymerase appeared to be Taq Polymerase/high yield from Jena Bioscience (Germany). The final annealing temperature was 52°C. In both nested PCR cycles, the same, so-called “catch-all” primers 16S27f and 16S1495r were used. These primers are slightly modified versions of the previously published eubacterial primers fD1 and rP2 [38]. 16S27f is the same as fD1 but with an additional G at the 5′ end. Modification of rP2 included CT addition to the 5′ end of 16S1495r and truncation of three nucleotides from the 3′ end (Table 1). The Cy5-labeled PCR products were precipitated in the presence of ammonium sulphate and ethanol [42] and the pellets were resuspended in the required volume of hybridization solution (see Methods).
Coupling of target Cy5-labeled amplicons with capture probes
The capture probes Bv, Be, Bg1, C1, Cv, Av, A3, F1v, F2v, Fa, F2, R1, Rv and Re were previously designed to bind Cy5-labeled PCR-generated fragments encoding the 16S rDNA genes of Borrelia spp., Coxiella spp., Anaplasma spp., Francisella spp. and Rickettsia spp., respectively [28]. Even though the hybridization conditions and post-hybridization washes had been changed in the present study, they still reacted specifically. To increase the ability of the assay to detect the targeted bacteria, new capture probes for Borrelia spp. (Bg2, Bsp, Bv2) and Candidatus N. mikurensis (NM) were designed (see Table 1). All capture probes were printed onto epoxy slides in triplets in final concentrations of 30 μM. A Cy5-labeled PCR product encoding the 16S rDNA (rrs) of Borrelia spp., Coxiella spp., Anaplasma spp., Rickettsia spp. and Francisella spp. was generated by amplifying purified DNA from bacterial stocks. These Cy5-amplicons were then hybridized with the capture probes. Cy5-amplicons encoding the 16S rDNA of Candidatus N. mikurenses were generated using DNA isolated from ticks. The positivity of the tick for Candidatus N. mikurenses was analyzed in a previous study by qPCR [43].
The ability of the individual capture probes to bind the same target DNA differs depending on the status of the Cy5-amplicons or the quality of the spotted probes or epoxy slides. Our results clearly show that the coupling of capture probes with Cy5-PCR fragments generated from DNA isolated from ticks was not as efficient as that with Cy5-PCR amplicons generated from purified DNA. Thus, the measured fluorescence intensity of the M probe (positive control) and positive capture probes had to be normalized after scanning. The fluorescence intensity is expressed as the mean feature pixel intensity of the positive spot at 635 nm minus the median background at 635 nm (F635 Mean – B635).
The fluorescence intensity of the M probe and the positive capture probes for Coxiella spp., Borrelia spp., Francisella spp., Rickettsia spp. and Anaplasma spp., gave much stronger positive signals than those obtained after the hybridization of a Cy5-labeled amplicon with DNA isolated from a tick coinfected with Borrelia spp. and Candidatus N. mikurensis (Figure 1A). In order to compare these different fluorescence values, the fluorescence intensity of each positive signal was expressed as a percentage of the fluorescence of the M probe. The DNA microarray for Candidatus N. mikurensis revealed a possible co-infection with Borrelia spp. but the percentage of fluorescence intensities was quite low, only 12% and 9% respectively of the fluorescence intensity of the M probe. Such a low signal was considered only ambiguously positive and required further analysis.
Figure 1.

Specificity of capture probes and target bacteria detected by the DNA microarray. (A) Fluorescence intensity at 635 nm (F634Mean-B635) of specific capture probes coupled to target Cy5-labeled amplicons. (B) Fluorescence intensity expressed as a percentage of the fluorescence intensity of the capture probe in relative to 100% of the fluorescence intensity of the M probe (positive control). All capture probes are listed in Table 1. The targeted bacteria were (CNe) Candidatus Neoehrlichia mikurensis, (Rc) Rickettsia spp., (Cb) Coxiella spp., (Bsp) Borrelia spp., (An) Anaplasma spp., (Fr) Francisella spp.
