

Abstract
Purpose
We recently demonstrated in a mouse model of glaucoma that endogenous epigenetic mechanisms can be activated by a repetitive hypoxic preconditioning (RHP) stimulus to provide robust retinal ganglion cell (RGC) protection. Although we also provided evidence that RHP prevents or delays the apoptotic demise of the RGC soma, the mechanisms responsible for signaling this epigenetic response, as well as the effectors of the glaucoma-tolerant phenotype at the somatic and axonal levels, remain unidentified. In the present study, we used conditional mutant mice lacking hypoxia-inducible factor-1α (HIF-1α) in RGCs (HIF-1α RGC-knockout [KO] mice) to test the hypothesis that RHP-mediated activation of this transcription factor in these cells protects them from glaucomatous injury.
Methods
Adult HIF-1α RGC-KO mice, generated by mating floxed HIF-1α mice with math5-Cre mice, were used. Experimental glaucoma was induced unilaterally in the HIF-1α RGC-KO mice and matched wild-types by elevating the intraocular pressure to 16–20 mmHg for 3 consecutive weeks, secondary to episcleral vein ligation. Mice of each genotype were randomized to either an RHP protocol (six total exposures to systemic hypoxia [11% oxygen], interspersed over a 2-week period, completed 3 days before ligation surgery) or to an untreated group. RGC soma and axon injury was quantified with Neuronal Nuclei (NeuN) immunohistochemistry in retinal flat mounts and SMI32 immunohistochemistry in cross sections of the post-laminar optic nerve, respectively.
Results
HIF-1α RGC-KO mice exhibited normal retinal function and morphology, and crosses of math5-Cre mice with floxed ROSA26 reporter mice confirmed Cre recombinase activity was confined to the RGC axons and soma. Untreated wild-type mice exhibited a 30±2% loss of RGC soma and a 31±3% loss of RGC axons after 3 weeks of intraocular hypertension (both p<0.05 versus fellow eye). The 90% and 81% improvement in soma and axon survival, respectively, observed in the wild-type mice treated with RHP (both p<0.05 versus the glaucoma eye in the untreated mice) was still observed to a near identical extent in the RHP-treated HIF-1α RGC-KO mice. RHP had no effect on the magnitude of intraocular pressure elevation in either the KO or wild-type groups, indicating that protection was realized in both genotypes in the face of ongoing intraocular hypertension.
Conclusions
These findings indicate that the robust, glaucomatous protection of the RGC soma and axons induced by RHP does not require HIF-1α-mediated transcription of survival genes and other adaptive responses within the RGCs themselves. Rather, we infer that RGC survival is augmented secondary to the activation of other hypoxia-sensitive transcription factors in RGCs and/or the action of diffusible HIF-1α target gene proteins released from neighboring retinal cells. Ideally, the involvement of such autocrine- and/or paracrine-based mechanisms would be confirmed in future studies, but distinct components of the integrated, pleiotropic, and multicellular basis of this endogenous epigenetic response may prove difficult to demonstrate experimentally, as we found in the present study.
Introduction
Glaucoma is the second leading cause of blindness in the world [1]. Therapeutic approaches used in clinical practice involve surgery or drugs designed to reduce the patient’s abnormally high intraocular pressure (IOP), the cardinal feature and risk factor of open-angle glaucoma. In theory, neuroprotective therapies designed to protect retinal ganglion cells (RGCs) from the damage induced by progressive and sustained increases in IOP could represent a third treatment strategy, but to date, no drug has demonstrated efficacy in clinical trials. Not surprisingly, given that the mechanisms of RGC injury in this neurodegenerative disease are likely to be multifactorial, act in a progressive fashion over protracted periods of time, and operate at the level of the soma and the axon, a single-drug therapeutic approach may never prove efficacious in practice.
In recent years, preclinical studies in the central nervous system (CNS) and other tissues have revealed that potent, endogenous mechanisms affording protection against acute injury can be induced by various mild preconditioning stressors [2-4]. The robust magnitude of protection in these animal models implies that the epigenetic response is mechanistically pleiotropic, and involves gene expression changes in neuronal, glial, and vascular cells. In the retina, considerable evidence has accumulated supporting the ability of brief, hypoxic, or ischemic preconditioning stimuli applied before [5,6] and even after [7,8] an ischemic insult to transiently protect ganglion and other retinal cells. We demonstrated that the duration of the resultant ischemic tolerance in the retina could be extended from days to weeks by repeatedly presenting a hypoxic preconditioning stimulus [9]. That finding led us to test whether this sustained phenotypic change would also protect RGCs in the setting of glaucoma. Indeed, using an inducible mouse model of open-angle glaucoma, we demonstrated significantly enhanced survival of the retinal ganglion cell (RGC) soma and axons with repetitive hypoxic preconditioning (RHP) [10].
The present study was undertaken to begin to ascertain the mediators of this innate protective response. Because hypoxia was the triggering stimulus, we turned our attention to the potential involvement of the oxygen-dependent transcription factor known as hypoxia-inducible factor-1α (HIF-1α), a well-established “master regulator” of hypoxic adaptation and survival [11,12] that is expressed throughout the retina [13-15], including RGCs [16,17]. Considerable evidence supports the involvement of HIF-1α in hypoxic preconditioning-induced protection of the brain [18-20] and the retina [9-17,21] from ischemic injury. Moreover, we previously documented a unique retinal HIF-1α protein signature, in which the magnitude and duration of expression are increased in response to multiple versus single hypoxic challenges. This expression pattern provides correlative support for the hypothesis that the long-lasting protection against RGC glaucomatous injury induced by RHP [9] depends on HIF-1α. To obtain direct causal data for this hypothesis, we leveraged a Cre-Lox approach to generate mice lacking HIF-1α in the RGCs, and then assessed the effects of the gene deletion on RHP-mediated protection. Results of those studies are detailed herein.
