

Background: UDP-glucose dehydrogenase (UGDH) mutants were engineered to perturb hexamer:dimer quaternary structure equilibrium.
Results: Dimeric species of UGDH have reduced activity in vitro and in supporting hyaluronan production by cultured cells.
Conclusion: Only dynamic UGDH hexamers support robust cellular function.
Significance: Manipulation of UGDH activity by hexamer stabilization may offer new therapeutic options in cancer and other pathologies.
Keywords: Dehydrogenase, Enzyme Catalysis, Glycobiology, Hyaluronate, Prostate Cancer, Protein Conformation, Protein Dynamics, Cardiac Defects, Human UGDH, Hyaluronan
Abstract
UDP-glucose dehydrogenase (UGDH) provides precursors for steroid elimination, hyaluronan production, and glycosaminoglycan synthesis. The wild-type UGDH enzyme purifies in a hexamer-dimer equilibrium and transiently undergoes dynamic motion that exposes the dimer-dimer interface during catalysis. In the current study we created and characterized point mutations that yielded exclusively dimeric species (obligate dimer, T325D), dimeric species that could be induced to form hexamers in the ternary complex with substrate and cofactor (T325A), and a previously described exclusively hexameric species (UGDHΔ132) to investigate the role of quaternary structure in regulation of the enzyme. Characterization of the purified enzymes revealed a significant decrease in the enzymatic activity of the obligate dimer and hexamer mutants. Kinetic analysis of wild-type UGDH and the inducible hexamer, T325A, showed that upon increasing enzyme concentration, which favors the hexameric species, activity was modestly decreased and exhibited cooperativity. In contrast, cooperative kinetic behavior was not observed in the obligate dimer, T325D. These observations suggest that the regulation of the quaternary assembly of the enzyme is essential for optimal activity and allosteric regulation. Comparison of kinetic and thermal stability parameters revealed structurally dependent properties consistent with a role for controlled assembly and disassembly of the hexamer in the regulation of UGDH. Finally, both T325A and T325D mutants were significantly less efficient in promoting downstream hyaluronan production by HEK293 cells. These data support a model that requires an operational dimer-hexamer equilibrium to function efficiently and preserve regulated activity in the cell.
Introduction
UDP-glucuronate is an essential cellular metabolite that serves as the precursor for multiple diverse and compartmentalized processes. Notably, its production is critical in tissues such as liver, prostate, and breast for endoplasmic reticulum-localized phase II detoxification of lipophilic hormones and xenobiotics by glucuronidation (1). In adult tissues, in wound healing, and during development, high levels of UDP-glucuronate are used to synthesize extracellular glycosaminoglycans such as heparan sulfate and hyaluronan (HA)4 (2), and the decarboxylation of UDP-glucuronate to UDP-xylose initiates proteoglycan production in the Golgi (3). In embryonic development, temporal and spatial elevation in UDP-glucuronate synthesis are indispensible for rapid extrusion of HA to the extracellular space to effect the dramatic morphological changes that generate anatomical structures such as cardiac valves (4, 5).
UDP-glucuronate is exclusively synthesized by UDP-glucose dehydrogenase (UGDH), which is an essential enzyme for circulatory system development in multiple organisms from nematodes to humans (4, 6–11). In prostate tumor cell lines, UGDH is a constitutively expressed but androgen-stimulated gene product, up-regulation of which can specifically drive excess steroid elimination through glucuronidation in hormone-dependent cells (12). In the developing heart, UGDH point mutations that alter the integrity of quaternary structure, normally hexameric, fail to support maximal HA production that is needed for cardiac valve formation (4). Clinical and cellular consequences associated with UGDH defective function implicate stable but dynamic association of subunits and suggest useful therapeutic applications would focus on sustaining robust UGDH activity.
Several reports have provided structural and enzymologic insights into UGDH function in multiple species (6, 7, 10, 11, 13–26). In all cases the enzyme oxidizes UDP-glucose through two NAD+-dependent electron transfers. Of particular interest to studies relating quaternary structure to cellular function is that bacterial UGDH adopts a strictly dimeric conformation in the presence or absence of its substrate and cofactor (27), whereas higher eukaryotic UGDH orthologues purify in a hexamer-dimer equilibrium that is stabilized to hexameric association under conditions representative of the cellular environment (4, 11, 16, 28). Kinetic data comparing wild-type UGDH to an obligate (inactive) hexamer mutant in which residue 132 was deleted (25) together with the interesting observation of an “open” conformation of the wild-type enzyme observed in co-crystals with the potent UGDH inhibitor UDP-xylose suggest that the transient capacity to dissociate and reorganize the hydrogen bond network at the interface between dimeric units is an important element of the normal catalytic cycle.