To determine if a sample with such a low fluorescence tests positive or negative, it was necessary to determine the limit of detection (LOD) of the DNA microarray. To do this, a series of 10-fold dilutions, ranging from 105 genome copies per μl down to 1 copy/μl, was prepared from DNA of the targeted tick-borne bacteria. These dilutions were then used as templates for nested PCRs and DNA microarrays. The fluorescence intensities of the capture probes were compared to that of the M probe (Figure 2). The fluorescence intensity of the Cv capture probe specific for Coxiella spp. was 120% when 105 copies were used as template DNA, 12% when 104 copies and 10% when 103 copies were used. 102 copies of template DNA apparently did not bind the capture probes and no fluorescence intensity could be measured; the 10 copies and 1 copy dilutions were also negative. So, the maximal dilution of template DNA, which is detectable by the DNA microarray is 103 copies/μl, which gives a fluorescence intensity 10% of that of the M probe. Therefore, a sample should be evaluated as positive when the mean fluorescence intensity is at least 10% of the mean fluorescence intensity of the M Probe. Thus, samples with values close to this cut off limit will require other analyses, such as some previously published qPCR protocols [20,43], to verify their results. An analysis using the protocols of Courtney et al.[20] and Jahfari et al. [43] confirmed that the signal at the 12% level for the Candidatus N. mikurensis specific probe, as well as the signal at 9% for the B. burgdorferi s.l. probe both represent positive samples (data not shown).
Figure 2.
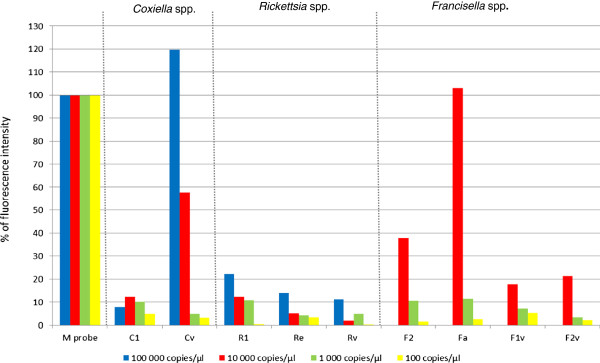
The DNA microarray limit of detection (LOD). LOD for Rickettsia spp., Coxiella spp., and Francisella spp. was determined as the highest dilution of genomic DNA that still tested as positive. 103 genome copies exhibited 10% of the fluorescence intensity of the M probe, while 102 genome copies produced no detectible signal; the LOD was therefore determined to be 103 copies and 10% of the positive control. All capture probes are listed in Table 1. The fluorescence intensity of the capture probes is expressed as a percentage of the fluorescence intensity of the M probe.
The specificity of the DNA microarray was tested using mixed genomic DNAs from many bacterial species, including DNA from tick-borne pathogens, but excluding the target DNA for which the chip was designed. No unspecific cross-reactivity between the capture probes and genomic DNAs was detected (data not shown).
Development of qPCR based on the variability of rrs specific for detection of Francisella spp., Rickettsia spp. and Coxiella spp
This newly developed DNA microarray represents our first line approach for the detection of tick-borne bacteria. To validate the results obtained with the DNA microarray, we used quantitative PCR. It was also necessary to verify the positive detection limits for Candidatus N. mikurensis (F635 Mean - B635 of 12%) and B. burgdorferi s.l. spp. (F635 Mean - B635 of 9%) with this method. Duplex qPCR for the simultaneous detection of A. phagocytophilum and Borrelia burgdorferi s.l. was developed by Courtney et al. [20]. They employed the msp2 gene and the 23S rDNA gene, respectively, for the detection of these two pathogenic species. A real-time PCR assay for the detection of Candidatus N. mikurensis was developed by Jahfari et al.[43]. These two protocols were used to confirm the Candidatus N. mikurensis results from the DNA microarray. In addition, two real-time PCRs based on two putative target genes for hypothetical proteins FTT0376 and FTT0523 were developed to distinguish the pathogenic subspecies Francisella tularensis (subsp. tularensis, holarctica and mediaasiatica) from nonpathogenic F. philomiragia or F. novicida[44]. Because FTT0376 and FTT0523 real-time PCRs detect only the pathogenic but not the nonpathogenic Francisella subspecies, this approach is not suitable as a confirmatory method for our DNA microarray analysis, since all Francisella species is our primary target. For this reason, we developed a qPCR employing the 16S rDNA (rrs) gene in order to detect all Francisella species, and two other qPCRs, to detect the Rickettsia spp. and Coxiella spp. The primers and oligonucleotide dual-labeled probes were designed using GenScript Real-time PCR (TaqMan) Primer Design online software [45] based on the alignment of the rrs genes of all tick-borne bacteria of interest. These probes and primers are specific for the hypervariable region of the rrs gene, which are different for every genus (Table 1). Their specificities were tested against mixed genomic DNA from all possible tick-borne bacteria and other bacterial genomic DNA present in our laboratory stocks.