Methods
Animals and experimental groups
Conditional, retinal cell-specific HIF-1α knockout (KO) mice were generated by mating HIF-1αflox/flox mice on a SV129 background (graciously supplied by Dr. David C. Beebe at Washington University; originally from Dr. Randall S. Johnson at University of California San Diego [22]) with math5-Cre+/? mice on a C57Bl/6J background (graciously supplied by Dr. Susan Culican at Washington University; originally from Dr. Russell Van Gelder [University of Washington]), which express at least one Cre recombinase allele under the control of the math5 promoter. Math5 is a transcription factor intimately involved in directing differentiation of retinal cell progenitors into RGCs and other retinal neurons during development. The math5 lineage is essential for RGC competence, but math5 progenitors also give rise to amacrine, horizontal, and photoreceptor cells [23,24]. Thus, in the offspring of the resultant Cre-lox cross, HIF-1αflox/flox;math5-Cre double mutants (which we refer to as HIF-1α RGC-KO mice), HIF-1α is knocked out in all RGCs. Littermate HIF-1αflox/flox;Cre−/− mice were used as wild-type (WT) controls in all studies. The primer sequences for genotyping were as follows: 5′-GCA TTA CCG GTC GAT GCA ACG AGT GAT GAG-3′ (Cre-forward), 5′-GAG TGA ACG-AAC CTG-GTC GAA ATC AGT GCG-3′ (Cre-reverse), 5′-ATA TGC TCT TAT GAA GGG-GCC TAT GGA GGC-3′ (for HIF-1α_flox5), and 5′-GAT CTT TCC GAG GAC CTG GAT TCA-ATT CCC-3′ (for HIF-1α_flox3).
To confirm the cell-specific expression and success of Cre recombinase in the Math5-Cre mice, we bred them to floxed ROSA26 reporter mice that express membrane bound tomato red fluorescence or green fluorescent protein (GFP) fluorescence before, or after, respectively, Cre recombinase-mediated excision (Gt(ROSA)26Sortm4(ACTB-tdTomato,EGFP)Luo/J [stock #007576]; Jackson Labs, Bar Harbor, ME). Retinal flat mounts were prepared from the offspring and examined with confocal microscopy.
Mice were randomized, by genotype, into either an untreated glaucoma group or a glaucoma group with prior repetitive hypoxic preconditioning (RHP; see below); thus, a total of four experimental groups were studied. We intended to process the tissue to measure the RGC soma and post-laminar optic nerve axon integrity endpoints in both eyes of every animal in every group. However, occasionally, technical problems precluded us obtaining one or the other endpoint. This resulted in the following groups/sizes: untreated WT mice (n=8 total, with n=6 for Neuronal Nuclei [NeuN], n=7 for SMI32, and n=5 for both), RHP-treated WT mice (n=11 total, with n=10 for NeuN, n=11 for SMI32, and n=10 for both), untreated HIF-1α RGC-KO mice (n=9 total, with n=7 for NeuN, n=7 for SMI32, and n=5 for both), and RHP-treated HIF-1α RGC-KO mice (n=7 total, with n=7 for NeuN, n=6 for SMI32, and n=6 for both).
Experimental glaucoma model and repetitive hypoxic preconditioning
All experimental methods and animal care procedures used were in accordance with National Institutes of Health and Association for Research in Vision and Ophthalmology guidelines for the care and use of laboratory animals, and approved by the Animal Studies Committee at Washington University. As described previously by our group [10,25], a sustained period of elevated IOP was induced by repeated ligation of the episcleral veins of one eye, allowing the fellow eye of each animal to serve as a non-glaucomatous control. Briefly, the ketamine/xylazine-anesthetized mice received topical application of 0.5% proparacaine hydrochloride, and incising the conjunctiva and Tenon’s capsule exposed the episcleral veins, three of which over 300° of the limbus were doubly ligated with 11–0 nylon suture (Alcon Surgical, Fort Worth, TX), and then severed. Core body temperature was maintained at 37 °C in all animals throughout the surgical procedure via a thermoregulated heating pad. If IOP was not elevated at 24 h or did not remain elevated at the 1- or 2-week time points, additional ligations of one or more presumably patent veins were performed at these time points. There were no significant differences between the four experimental groups in the absolute number of ligations performed at any given follow-up time point (24 h to 3 weeks), or in the relative number of ligations performed, expressed as a proportion of the total number of animals in the group, at any given follow-up time point. Following the topical antibiotic, mice were recovered in their home cages during the 3-week period of experimental glaucoma. IOP determinations were made between 10 a.m. and 2 p.m. in both eyes before and at weekly intervals following the induction of unilateral intraocular hypertension using the TonoLab Rebound Tonometer system (Colonial Medical Supply, Franconia, NH), as described previously by us [10,25] and others [26].
Before glaucoma was induced, animals of each genotype were randomized to RHP or normoxic control groups, as described previously [9,10]. Briefly, conscious mice were exposed for 2 h to a single period of systemic hypoxia (11% oxygen) three times per week (approximately every other day) for 2 consecutive weeks. The initial episcleral vein ligation surgery was then performed 3 days after the last hypoxic exposure.
Morphological and functional analyses of hypoxia-inducible factor-1α retinal ganglion cell-knockout mice
Morphological and functional endpoints in the HIF-1α RGC-KO mice were assessed to identify potential abnormalities from the CreLox-mediated loss of the HIF-1α gene in retinal cells expressing Math5. In one morphological analysis, the adult mice were euthanized, eyes were perfusion-fixed, embedded in paraffin, and 5-μm-thick serial sections were obtained and stained with hematoxylin and eosin to examine the retinal morphology. We also immunostained for HIF-1α in separate sections of the retinas from the adult mice of both genotypes, after we euthanized the animals during exposure to either normoxic room air or immediately after exposure to a single bout of hypoxia (2 h, 11% oxygen). After deparaffinization, rehydration, and antigen retrieval, nonspecific binding was blocked with prediluted 10% normal goat serum (S-1000, Vector Laboratories, Burlingame, CA) for 20 min at room temperature. Sections were then incubated overnight at 4 °C with primary antibodies (HIF-1α [#448SS, Lot#A, 1:100, Novus Biologicals, Littleton, CO] and brn3 [#SC6026, 1:25, Santa Cruz Biotechnology, Dallas, TX]), washed with PBS (Sigma-Aldrich, St. Louis, MO; 0.138 M NaCl, 0.0027 M KCl, pH 7.4), and then incubated with secondary antibodies (Alexa Fluor 488 donkey antirabbit immunoglobulin [IgG; #A21206, 1:200, Invitrogen, Grand Island, NY] and Alexa Fluor 594 donkey antigoat IgG [A11058, 1:200, Invitrogen], respectively) for 1 h at room temperature. Nuclei were counterstained with 4',6-diamidino-2-phenylindole dihydrochloride (#H-1200, Vector Laboratories). Slides were washed and mounted with fluorescence-preserving mounting medium. Negative control slides were run without each primary antibody to ascertain the background autofluorescence levels of the respective secondary antibodies. A BX60 Olympus microscope (Olympus, Center Valley, PA) and a Zeiss AxioCam digital (Zeiss, Göttingen, Germany) camera were used to capture images.