For the current study we sought to determine the effects of quaternary structure on intrinsic and cellular functional properties of UGDH. Crystallographic data are available for several wild-type or point mutant UGDH forms (PDB codes 3ITK (29) and 3KHU (30)), including an apo structure as well as ternary forms with each permutation of the abortive complex (NAD+ cofactor and UDP-glucuronate product (PDB code 2QG4); NADH cofactor and UDP-glucose substrate (PDB code 2Q3E) (29)). These structural data provide a high resolution view of the non-covalent bonding network that sustains the subunit interface in the context of the hexamer (Fig. 1). We used these coordinates to rationally generate point mutations expected to perturb the interface and alter the hexamer-dimer equilibrium without impacting catalytic activity directly. With these tools, we additionally evaluated whether the hexameric conformation is required for cellular function and how its integrity relates to steady state activity. Our results demonstrate for the first time that the UGDH hexamer is more competent to support high levels of HA production than the dimer and that intrinsic stability of the enzyme is conformationally dependent.
FIGURE 1.
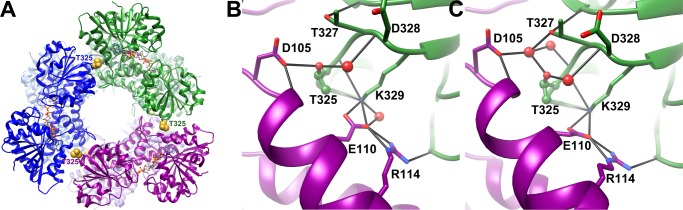
Overall diagram of UGDH and the molecular interactions at the subunit interface. A, the human hexameric enzyme is depicted in a ribbon representation of the ternary complex. Active sites contain the substrate UDP-glucose and the reduced cofactor NADH, which are shown in ball and stick form. Residue Thr-325, which was mutagenized in this study, is shown in space-filling form and colored gold. Individual subunits are colored in dimeric pairs of dark/light blue, dark/light green, and dark/light purple. The close-up views show proximal ordered water molecules and hydrogen-bonded amino acid side chains that interact to maintain integrity of the subunit interface. Black lines indicate hydrogen bonds, nitrogen atoms are colored blue, and oxygen atoms are red. Ordered water molecules are depicted as red spheres. B, apoprotein interface, PDB 3ITK. C, abortive ternary complex interface; PDB 2Q3E. Figure representations were made in Chimera (31).
EXPERIMENTAL PROCEDURES
Generation and Purification of UGDH Point Mutants
Molecular graphics and analyses were performed with the UCSF Chimera package (31), developed by the National Institutes of Health-supported Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. Point mutants of human UGDH were generated from the wild-type construct, UGDH-pET28a (for protein purification), using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol. Sequences were verified by Eurofins MWG Operon (Huntsville, AL). Wild-type UGDH and point mutants were expressed in Escherichia coli strain Rosetta2(DE3)pLysS (EMD Biosciences, Inc., San Diego, CA), induced overnight at 18 °C, and purified as N-terminal His6 fusions essentially as previously described (11). After nickel affinity chromatography, proteins were dialyzed against 20 mm Tris-HCl, pH 7.4, and 1 mm DTT. Protein was stored at a concentration of 2 mg/ml.
Enzymatic Activity Measurement and Kinetic Characterizations
Activity of the point mutants was assayed by monitoring the change in absorbance at 340 nm that accompanies reduction of NAD+ to NADH as reported previously (11). For standard measurement of enzymatic activity, the assay was done in a 96-well plate format at room temperature for 5 min in 0.1 m sodium phosphate buffer, pH 7.4. T325A and T325D mutants exhibited saturable kinetics, whereas the activity of the Δ132 mutant was found to be negligible. Michaelis constants Km and Vmax, for UDP-glucose and NAD+, were determined independently by holding NAD+ constant at 10 mm and varying UDP-glucose from 0 to 1 mm. Similarly, NAD+ measurements were made by holding UDP-glucose constant at 1 mm and varying NAD+ from 0 to 10 mm. Specific activities were calculated from absorbance values using the molar extinction coefficient for NADH of 6220 m−1cm−1. Triplicate determinations for each concentration increment were plotted with PRISM (GraphPad Software, Inc., San Diego, CA). Km and Vmax were calculated by fitting the data to the Michaelis-Menten equation and assuming a single binding site per subunit for substrate and cofactor.