Because of the slight variation in the hypervariable region of the Francisella spp. rrs sequences, two forward primers were designed. Forward primer FrqPCRF preferentially binds F. piscicida and F. philomiragia, while forward primer FrqPCRF2 is specific for F. tularensis subsp. holarctica, mediaasiatica, tularensis and novicida. All qPCRs were specific when tested against mixed genomic DNA from other bacteria.
Determination of qPCR detection limit
Limits of detection for all three qPCRs were determined based on the maximum dilution of genomic DNA from the target bacteria which still tested positive. To develop a qPCR specific for Coxiella spp. and Rickettsia spp., their genomic DNAs were diluted in a series of 10-fold dilutions, from 105 copies/μl to 1 copy/μl. Due to the low concentration of genomic DNA in Francisella spp., the starting copy number of the series was only 104 copies/μl. The absolute quantifications of the detectable genome copy number from Francisella spp., Coxiella spp. and Rickettsia spp. is shown in Figure 3. The standard curve amplification efficiencies (E), regression coefficients (R2), slopes (s) and y-intercept (y-int) are listed in Table 2. The amplification efficiencies of all qPCRs were 95%, 102% and 96% for Rickettsia spp., Coxiella spp. and Francisella spp., respectively. The sensitivity of qPCR for Rickettsia spp. was determined to be 102 genome copies; for Coxiella spp., 10 genome copies and for Francisella spp., only 1 genome copy. Considering the mean sizes of the genomes (2.2 Mb for Coxiella spp., 2 Mb for Francisella spp. and 1.2 Mb for for Rickettsia spp.), the copy numbers determined for the LOD correspond to approximately 22 fg of gDNA for Coxiella spp., 2.1 fg gDNA for Francisella spp. and 140 fg gDNA for Rickettsia spp.
Figure 3.

Absolute quantification of the detectable genome copy numbers from tick-borne bacteria. Quantitative PCRs were developed for Francisella spp. (blue curve), Coxiella spp. (red curve) and Rickettsia spp. (green curve). The trendlines and R2 values were generated using Microsoft Excel based on the average of the cycle of quantification values (Cq) and the genome copy numbers.
Table 2.
The parameters of the standard curves of qPCRs
| Template gDNA | s | R2 | E | Y-int |
|---|---|---|---|---|
|
Rickettsia spp. |
−3,452 |
0,984 |
94,8% |
46,75 |
|
Coxiella spp. |
−3,266 |
0,999 |
102,4% |
41,2 |
| Francisella spp. | −3,435 | 0,999 | 95,5% | 38,294 |
Discussion
Broad epidemiological studies of veterinary or human importance and clinical diagnostic laboratories require the application of high-throughput, large scale assays allowing the simultaneous detection of all possible microorganisms present in a given sample. Such methods can involve broad range PCRs, multiplex quantitative PCR, molecular beacons or DNA microarrays. Usually these methods employ universal genes, such as the 16S rDNA rrs gene, the 23S rDNA gene, or, occasionally, genes specific for each bacterial genus or species identified in previous studies. In the last decade, DNA microarrays have become one of the most powerful approaches for the simultaneous detection of several bacterial species. They have many possible applications, including identifying bioterror agents [46] and detecting causative pathogens in clinical samples or epidemiological studies [31,47,48]. In clinical diagnostics, it is essential to know all possible co-infections of the patient in order to consider all potential complications, and thus prescribe an effective treatment. Along with other methods, DNA microarrays allow the detection of all agents of a multiple infection in one step.
The major focus of this study was to develop an easy to use, second generation low-density DNA microarray for the simultaneous detection of the many kinds of bacteria present in tick samples. This technique has potential applications in clinical and veterinary laboratories and is also suitable for broad epidemiological studies. The DNA microarray consists of genus-specific capture probes for Borrelia spp., Coxiella spp., Anaplasma spp., Francisella spp. and Rickettsia spp. that were designed for the first generation DNA microarray [28] along with new capture probes Bsp, Bg2 and Bv2 which were designed to increase the possibility of detecting all Borrelia species. In addition, a specific NM probe was designed to detect the newly emerged tick-borne bacterium Candidatus N. mikurensis. The modified solution compositions, hybridization conditions and post-hybridization washes did not affect specificity either of the original or the new capture probes. Capture probe specificity was analyzed by hybridization with Cy5-dUTP labeled PCR fragments generated by PCR on a template containing mixed genomic DNA from other, non-target bacteria.