For assessments of retinal function, scotopic flash electroretinograms were obtained simultaneously for both eyes of normothermic, adult WT, and HIF-1α RGC-KO mice, as described in detail in our previous studies [6,9]. Briefly, animals were dark-adapted overnight, anesthetized with chloral hydrate (350 mg/kg; Sigma-Aldrich) and xylazine (4 mg/kg; Burns Veterinary Supply, Inc., Rockville Center, NY), and the pupils were dilated with 1% tropicamide and 2.5% phenylephrine, while the cornea was kept moist with 1% carboxymethylcellulose. Signals generated from a 2-mm stainless steel loop electrode positioned on the corneal surface, relative to the reference electrode placed subcutaneously behind the ear and a ground electrode under the animal, in response to brief (10 µsec) white-light flashes (0.1 Hz) were amplified and digitized with dedicated hardware and software. The a-wave and b-wave amplitudes at a stimulus intensity of 0.88 log cd-s/m2 were then calculated for each eye by a blinded observer, to obtain a single a-wave and b-wave amplitude for each animal.
Quantification of retinal ganglion cell axonal integrity
As indices of RGC axon health and survival after 3 weeks of experimental glaucoma, axon integrity in the WT and HIF-1α RGC-KO mice was quantified in the post-laminar optic nerve (approximately 50–150 µm behind the globe) with confocal immunofluorescence microscopy of SMI32-immunopositive axons, as described previously by us [10,25] and others [27-29]. Briefly, after the animals were euthanized by transcardial perfusion, optic nerve segments were fixed overnight in 4% paraformaldehyde, thrice rinsed with PBS, transferred to 10% formalin, and then paraffin-embedded. Four-µm-thick sections perpendicular to the long axis of the nerve were collected, deparaffinized, rehydrated, and subjected to antigen retrieval (a 10-min immersion in boiling antigen unmasking solution [H3300, Vector Laboratories]). After a 20-min incubation in blocking buffer, sections were incubated overnight at 4 °C with primary antibodies against the non-phosphorylated neurofilament heavy subunit SMI-32 (1:1,000; mouse monoclonal; Covance, Emeryville, CA) and glial fibrillary acidic protein (1:200; rabbit polyclonal; G4546, Sigma), followed by room temperature incubation for 1 h with secondary antibody (1:400, goat antimouse IgG linked to Alexa Fluor 488, A11001, Molecular Probes (Grand Island, NY), for SMI-32; and 1;400, goat antirabbit IgG linked to Alexa Fluor 546, A11010, Molecular Probes, for glial fibrillary acidic protein). VectaShield Hardset Mounting Medium with 4',6-diamidino-2-phenylindole dihydrochloride (H-1200, Vector Laboratories) was used for counterstaining nuclei. Confocal images (Carl Zeiss, LSM 5 PASCAL; 40X and 63X oil immersion objective [1.25 NA]) were obtained at excitation wavelengths of 405, 488, and 560 nm over an area of 146 µm × 146 µm. Total fluorescence intensities of SMI32-positive axons within the nerve border were determined by binarizing the image after background subtraction to identify the total surface area in the nerve covered by SMI32 above the threshold, calculating the total fluorescence intensity of the resulting image, and then normalizing to the total area of SMI32-positive signal, as detailed previously [25].
Quantification of retinal ganglion cell soma survival
After 3 weeks of sustained intraocular hypertension, the WT and HIF-1α RGC-KO mice in each group were euthanized, and the surviving RGC soma were quantified in retinal flat mounts using NeuN immunohistochemistry, as described previously by us [25] and others [30-32]. Briefly, mice were euthanized by transcardial perfusion, and the retinas were harvested from each eye, washed three times in Tris-buffered saline with Tween-20 (TBST) buffer (50 mM Tris, 150 mM NaCl, and 0.05% Tween-20), and permeabilized for 30 min with 0.3% Triton X-100 (Sigma T-9284) in TBS buffer. After blocking nonspecific binding with 1% BSA (Jackson ImmunoResearch, West Grove, PA), 0.15% Triton X-100, and 10% normal goat serum (Jackson ImmunoResearch) in TBST for 30 min at room temperature, the retinas were incubated overnight at 4 °C with NeuN (1:500; monoclonal anti-NeuN; MAB377, Chemicon/Millipore, Billerica, MA). Secondary antibody (1:400; goat antimouse IgG linked to Alexa Fluor 568, A11004, Invitrogen) was applied for 1 h at room temperature following a TBST wash, and the retinas were then flat-mounted with 50% glycerol (G-5516, Sigma) and coverslipped.
Eight distinct 221,600 μm2 (460 µm × 460 µm) regions of the peripheral retina (two per quadrant) were examined with confocal fluorescence microscopy (Carl Zeiss, LSM 5 PASCAL; 20X objective; 0.5 NA; excitation wavelength=560 nm). A blinded observer initially established fluorescence thresholds for each experimental/fellow eye pair and applied manual corrections of scanned images to remove signal artifacts; thereafter, AnalySIS software (Soft Image System GmbH, Munster, Germany) was used to automatically count the NeuN-positive RGC soma, which included all homogeneously stained and round NeuN-positive cell bodies.
Statistical analysis
Results are provided as means±standard error of the mean (SEM). Nonparametric signed-rank and rank-sum tests were used to identify significant differences between measures from paired eyes in the same animal and from eyes in different animal groups, respectively. Differences in IOP within and between animals in the four experimental groups over the 3-week period of intraocular hypertension, as well as differences in the number of episcleral vein ligations performed between the four experimental groups, were identified with one-way repeated measures ANOVA. In all instances, p<0.05 was accepted as significant.
Results
Characterization of the hypoxia-inducible factor-1α retinal ganglion cell-knockout mice
Several measurements were undertaken in the HIF-1α RGC-KO mice to confirm normal morphology and function, as well as to characterize the cell specificity and efficiency of the HIF-1α knockdown. First, we crossed math5-Cre mice with a line of floxed ROSA26 reporter mice that endogenously express a tomato red fluorescent protein in the membrane of all cells. Because they also incorporate a GFP reporter in this locus with an upstream loxP-flanked stop cassette, membranes fluoresce green following Cre-mediated recombination in the resulting double mutant offspring, if the cells express math5-Cre. As can be seen in Figure 1, RGC axons coursing through the nerve fiber layer of the mid-peripheral and central retina are brightly fluorescent, but not retinal blood vessels, indicative of a functional Cre transgene, and consistent with its localization to all RGCs. A similar expression pattern was observed in the peripheral retina (data not shown). The fluorescent cell membranes of some underlying soma are also evident in the photomicrograph, implicating them as Cre-expressing RGCs. No GFP fluorescence was observed anywhere in the retina in F1 offspring from Cre-negative mice crossed with ROSA26 reporter mice (data not shown).