The sensitivity of the first generation DNA microarray was not tested [28], and thus it was necessary to test the sensitivity of the second generation DNA microarray by determining the limit of detection (LOD). The LOD of our second generation DNA microarray was determined to be the highest dilution of the target genomic DNA that still tested as positive. The LOD was determined to be 103 target genome copies based on hybridization with specific capture probes at different dilutions. Given the mean genome sizes of the targeted bacteria, the limit of detection is about ~ 1–2 pg of genomic DNA. This sensitivity is comparable to that of the low-density DNA microarrays developed to detect tick-borne bacteria such as Borrelia spp. [31] or other array techniques developed to detect potential biological weapons, with detection limits ~102-104 target genome copies [29,30].
Differences were observed in the fluorescence signal intensities produced by the DNA microarray, depending on whether the target DNA was directly purified from bacteria, or was isolated from an infected tick. These differences are likely due to the presence of junk DNA from the tick and a low concentration of the target bacterial DNA. The crucial steps of PCR amplification and Cy5-labeling on such targets are also more complicated. Both of these factors can lead to a low level of fluorescence intensity following hybridization, making interpretation of a positive signal difficult. This situation was observed when genomic DNA isolated from a tick co-infected with Candidatus N. mikurensis and Borrelia spp. was used as a template for DNA microarray analysis. The mean fluorescence of the capture probes was at the detection limit. It was therefore necessary to develop qPCRs using dual labeled TaqMan probes (Table 1). Since duplex qPCR protocols have been developed to detect A. phagoctytophilum and B. burgdorferi s.l. [20], and real-time PCR protocols exist for Candidatus N. mikurensis [43], the presence or absence of these pathogens could be verified. Quantitative PCR protocols were developed to detect Rickettsia spp., Coxiella spp. and Francisella spp. Oligonucleotide probes were designed according to the hypervariable regions of the rrs gene, which are specific for each bacterial genus; both the specificity and sensitivity of the procedure were evaluated. As for the DNA microarray, the qPCR specificity was determined by amplifying the target gene on a template containing mixed genomic, non-target DNAs other than the genomic DNA of the target bacteria. No cross-reactivity with any of these gDNAs was observed. To test the sensitivity of each qPCR, a series of 10-fold dilutions of the target genomic DNAs from Rickettsia spp., Coxiella spp. and Francisella spp. were employed in the amplification, with the final efficiencies of 95%, 102% and 96%, respectively. The highest dilution that was evaluated as positive was 102 genome copies from Rickettsia spp., 10 genome copies from Coxiella spp. and only 1 genome copy from Francisella spp.; these corresponded to approximately 140 fg of Rickettsia spp. genomic DNA, 22 fg of Coxiella spp. genomic DNA and 2.1 fg of Francisella spp. genomic DNA. The limits of detection for all three qPCRs assays were at the breakpoint of qPCR detection and were very similar to those of multiplex qPCR for C. burnetii targeted to the com1, icd and IS1111 genes [49], real-time PCR for F. piscicida targeted to the rrs gene [39], and qPCR developed to detect a Rickettsia-like microorganism, which is responsible for strawberry disease in fish [40]. The qPCRs for the detection of all three pathogens appeared to be more sensitive than the DNA microarray. The qPCR for the detection of Francisella spp. was more sensitive than that of either Coxiella spp. or Rickettsia spp. This may be due to the existence of three copies of the rrs gene in the Francisella genomes present in the Ribosomal RNA Operon Copy Number Database [50] compared to only one copy of the rrs gene in Rickettsia prowazekii[51] and C. burnetii[52].
It should be noted that many ticks harbor non-pathogenic bacteria, including Coxiella-like [53-56], Rickettsia-like [57-59] and Francisella-like [60,61] endosymbionts, which are in a mutualistic relationship with the tick. These primary endosymbionts can provide nutrition to the host [62]. They likely evolved over a long time and they are characterized with a reduced genome [63]. However, they still retain ribosomal RNA genes which have a relatively high level of similarity to those found in pathogenic organisms. For example, the rrs gene of the Amblyoma-associated, Coxiella-like endosymbiont has 93% indentity to the C. burnetii rrs[64]. The role of the secondary endosymbionts is unknown, but they can serve as protection against other pathogens [65]. The DNA microarray developed here, together with the qPCRs, is targeted to the hypervariable regions of rrs genes. A positive signal generated using this approach therefore does not necessarily indicate that the host vector contains a pathogenic bacteria. A sequence analysis of the final result is needed to distinguish between endosymbiont and pathogen in these samples.