Figure 1.
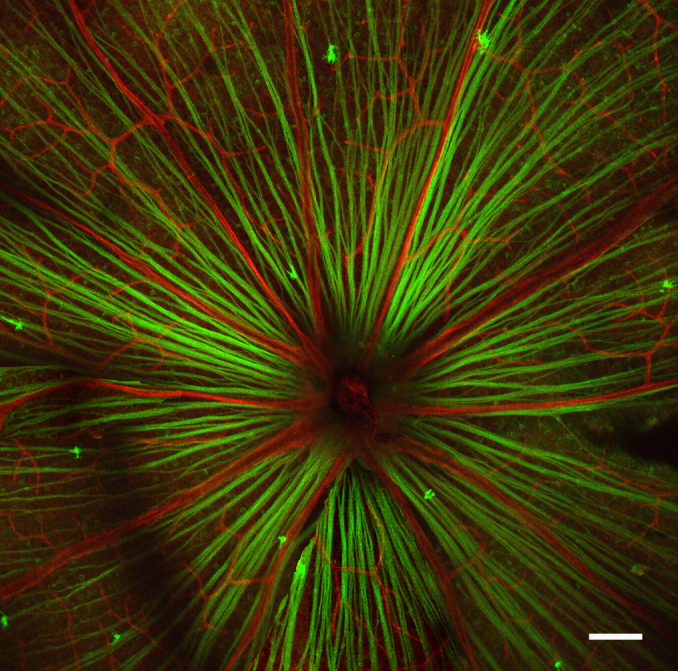
Math5-Cre reporter mice. Confocal fluorescence microscopy image of a retinal flat mount from an animal derived from crossing ROSA26 green fluorescent protein-reporter mice with math5-Cre mice. Note the presence of green fluorescence protein (GFP) in the retinal ganglion cell axons and some underlying soma in the ganglion cell layer, reflecting the presence of math5-Cre in these cells, but the lack of math5-Cre expression in retinal vascular cells (red). Scale bar=100 µm.
As shown in Figure 2, HIF-1α immunohistochemistry on retinal cross sections in WT mice revealed HIF-1α-positive cells throughout the retinas even under normoxic control conditions, including cells in the ganglion cell and inner nuclear layers, as well as in photoreceptors (although much of the signal in the photoreceptor layer was the result of antibody autofluorescence; data not shown). In response to hypoxia, HIF-1α-positive expression was greatly enhanced throughout the tissue. Coimmunostaining for the RGC-specific antigen brn3 revealed both HIF-1α-positive and brn3-positive cells in the ganglion cell layer, indicative of RGCs, as well as HIF-1α-positive and brn3-negative cells, indicative of either displaced amacrine cells or brn3-negative RGCs. In the HIF-1α RGC-KO mice, a similar baseline and hypoxia-driven increase in HIF-1α expression was evident throughout the retina, including within the ganglion cell layer. However, upon closer examination, it was clear that, although HIF-1α-positive cells could be identified in this layer, no brn3-positive cells coimmunostained for HIF-1α in sections from either normoxic or hypoxic mice, implicating the former as displaced amacrine cells. These results confirm that brn3-positive RGCs in the HIF-1α RGC-KO mice lacked HIF-1α protein expression, but we cannot say with certainty from these specific results that all RGCs lacked the HIF-1α protein.
Figure 2.

Hypoxia-inducible factor-1α (HIF-1α) immunohistochemistry in normoxic and hypoxic wild-type and HIF-1α retinal ganglion cell-knockout mice. HIF-1α expression is shown in representative wild-type (WT; A, B) and HIF-1α retinal ganglion cell-knockout (RGC-KO) mice (C, D) under normoxic (A, C) and hypoxic (B, D) conditions. The HIF-1α (green), brn3 (red), 4',6-diamidino-2-phenylindole dihydrochloride (DAPI; blue), and “merged” images are shown in each row, left to right. In the images from the WT mice (A, B), arrowheads identify examples of HIF-1α and brn3 copositive RGCs in the ganglion cell layer (inset in the merged image shows one example at higher magnification), and arrows identify HIF-1α-positive but brn3-negative amacrine cells (or brn3-negative RGCs) in the ganglion cell layer. In the images from the HIF-1α RGC-KO mice (C, D), arrowheads identify examples of HIF-1α-negative but brn3-positive RGCs in the ganglion cell layer (inset in the merged image shows one example at higher magnification), and arrows identify HIF-1α-positive but brn3-negative amacrine cells in the ganglion cell layer. Scale bar=20 µm.
The hematoxylin and eosin−stained retinas from the adult HIF-1α RGC-KO mice looked unremarkable and essentially identical to those from WT mice. As shown in the representative cross sections (Figure 3), the relative cell densities in the different retinal layers and the relative thicknesses of the different retinal layers were similar from the peripheral to the central retina in each genotype, suggesting normal development of the retina in the HIF-1α RGC-KO mutants. Functionally, we observed normal a-wave and b-wave amplitudes in the resting scotopic flash electroretinograms measured in adult HIF-1α RGC-KO mice that were indistinguishable from those recorded in the WT mice (Figure 4).
Figure 3.

Hematoxylin and eosin–stained cross sections of retinas from wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice. No gross differences in cell body densities or layer thicknesses were noted between the two genotypes across the retina, as exemplified by these two representative images (n=3 each) from the mid-central retina from the wild-type (WT; left) and hypoxia-inducible factor-1α retinal ganglion cell-knockout (HIF-1α RGC-KO; right) mice. Scale bar=50 µm.
Figure 4.