Conclusion
We have developed a sophisticated detection system for the simultaneous detection of bacteria present in reservoir hosts, tick-vectors, and clinical specimens, based on a second generation DNA microarray employing the genus-specific, hypervariable regions of the rrs gene. These hypervariable sequences were used to design capture probes for the DNA microarray as well as primers and TaqMan probes for qPCRs. The qPCRs can be used to verify the positive results from the DNA microarray. Both methods display a high level of specificity and sensitivity. The limit of detection for the DNA microarray was 103 genome copies. Quantitative PCRs were developed for Rickettsia spp, Coxiella spp. and Francisella spp. and the limits of detection were determined. Finally, previously developed and published qPCR procedures are available for the verification of presence of the other bacteria involved in this study.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The study was designed by JM and IB. The DNA samples were provided by MD. JM and MD performed laboratory experiments. JM and IB conducted genetic analysis and evaluated microarray and qPCR data. The final manuscript was written by JM, IB and MD. All authors read and approved the final version of the manuscript.
Contributor Information
Jana Melničáková, Email: jana.melnicakova@savba.sk.
Marketa Derdáková, Email: marketa.derdakova@savba.sk.
Imrich Barák, Email: imrich.barak@savba.sk.
Acknowledgements
We thank Jacob Bauer for helpful comments. This publication is the result of the project implementation: Development of diagnostic procedures for detection of tick-borne pathogens and procedures for the preparation of vaccines against ticks (ITMS code: 26240220044) supported by the Research & Development Operational Programme funded by the ERDF and by the Slovak Research and Development Agency under contract No. APVV – 0267–10.
References
- Franke J, Hildebrandt A, Dorn W. Exploring gaps in our knowledge on Lyme borreliosis spirochaetes–updates on complex heterogeneity, ecology, and pathogenicity. Ticks Tick Borne Dis. 2013;4:11–25. doi: 10.1016/j.ttbdis.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Van Dam AP, Kuiper H, Vos K, Widjojokusumo A, de Jongh BM, Spanjaard L, Ramselaar AC, Kramer MD. Different genospecies of Borrelia burgdorferi are associated with distinct clinical manifestations of Lyme borreliosis. Clin Infect Dis. 1993;17:708–717. doi: 10.1093/clinids/17.4.708. [DOI] [PubMed] [Google Scholar]
- Gern L. Borrelia burgdorferi sensu lato, the agent of lyme borreliosis: life in the wilds. Parasite. 2008;15:244–247. doi: 10.1051/parasite/2008153244. Review. [DOI] [PubMed] [Google Scholar]
- Margos G, Vollmer SA, Ogden NH, Fish D. Population genetics, taxonomy, phylogeny and evolution of Borrelia burgdorferi sensu lato. Infect Genet Evol. 2011;11:1545–1563. doi: 10.1016/j.meegid.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunikis J, Tsao J, Garpmo U, Berglund J, Fish D, Barbour AG. Typing of Borrelia relapsing fever group strains. Emerg Infect Dis. 2004;10:1661–1664. doi: 10.3201/eid1009.040236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler JS, Barbet AF, Bekker CPJ, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR. Reorganization of Genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales; unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia, and Ehrlichia with Neorickettsia; description of six new species combinations; and designation of Ehrlichia equi and “HGE agent” as subjective synonyms of Ehrlichia phagocytophilum. Int J Syst Evol Microbiol. 2001;51:2145–2165. doi: 10.1099/00207713-51-6-2145. [DOI] [PubMed] [Google Scholar]
- Woldehiwet Z. The natural history of Anaplasma phagocytophilum. Vet Parasitol. 2010;167:108–122. doi: 10.1016/j.vetpar.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Rikihisa Y, Isogai E, Takahashi M, Misumi H, Suto C, Shibata S, Zhang C, Tsuji M. Ultrastructure and phylogenetic analysis of ‘Candidatus Neoehrlichia mikurensis’ in the family Anaplasmataceae, isolated from wild rats and found in Ixodes ovatus ticks. Int J Syst Evol Microbiol. 2004;54:1837–1843. doi: 10.1099/ijs.0.63260-0. [DOI] [PubMed] [Google Scholar]