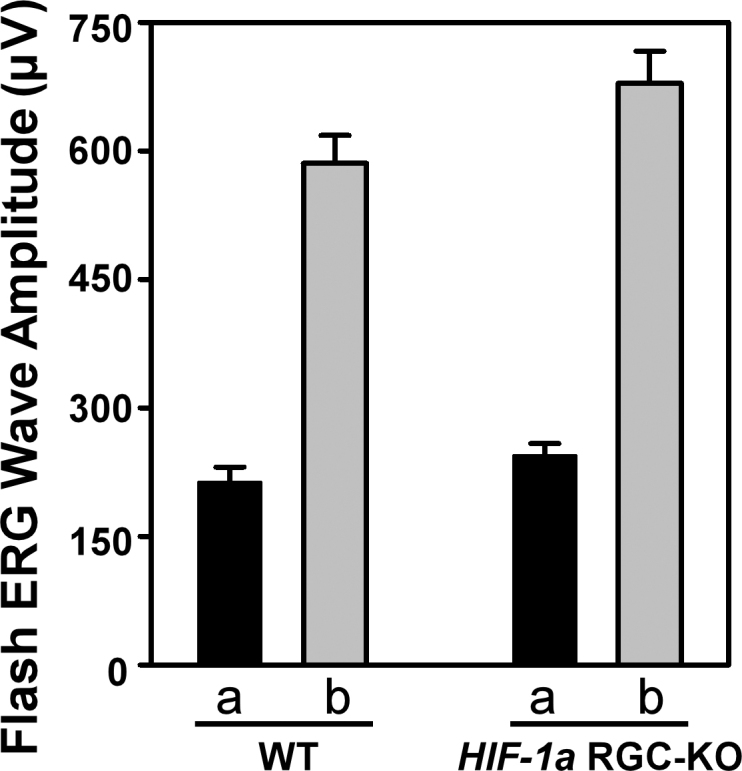
Flash electroretinogram wave amplitudes in wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice. Amplitudes of a-waves (filled bars) and b-waves (shaded bars) of resting scotopic flash electroretinograms measured in wild-type (WT; left; n=8) and hypoxia-inducible factor-1α retinal ganglion cell-knockout (HIF-1α RGC-KO; right; n=8) mice are shown. Means±standard error of the mean (SEM).
Intraocular pressure
As shown previously by our group [10,25], our episcleral vein ligation protocol resulted in consistent and sustained elevations in IOP in all four experimental groups, starting 24 h after the initial surgery and lasting for 3 weeks (when all endpoints were measured; Figure 5). Baseline IOP values did not differ between the eyes in any given group, or between groups; baseline IOPs ranged from 9.8±0.6 to 10.8±0.9 mmHg in fellow eyes, and from 10.4±0.5 to 12.0±0.6 mmHg in experimental eyes. The increases in IOP measured in the experimental eyes at all four time points were significantly greater (p<0.001 to p=0.039) at each time point than the respective baseline IOP value for that eye, and significantly greater (p<0.001 to p=0.016) than the respective IOP in the corresponding fellow eye at the same time point. IOPs in the experimental group ranged from 16.5±1.4 to 16.8±1.5 mmHg at 24 h, from 19.0±1.5 to 20.0±2.0 mmHg at 1 week, from 17.0±0.7 to 19.0±1.0 mmHg at 2 weeks, and from 15.9±1.1 to 19.3±0.9 mmHg at 3 weeks of intraocular hypertension. There were no statistically significant differences in the extent of IOP elevation in the experimental eyes among the four groups at any time point. In sum, the experimental eyes in our study started with the same IOPs, and experienced the same elevation in IOP, independent of genotype or prior RHP treatment.
Figure 5.

Intraocular pressure in wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice with and without repetitive hypoxic preconditioning. Intraocular pressure (IOP) in the experimental eyes (filled symbols) and respective fellow eyes (open symbols) in the wild-type (WT; red; n=10) and hypoxia-inducible factor-1α retinal ganglion cell-knockout (HIF-1α RGC-KO; purple; n=8) groups without repetitive hypoxic preconditioning (RHP), and the WT (green; n=8) and HIF-1α RGC-KO (blue; n=14) with prior RHP over the 3-week period of intraocular hypertension are shown. All IOP values for the experimental eyes in each group at 24 h and 1, 2, and 3 weeks were significantly different from their respective baselines and from their fellow eyes at each time point, but no statistically significant differences were noted among the four groups. Mean±standard error of the mean (SEM).
Retinal ganglion cell soma protection by repetitive hypoxic preconditioning in wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice
As shown in Figure 6, consistent with what we reported previously in other mouse strains [10,25], 3 weeks of experimental glaucoma led to significant RGC soma loss in the WT mice; specifically, 30±2% (p=0.016; n=7) of NeuN-positive soma were lost in non-preconditioned animals, relative to respective fellow eyes. RHP completely prevented (p<0.001) RGC soma loss in the WT mice (n=7), with 97±3% of NeuN-positive cells still remaining (not significantly different [p=0.375] from fellow eyes), a robust protective effect in line with what we reported earlier for RHP, using brn3 as an RGC immunolabel [10]. Loss of NeuN-positive cells in response to 3 weeks of intraocular hypertension was similar (p=0.445) in the non-preconditioned HIF-1α RGC-KO mice (28±7% of NeuN-positive cells lost; n=6; p=0.031) and the non-preconditioned WT mice. However, RHP also protected the RGC soma in the HIF-1α RGC-KO mice (n=10) significantly (p=0.004), with 93±9% of the NeuN-positive soma surviving (not significantly different from fellow eyes [p=0.164]; Figure 6). Thus, HIF-1α RGC-KO mice, despite lacking the HIF-1a gene in RGCs, were still protected from glaucoma-induced RGC soma loss by our RHP stimulus.
Figure 6.

Retinal ganglion cell soma protection in wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice with experimental glaucoma by prior repetitive hypoxic preconditioning. Quantification of NeuN–positive soma in the retinal ganglion cell layer following 3 weeks of intraocular hypertension in the four experimental groups, normalized to fellow eyes (black bars) are shown. Note that repetitive hypoxic preconditioning (RHP) provided protection against the glaucomatous loss of the retinal ganglion cell (RGC) soma in wild-type (WT; green) and hypoxia-inducible factor-1α retinal ganglion cell-knockout (HIF-1α RGC-KO; blue) mice. *p<0.05 versus fellow eye; #p<0.05 versus non-preconditioned glaucoma in the same genotype. Means±standard error of the mean (SEM).
Retinal ganglion cell axon protection by repetitive hypoxic preconditioning in wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice
In line with our previous studies of experimental glaucoma [10,25], Figure 7 illustrates that sustained elevations in IOP for 3 weeks in the WT mice resulted in a significant loss of axons at the post-laminar optic nerve level that was robustly and significantly attenuated with RHP. Specifically, a 31±3% reduction (p=0.016) in SMI32 fluorescence intensity was recorded in the non-preconditioned WT mice (n=7), but in the RHP-treated mice (n=6), this loss was significantly (p=0.001) abrogated, with 94±2% of the SMI32-positive axons remaining, not significantly different (p=0.219) from fellow eyes. Importantly, as observed for the RGC soma, RHP also protected the HIF-1α RGC-KO mice against glaucomatous RGC axon loss. In particular, the non-preconditioned HIF-1α RGC-KO mice (n=7) exhibited a 23±3% loss (p=0.016 versus fellow eyes) in SMI32 fluorescence intensity after 3 weeks of experimental glaucoma, whereas 95±2% of the SMI32-positive axons remained in the RHP-treated HIF-1α RGC-KO mice (n=11; p=0.001 versus non-preconditioned HIF-1α RGC-KO mice). Both responses closely paralleled those measured in the WT mice. Thus, RHP protected RGC axons from glaucomatous injury despite the Cre-Lox-mediated deletion of the HIF-1a gene in RGCs.
Figure 7.

Retinal ganglion cell axon protection in wild-type and hypoxia-inducible factor-1α retinal ganglion cell-knockout mice with experimental glaucoma by prior repetitive hypoxic preconditioning. Quantification of SMI32-positive axons in the post-laminar optic nerve following 3 weeks of intraocular hypertension in the four experimental groups is shown, normalized to fellow eyes (black bars). Note that repetitive hypoxic preconditioning (RHP) provided protection against the glaucomatous loss of retinal ganglion cell (RGC) axons in the wild-type (WT; green) and hypoxia-inducible factor-1α retinal ganglion cell-knockout (HIF-1α RGC-KO; blue) mice. *p<0.05 versus fellow eye; #p<0.05 versus non-preconditioned glaucoma in the same genotype. Means±standard error of the mean (SEM).
Discussion
Results of the present study in RGC-specific, conditional HIF-1α-null mice indicate that the endogenous epigenetic mechanisms induced by RHP, which we showed previously provided RGCs somatic and axonal protection against glaucomatous injury [10], do not depend on autocrine-based, HIF-1α-mediated transcriptional activation within RGCs themselves. Given the robustness of the protection induced by RHP against glaucomatous [10] and ischemic [9] injury, it is not unlikely that RHP promotes a pleiotropic epigenetic response that is not only operative at the somatic and axonal levels of the cell but also mechanistically redundant—all of which may have contributed to the lack of phenotype we observed in the HIF-1α RGC-KO mice.
That HIF-1α is a canonical transcriptional regulator of survival-enhancing genes throughout the body is well established [11,12,18,33]. Considerable physiologic [9,17,34] and pharmacologic [13,21,35,36] evidence exists showing that prior augmentation of HIF-1α expression reduces pathology in models of retinal ischemia [9,17,21,35], oxygen-induced retinopathy [13], and phototoxicity [34,36]. Moreover, there is ample evidence that hypoxia drives HIF-1α expression in the retina in general [14,15] and in RGCs in particular [16,17]. In fact, in an earlier study [9], we documented distinct, time-dependent changes in retinal HIF-1α expression in response to a single bout of systemic hypoxia versus the multiple exposures to hypoxia that defined the RHP stimulus we used in the present investigation. Nevertheless, this uniquely protracted expression pattern, while consistent with the long-lasting cytoprotective phenotype exhibited by RGCs in RHP-treated mice subjected to ischemia [9] or glaucoma [10], provides only correlative evidence for HIF-1α’s participation in the observed protection. Thus, we pursued this causal study, leveraging RGC-specific HIF-1α knockout mice to test our hypothesis that this transcription factor would be a critical mediator of a repetitive hypoxia-induced, glaucomatous injury-resistant RGC phenotype [10].
Several investigators, including many in the field of retinal neurodegeneration [37], have leveraged Cre-Lox genetics to create mice that allow the documentation of causality regarding the injury or protection mechanisms operative in a given retinal cell type. To knock out HIF-1α selectively in RGCs, we mated floxed HIF-1α mice to Math5-Cre mice. Math5 is a transcription factor that determines RGC competence during RGC differentiation [23,38], and thus is present in every RGC, as well as in some horizontal and amacrine cells and some photoreceptors [24]. Thus, all RGCs in the retinas of the resulting progeny should lack HIF-1α. To validate this, we also crossed Math5-Cre mice with Rosa26 reporter mice in which a membrane-targeted, red fluorescent protein is floxed; Cre-mediated recombination results in a green fluorescent product, which we observed in flat-mounted retinas as uniformly distributed in the RGC axons and underlying soma (Figure 1), consistent with the expected RGC localization of a functional Math5-Cre. Using histology (Figure 3) and scotopic electroretinography (Figure 4), we validated that the HIF-1α RGC-KO mice used in our studies exhibited no structural or functional abnormalities. Finally, we obtained immunohistochemical evidence in our HIF-1α RGC-KO mice that HIF-1α protein expression in RGCs is lacking under baseline and hypoxic conditions (Figure 2). This series of successful validation measures supported our intended use of these mice to test the hypothesis that RGC HIF-1α contributes to the enhanced survival of these cells in our glaucoma tolerance model [10]. That said, an important caveat of our study is that we did not provide quantitative data that would more clearly document the efficiency of the Cre-mediated excision of the HIF-1α gene in RGCs; our results from reporter mice and from our immunohistochemical analyses only qualitatively address this concern.
Overall, we found that RHP still promoted protection of the RGC soma and axons in HIF-1α RGC-KO mice, to levels as robust as that seen in their wild-type counterparts, indicating that the transcriptional activity of HIF-1α within RGCs is not required for the ability of RHP to protect either the soma or the axons of these cells from glaucomatous injury. However, our results do not provide evidence for alternative explanations for how the RGC-protective phenotype is realized independent of HIF-1α. One possibility is that HIF-1α-derived cytoprotective mediators, such as erythropoietin, heme oxygenase-1, VEGF, and adrenomedullin, and the protein products of many other prosurvival genes whose transcription is activated by HIF-1α are produced and released into the extracellular space by neighboring retinal cells and then act through their respective receptors on the RGCs to exert protection. How such a diffusible mediator within the retina protects the more distant RGC axon, the demise of which is thought to occur by mechanisms distinct from the RGC soma [39,40], represents an additional unknown. Another possibility is that, since hypoxia can drive changes in gene expression through other hypoxia-sensitive transcription factors (e.g., nuclear factor (erythroid-derived 2)-like 2 [Nrf-2], nuclear factor kappa B [NFĸB], tumor protein 53 [p53], signal transducer and activator of transcription 3 [STAT-3], activator protein 1 [AP-1]) [41,42], RGC resistance to glaucomatous injury may be realized in an autocrine or paracrine manner through these HIF-1α-independent transcriptional mechanisms. Finally, the repetitive hypoxic stimulus may induce in RGCs, or other neighboring retinal or vascular cells, the hypoxia-responsive isoform HIF-2α; functional roles for this transcription factor and its gene products have already been described in this tissue [43-46]. Further studies are needed to identify and confirm the involvement of these epigenetic modulators. More than likely, the robust protection afforded by RHP is mechanistically multifactorial, and represents an integrated, multicellular response designed to ensure the viability of the entire tissue.