- Welinder-Olsson C, Kjellin E, Vaht K, Jacobsson S, Wennerås C. First case of human “Candidatus Neoehrlichia mikurensis” infection in a febrile patient with chronic lymphocytic leukemia. J Clin Microbiol. 2010;48:1956–1959. doi: 10.1128/JCM.02423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr JS, Bloemberg GV, Ritter C, Hombach M, Lüscher TF, Weber R, Keller PM. Septicemia caused by tick-borne bacterial pathogen Candidatus Neoehrlichia mikurensis. Emerg Infect Dis. 2010;16:1127–1129. doi: 10.3201/eid1607.091907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekova S, Vydra J, Kabickova H, Frankova S, Haugvicova R, Mazal O, Cmejla R, Hardekopf DW, Jancuskova T, Kozak T. Candidatus Neoehrlichia mikurensis infection identified in 2 hematooncologic patients: benefit of molecular techniques for rare pathogen detection. Diagn Microbiol Infect Dis. 2011;69:266–270. doi: 10.1016/j.diagmicrobio.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Von Loewenich FD, Geissdörfer W, Disqué C, Matten J, Schett G, Sakka SG, Bogdan C. Detection of “Candidatus Neoehrlichia mikurensis” in two patients with severe febrile illnesses: evidence for a European sequence variant. J Clin Microbiol. 2010;48:2630–2635. doi: 10.1128/JCM.00588-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöstedt A. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann N Y Acad Sci. 2007;1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- Rath N, Rath A. Rickettsial Infections: Indian perspective. Indian Pediatr. 2010;47:157–164. doi: 10.1007/s13312-010-0024-3. [DOI] [PubMed] [Google Scholar]
- Beati L, Péter O, Burgdorfer W, Aeschlimann A, Raoult D. Confirmation that Rickettsia helvetica sp. nov. is a distinct species of the spotted fever group of rickettsiae. Int J Syst Bacteriol. 1993;43:521–526. doi: 10.1099/00207713-43-3-521. [DOI] [PubMed] [Google Scholar]
- Beati L, Meskini M, Thiers B, Raoult D. Rickettsia aeschlimannii sp. nov., a new spotted fever group rickettsia associated with Hyalomma marginatum ticks. Int J Syst Bacteriol. 1997;47:548–554. doi: 10.1099/00207713-47-2-548. [DOI] [PubMed] [Google Scholar]
- Cowan G. Rickettsial diseases: the typhus group of fevers–a review. Postgrad Med J. 2000;76:269–272. doi: 10.1136/pmj.76.895.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola B, Raoult D. Laboratory diagnosis of rickettsioses: current approaches to diagnosis of old and new rickettsial diseases. J Clin Microbiol. 1997;35:2715–2727. doi: 10.1128/jcm.35.11.2715-2727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Jiang JF, Liu W, Zheng YC, Huo QB, Tang K, Zuo SY, Liu K, Jiang BG, Yang H, Cao WC. Human Infection with Candidatus Neoehrlichia mikurensis, China. Emerg Infect Dis. 2012;18:1636–1639. doi: 10.3201/eid1810.120594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney JW, Kostelnik LM, Zeidner NS, Massung RF. Multiplex real-time PCR for detection of Anaplasma phagocytophilum and Borrelia burgdorferi. J Clin Microbiol. 2004;42:3164–3168. doi: 10.1128/JCM.42.7.3164-3168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymaszewska A. PCR for detection of tick-borne Anaplasma phagocytophilum pathogens: a review. Vet Med. 2011;56:529–536. [Google Scholar]
- Marmion BP, Storm PA, Ayres JG, Semendric L, Mathews L, Winslow W, Turra M, Harris RJ. Long-term persistence of Coxiella burnetii after acute primary Q fever. QJM. 2005;98:7–20. doi: 10.1093/qjmed/hci009. [DOI] [PubMed] [Google Scholar]
- Subramanian G, Sekeyova Z, Raoult D, Mediannikov O. Multiple tick-associated bacteria in Ixodes ricinus from Slovakia. Ticks Tick Borne Dis. 2012;3:406–410. doi: 10.1016/j.ttbdis.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Postic D, Assous M, Grimont PAD, Baranton G. Diversity of Borrelia burgdorferi sensu lato evidenced by restriction fragment length polymorphism of rrf (5S)-rrl(23S) intergenic spacer amplicons. Int J Syst Bacteriol. 1994;44:743–752. doi: 10.1099/00207713-44-4-743. [DOI] [PubMed] [Google Scholar]