Results of the present investigation indicate that the complex mechanisms of innate retinal neuroprotection may not easily be revealed by the deletion of a single gene in a single retinal cell type. In fact, in a similarly designed study, the cell-specific, Cre-mediated deletion of HIF-1α in photoreceptors also was without effect on the photoreceptor-protective response induced by hypoxic preconditioning [34]. Similarly, Müller cell-specific knockdown of the interleukin-6 receptor gp130 did not negatively influence the ability of cyclic light to precondition against subsequent phototoxicity [47]. Taken together, our findings and the results of these studies suggest two, potentially inclusive, possibilities for the RHP-mediated protection of RGCs: (1) Survival factors released by neighboring cells, whether neuronal or glial or endothelial, subserve a mechanism that helps ensure pan-retinal protection, and/or (2) multiple, functionally redundant stress-responsive signaling pathways exist in retinal cells, with HIF-1α-mediated transcription being just one, that increase the likelihood that an adaptive, epigenetic responses of the cell will be realized. That the retinal messenger RNA levels of several other HIF-1α-independent prosurvival genes known to be induced by hypoxia were not affected by photoreceptor-specific HIF-1α gene knockdown [48] supports the former hypothesis; however, the list of potential cytoprotective mediators is likely to be much longer than those measured.
In summary, the in vivo glaucomatous protection provided the RGC soma and axons of wild-type mice by RHP [10] persists even in mutant mice lacking the HIF-1α gene in their RGCs. This implies an epigenetic response to the RHP stimulus by RGCs that is HIF-1α-independent or a paracrine-based mechanism of RGC protection secondary to RHP-stimulated HIF-1α gene target protein elaboration and release from neighboring retinal cells. In fact, neither of these possibilities is mutually exclusive. Our findings are consistent with an endogenous, multicellular, pleiotropic mechanism of RGC protection that may not be revealed by deletion of a single gene in a specific retinal cell type. Translationally speaking, the quest to pharmacologically induce such a protective response in glaucoma patients may require multiple drugs to coordinately activate the many signaling pathways regulating a broad set of cell-specific epigenetic responses. Attempts to administer any number of the many different effector proteins in an effort to mimic the glaucoma-tolerant phenotype may also present an insurmountable challenge. Thus, ultimately, exposing patients to a physiologic stimulus, such as intermittent hypoxia, may be the most efficacious therapeutic approach.
Acknowledgments
This study was supported by NIH R01EY18607 (JMG, YZ), AHAF National Glaucoma Foundation (JMG, YZ), EY02687 (Dept of Ophthalmology, Washington University), NIH Neuroscience Blueprint Interdisciplinary Core Grant P30 NS057105 (Washington University), the HOPE Center for Neurologic Disorders (Washington University), and the Spastic Paralysis Research Foundation of the Illinois–Eastern Iowa District of Kiwanis International.
References
- 1.Quigley HA. Glaucoma. Lancet. 2011;377:1367–77. doi: 10.1016/S0140-6736(10)61423-7. [DOI] [PubMed] [Google Scholar]
- 2.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–48. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 3.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meller R, Simon RP. Tolerance to Ischemia - an increasingly complex biology. Transl Stroke Res. 2013;4:40–50. doi: 10.1007/s12975-012-0246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth S, Li B, Rosenbaum PS, Gupta H, Goldstein IM, Maxwell KM, Gidday JM. Preconditioning provides complete protection against retinal ischemic injury in rats. Invest Ophthalmol Vis Sci. 1998;39:777–85. [PubMed] [Google Scholar]
- 6.Zhu Y, Ohlemiller KK, McMahan BK, Gidday JM. Mouse models of retinal ischemic tolerance. Invest Ophthalmol Vis Sci. 2002;43:1903–11. [PubMed] [Google Scholar]
- 7.Dreixler JC, Shaikh AR, Alexander M, Savoie B, Roth S. Post-ischemic conditioning in the rat retina is dependent upon ischemia duration and is not additive with ischemic pre-conditioning. Exp Eye Res. 2010;91:844–52. doi: 10.1016/j.exer.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez DC, Bordone MP, Chianelli MS, Rosenstein RE. Retinal neuroprotection against ischemia-reperfusion damage induced by postconditioning. Invest Ophthalmol Vis Sci. 2009;50:3922–30. doi: 10.1167/iovs.08-3344. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Zhang Y, Ojwang BA, Brantley MA, Jr, Gidday JM. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: Role of HIF-1alpha and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–43. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y, Zhang L, Schmidt JF, Gidday JM. Glaucoma-induced degeneration of retinal ganglion cell soma and axons prevented by hypoxic preconditioning: A model of 'glaucoma tolerance'. Mol Med. 2012;18:697–706. doi: 10.2119/molmed.2012.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sears JE, Hoppe G, Ebrahem Q, Anand-Apte B. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc Natl Acad Sci USA. 2008;105:19898–903. doi: 10.1073/pnas.0805817105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crosson LA, Kroes RA, Moskal JR, Linsenmeier RA. Gene expression patterns in hypoxic and post-hypoxic adult rat retina with special reference to the NMDA receptor and its interactome. Mol Vis. 2009;15:296–311. [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes JM, Groot AJ, van der Groep P, Sersansie R, Vooijs M, van Diest PJ, van Noorden CJ, Schlingemann RO, Klaassen I. Active HIF-1 in the normal human retina. J Histochem Cytochem. 2010;58:247–54. doi: 10.1369/jhc.2009.953786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozaki H, Yu AY, Della N, Ozaki K, Luna JD, Yamada H, Hackett SF, Okamoto N, Zack DJ, Semenza GL, Campochiaro PA. Hypoxia inducible factor-1alpha is increased in ischemic retina: temporal and spatial correlation with VEGF expression. Invest Ophthalmol Vis Sci. 1999;40:182–9. [PubMed] [Google Scholar]
- 17.Peng P-H, Huang H-S, Lee Y-J, Chen Y-S, Ma M-C. Novel role for the delta-opioid receptor in hypoxic preconditioning in rat retinas. J Neurochem. 2009;108:741–54. doi: 10.1111/j.1471-4159.2008.05807.x. [DOI] [PubMed] [Google Scholar]
- 18.Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–48. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- 19.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–32. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taie S, Ono J, Iwanaga Y, Tomita S, Asaga T, Chujo K, Ueki M. Hypoxia-inducible factor-1 alpha has a key role in hypoxic preconditioning. J Clin Neurosci. 2009;16:1056–60. doi: 10.1016/j.jocn.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–8. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- 22.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Ding K, Pan L, Deng M, Gan L. Math5 determines the competence state of retinal ganglion cell progenitors. Dev Biol. 2003;264:240–54. doi: 10.1016/j.ydbio.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Feng L, Xie Z-h, Ding Q, Xie X, Libby RT, Gan L. MATH5 controls the acquisition of multiple retinal cell fates. Mol Brain. 2010;3:3–36. doi: 10.1186/1756-6606-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Zhang L, Sasaki Y, Milbrandt J, Gidday JM. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Invest Ophthalmol Vis Sci. 2013;54:25–36. doi: 10.1167/iovs.12-10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pease ME, Hammond JC, Quigley HA. Manometric calibration and comparison of TonoLab and TonoPen tonometers in rats with experimental glaucoma and in normal mice. J Glaucoma. 2006;15:512–9. doi: 10.1097/01.ijg.0000212276.57853.19. [DOI] [PubMed] [Google Scholar]
- 27.Dieterich DC, Trivedi N, Engelmann R, Gundelfinger ED, Gordon-Weeks PR, Kreutz MR. Partial regeneration and long-term survival of rat retinal ganglion cells after optic nerve crush is accompanied by altered expression, phosphorylation and distribution of cytoskeletal proteins. Eur J Neurosci. 2002;15:1433–43. doi: 10.1046/j.1460-9568.2002.01977.x. [DOI] [PubMed] [Google Scholar]
- 28.Jakobs TC, Libby RT, Ben Y, John SWM, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–25. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raymond ID, Pool AL, Vila A, Brecha NCA. Thy1-CFP DBA/2J mouse line with cyan fluorescent protein expression in retinal ganglion cells. Vis Neurosci. 2009;26:453–65. doi: 10.1017/S095252380999023X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beirowski B, Babetto E, Coleman MP, Martin KR. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci. 2008;28:1166–79. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- 31.Dijk F, Bergen AA, Kamphuis W. GAP-43 expression is upregulated in retinal ganglion cells after ischemia/reperfusion-induced damage. Exp Eye Res. 2007;84:858–67. doi: 10.1016/j.exer.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, Vetter ML, Marsh-Armstrong N, Horner PJ. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28:2735–44. doi: 10.1523/JNEUROSCI.4443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aragonés J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–80. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 34.Grimm C, Wenzel A, Groszer M, Mayser H, Seeliger M, Samardzija M, Bauer C, Gassmann M, Reme CE. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat Med. 2002;8:718–24. doi: 10.1038/nm723. [DOI] [PubMed] [Google Scholar]
- 35.Zhu Y, Zhang L, Gidday JM. Deferroxamine preconditioning promotes long-lasting retinal ischemic tolerance. J Ocul Pharmacol Ther. 2008;24:527–35. doi: 10.1089/jop.2008.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren H, Liu N-Y, Song X-F, Ma Y-S, Zhai X-Y. A novel specific application of pyruvate protects the mouse retina against white light damage: differential stabilization of HIF-1α and HIF-2α. Invest Ophthalmol Vis Sci. 2011;52:3112–8. doi: 10.1167/iovs.10-5605. [DOI] [PubMed] [Google Scholar]
- 37.Le Y-Z. Conditional gene targeting: dissecting the cellular mechanisms of retinal degenerations. J Ophthalmol. 2011;2011:806783. doi: 10.1155/2011/806783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang SW, Kim BS, Ding K, Wang H, Sun D, Johnson RL, Klein WH, Gan L. Requirement for math5 in the development of retinal ganglion cells. Genes Dev. 2001;15:24–9. doi: 10.1101/gad.855301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinnon SJ, Schlamp CL, Nickells RW. Mouse models of retinal ganglion cell death and glaucoma. Exp Eye Res. 2009;88:816–24. doi: 10.1016/j.exer.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450:363–71. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 42.Kenneth NS, Rocha S. Regulation of gene expression by hypoxia. Biochem J. 2008;414:19–29. doi: 10.1042/BJ20081055. [DOI] [PubMed] [Google Scholar]
- 43.Sheridan CM, Pate S, Hiscott P, Wong D, Pattwell DM, Kent D. Expression of hypoxia-inducible factor-1alpha and −2alpha in human choroidal neovascular membranes. Graefes Arch Clin Exp Ophthalmol. 2009;247:1361–7. doi: 10.1007/s00417-009-1133-3. [DOI] [PubMed] [Google Scholar]
- 44.Weidemann A, Krohne TU, Aguilar E, Kurihara T, Takeda N, Dorrell MI, Simon MC, Haase VH, Friedlander M, Johnson RS. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia. 2010;58:1177–85. doi: 10.1002/glia.20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mowat FM, Luhmann UFO, Smith AJ, Lange C, Duran Y, Harten S, Shukla D, Maxwell PH, Ali RR, Bainbridge JW. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS ONE. 2010;5:e11103. doi: 10.1371/journal.pone.0011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morita M, Ohneda O, Yamashita T, Takahashi S, Suzuki N, Nakajima O, Kawauchi S, Ema M, Shibahara S, Udono T, Tomita K, Tamai M, Sogawa K, Yamamoto M, Fujii-Kuriyama Y. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003;22:1134–46. doi: 10.1093/emboj/cdg117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueki Y, Le Y-Z, Chollangi S, Muller W, Ash JD. Preconditioning-induced protection of photoreceptors requires activation of the signal-transducing receptor gp130 in photoreceptors. Proc Natl Acad Sci USA. 2009;106:21389–94. doi: 10.1073/pnas.0906156106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiersch M, Lange C, Joly S, Heynen S, Le YZ, Samardzija M, Grimm C. Retinal neuroprotection by hypoxic preconditioning is independent of hypoxia-inducible factor-1 alpha expression in photoreceptors. Eur J Neurosci. 2009;29:2291–302. doi: 10.1111/j.1460-9568.2009.06781.x. [DOI] [PubMed] [Google Scholar]