- Derdakova M, Beati L, Pet’ko B, Stanko M, Fish D. Genetic variability within Borrelia burgdorferi sensu lato genospecies established by PCR-single-strand conformation polymorphism analysis of the rrfA-rrlB intergenic spacer in Ixodes ricinus ticks from the Czech Republic. Appl Environ Microbiol. 2003;69:509–516. doi: 10.1128/AEM.69.1.509-516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Small J, Call DR, Brockman FJ, Straub TM, Chandler DP. Direct detection of 16S rRNA in soil extracts by using oligonucleotide microarrays. Appl Environ Microbiol. 2001;67:4708–4716. doi: 10.1128/AEM.67.10.4708-4716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovic D, Barák I. Oligo-chip based detection of tick-borne bacteria. FEMS Microbiol Lett. 2005;243:473–478. doi: 10.1016/j.femsle.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Wilson WJ, Erler AM, Nasarabadi SL, Skowronski EW, Imbro PM. A multiplexed PCR-coupled liquid bead array for the simultaneous detection of four biothreat agents. Mol Cell Probes. 2005;19:137–144. doi: 10.1016/j.mcp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Gans J, Graves SW, Green L, Taylor L, Kim HB, Kunde YA, Leonard PM, Li PE, Mark J, Song J, Vuyisich M, White PS. A rapid multiplex assay for nucleic acid-based diagnostics. J Microbiol Methods. 2010;80:155–163. doi: 10.1016/j.mimet.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Houck JA, Hojgaard A, Piesman J, Kuchta RD. Low-density microarrays for the detection of Borrelia burgdorferi s.s. (the Lyme disease spirochete) in nymphal Ixodes scapularis. Ticks Tick Borne Dis. 2011;21:27–36. doi: 10.1016/j.ttbdis.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Barbaro M, Bonfiglio A, Raffo L, Alessandrini A, Facci P, Barák I. Fully electronic DNA hybridization detection by a standard CMOS biochip. Sens Actuators B. 2006;118:41–46. doi: 10.1016/j.snb.2006.04.010. [DOI] [Google Scholar]
- Barbaro M, Bonfiglio A, Raffo L, Alessandrini A, Facci P, Barák I. A CMOS, fully integrated sensor for electronic detection of DNA hybridization. IEEE Electron Device Letters. 2006;27:595–597. [Google Scholar]
- Barlaan EA, Sugimori M, Furukawa S, Takeuchi K. Electronic microarray analysis of 16S rDNA amplicons for bacterial detection. J Biotechnol. 2005;115:11–21. doi: 10.1016/j.jbiotec.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM, Tiedje JM. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 2003;31:442–443. doi: 10.1093/nar/gkg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Integrated DNA Technologie’s OligoAnalyzer 3.1 http://eu.idtdna.com/analyzer/applications/oligoanalyzer/default.aspx21857903
- Basic Local Alignment Search Tool. http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&BLAST_PROGRAMS=megaBlast&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome.
- Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottem KF, Nylund A, Isaksen TE, Karlsbakk E, Bergh Ø. Occurrence of Francisella piscicida in farmed and wild Atlantic cod, Gadus morhua L., in Norway. J Fish Dis. 2008;31:525–534. doi: 10.1111/j.1365-2761.2008.00930.x. [DOI] [PubMed] [Google Scholar]
- Lloyd SJ, LaPatra SE, Snekvik KR, Cain KD, Call DR. Quantitative PCR demonstrates a positive correlation between a Rickettsia-like organism and severity of strawberry disease lesions in rainbow trout, Oncorhynchus mykiss (Walbaum) J Fish Dis. 2011;34:701–709. doi: 10.1111/j.1365-2761.2011.01285.x. [DOI] [PubMed] [Google Scholar]
- DNA copy number calculation. http://www.thermoscientificbio.com/webtools/copynumber/
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A laboratory manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Jahfari S, Fonville M, Hengeveld P, Reusken C, Scholte EJ, Takken W, Heyman P, Medlock J, Heylen D, Kleve J, Sprong H. Prevalence of Neoehrlichia mikurensis in ticks and rodents from North-west Europe. Parasit Vectors. 2012;5:74. doi: 10.1186/1756-3305-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JL, Chatwell N, Christensen D, Diaper H, Minogue TD, Parsons TM, Walker B, Weller SA. Development of real-time PCR assays for the specific detection of Francisella tularensis ssp. tularensis, holarctica and mediaasiatica. Mol Cell Probes. 2010;24:72–76. doi: 10.1016/j.mcp.2009.10.004. [DOI] [PubMed] [Google Scholar]
- GenScript Real-time PCR (TaqMan) Primer Design. https://www.genscript.com/ssl-bin/app/primer.
- Janse I, Bok JM, Hamidjaja RA, Hodemaekers HM, van Rotterdam BJ. Development and comparison of two assay formats for parallel detection of four biothreat pathogens by using suspension microarrays. PLoS One. 2012;7:e31958. doi: 10.1371/journal.pone.0031958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaizar J, Rementeria A, Porwollik S. DNA microarray technology: a new tool for the epidemiological typing of bacterial pathogens? FEMS Immunol Med Microbiol. 2006;47:178–189. doi: 10.1111/j.1574-695X.2006.00081.x. [DOI] [PubMed] [Google Scholar]
- Monecke S, Jatzwauk L, Weber S, Slickers P, Ehricht R. DNA microarray-based genotyping of methicillin-resistant Staphylococcus aureus strains from Eastern Saxony. Clin Microbiol Infect. 2008;14:534–545. doi: 10.1111/j.1469-0691.2008.01986.x. [DOI] [PubMed] [Google Scholar]
- de Bruin A, de Groot A, de Heer L, Bok J, Wielinga PR, Hamans M, van Rotterdam BJ, Janse I. Detection of Coxiella burnetii in complex matrices by using multiplex quantitative PCR during a major Q fever outbreak in The Netherlands. Appl Environ Microbiol. 2011;77:6516–6523. doi: 10.1128/AEM.05097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribosomal RNA Operon Copy Number Database. http://rrndb.umms.med.umich.edu/search.php.
- Pang H, Winkler HH. Transcriptional analysis of the 16s rRNA gene in Rickettsia prowazekii. J Bacteriol. 1996;178:1750–1755. doi: 10.1128/jb.178.6.1750-1755.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afseth G, Mallavia LP. Copy number of the 16S rRNA gene in Coxiella burnetii. Eur J Epidemiol. 1997;13:729–731. doi: 10.1023/A:1007384717771. [DOI] [PubMed] [Google Scholar]
- Bernasconi MV, Casati S, Péter O, Piffaretti JC. Rhipicephalus ticks infected with Rickettsia and Coxiella in Southern Switzerland (Canton Ticino) Infect Genet Evol. 2002;2:111–120. doi: 10.1016/S1567-1348(02)00092-8. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park HS, Jang WJ, Koh SE, Park TK, Kang SS, Kim BJ, Kook YH, Park KH, Lee SH. Identification of the Coxiella sp detected from Haemaphysalis longicornis ticks in Korea. Micro Immun. 2004;48:125–130. doi: 10.1111/j.1348-0421.2004.tb03498.x. [DOI] [PubMed] [Google Scholar]
- Mediannikov O, Ivanov L, Nishikawa M, Saito R, Sidelnikov YN, Zdanovskaya NI, Tarasevich IV, Suzuki H. Molecular evidence of Coxiella-like microorganism harbored by Haemaphysalis concinnae ticks in the Russian Far East. Ann N Y Acad Sci. 2003;990:226–228. doi: 10.1111/j.1749-6632.2003.tb07367.x. [DOI] [PubMed] [Google Scholar]
- Clay K, Klyachko O, Grindle N, Civitello D, Oleske D, Fuqua C. Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol Ecol. 2008;17:4371–4381. doi: 10.1111/j.1365-294X.2008.03914.x. [DOI] [PubMed] [Google Scholar]
- Mattila JT, Burkhardt NY, Hutcheson HJ, Munderloh UG, Kurtti TJ. Isolation of cell lines and a rickettsial endosymbiont from the soft tick Carios capensis (Acari: Argasidae: Ornithodorinae) J Med Entomol. 2007;44:1091–1101. doi: 10.1603/0022-2585(2007)44[1091:IOCLAA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Burgdorfer W, Hayes SF, Mavros AJ. In: Rickettsiae and rickettsial diseases. Burgdorfer W, Anacker RL, editor. New York: Academic Press, Inc; 1981. Non-pathogenic rickettsiae in D. andersoni, a limiting factor for the distribution of Rickettsia rickettsii; pp. 585–594. [Google Scholar]
- Noda H, Munderloh UG, Kurti TJ. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997;63:3926–3932. doi: 10.1128/aem.63.10.3926-3932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoles G. Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J Med Entomol. 2004;41:277–286. doi: 10.1603/0022-2585-41.3.277. [DOI] [PubMed] [Google Scholar]
- Sun LV, Scoles GA, Fish D, O’Neill SL. Francisella-like endosymbionts of ticks. J Invertebr Pathol. 2000;76:301–303. doi: 10.1006/jipa.2000.4983. [DOI] [PubMed] [Google Scholar]
- Dale C, Moran NA. Molecular interactions between bacterial symbionts and their hosts. Cell. 2006;126:453–465. doi: 10.1016/j.cell.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, Moran NA, Hattori M. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- Klyachko O, Stein BD, Grindle N, Clay K, Fuqua C. Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl Environ Microbiol. 2007;73:6584–6594. doi: 10.1128/AEM.00537-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Degnan PH, Santos SR, Dunbar HE, Ochman H. The players in a mutualistic symbiosis: insects, bacteria, viruses, and virulence genes. Proc Natl Acad Sci USA. 2005;102:16919–16926. doi: 10.1073/pnas.0507029102. [DOI] [PMC free article] [PubMed] [Google Scholar]