Stability of Enzyme Activity in Vitro
Wild-type and mutant UGDH (1 mg/ml each in 0.1 m sodium phosphate, pH 7.4) was incubated in the absence and presence of saturating NAD+ (10 mm) and UDP-glucuronate (1 mm). After a 1-min equilibration at 37 °C, aliquots were assayed for initial activity as above with saturating NAD+ and UDP-glucose and a final enzyme concentration of 670 nm. For each time point, absorbances were measured in triplicate divided by the average absorbance at time 0 and plotted as the percent relative activity over time. Using PRISM software, the graphs were fitted to the one phase exponential decay equation to calculate half-lives at 37 °C.
Molecular Mass Determination
Oligomeric state of the apoenzyme was determined by size exclusion chromatography as previously described (11). Each sample was precleared by centrifugation before loading. To determine the effect of substrate and cofactor inclusion on oligomeric state, purified recombinant UGDH wild-type and point mutants were individually loaded on a Superdex 200 HR 10/300 column (GE Healthcare) using a 250-μl loop and separated by FPLC in 1× PBS in the presence of 1 mm UDP-glucose and/or 5 mm NAD+ at a flow rate of 0.5 ml/min. Substrate and cofactor alone or together were included in each 250-μl load sample at the same final concentrations. Because of the interference from cofactor in UV absorbance readings from the column fractions of enzyme complexes, protein content of these samples was measured at 595 nm upon the addition of Bradford reagent. The standards used were: thyroglobulin, 699 kDa; ferritin, 440 kDa; albumin, 67 kDa; β lactoglobulin, 35 kDa; RNase A, 13.7 kDa. Resolution was sufficient to calculate molecular masses unambiguously in multiples of 57 kDa (the calculated molecular mass of monomeric His-tagged UGDH).
Thermal Stability Measurement
Purified recombinant UGDH wild-type and point mutants were analyzed using a modified version of the Thermofluor Stability Assay as described by Ericsson et al. (32). Solutions of the wild-type and mutant apoenzymes (∼5–20 μg of protein) were prepared in 1× PBS containing Sypro Orange dye (Invitrogen, 1:500 dilution). Samples containing substrate and/or cofactor were prepared in the same manner with UDP-glucose and NAD+ added to a final concentration of 1 and 5 mm, respectively. All samples were handled at room temperature throughout the preparation and transferred to an iCycler MyiQ Thermocycler (Bio-Rad) for analysis. The assay protocol was executed with the block initially at 20 °C. After the addition of samples, the temperature was raised over a period of 76 min from 20 to 95 °C in 0.5 °C increments. The change in fluorescence was monitored for each sample at an excitation and emission of 490 and 575 nm, respectively. Transitions representing the melting temperature, Tm, were obtained from the derivative of fluorescence with respect to temperature for at least 3–10 replicates and plotted as the mean ± S.D.
Proteolytic Sensitivity of UGDH
Purified recombinant UGDH wild-type and point mutants (10 μg protein) were digested with 10 ng of trypsin in 1× PBS for 2.5 h in the absence or presence of 1 mm UDP-glucose, 1 mm UDP-glucuronate, 5 mm NAD+, 5 mm NADH, or abortive ternary complexes (at saturating conditions). Samples were analyzed by reducing SDS-PAGE.
Cell Culture and Transfection
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (Fisher) and seeded in a 6-well plate 24 h before transfection. Cells were transiently co-transfected at 80% confluence using FuGENE HD (Roche Applied Science) with hyaluronan synthase 3 (HAS3) and wild-type or mutant UGDH constructs in the pIRES2-EGFP eukaryotic expression vector. Cells were harvested 48 h post-transfection in 1× radioimmune precipitation assay (RIPA) buffer and incubated on ice for 10 min. The whole cell lysate was separated by centrifugation.
HA Quantification and Western Analysis
HA content of cell culture-conditioned media was determined by a competitive binding assay (23). Human umbilical cord HA (Sigma) was coupled to Covalink 96-well plates at 100 μg/ml in 1× PBS in the presence of 25 μg of SulfoNHS/EDC. Plates were rocked for 1.5 h at room temperature then washed twice with Pierce SuperBlock before sample incubation. Serial dilutions of cell culture media were incubated with biotinylated HA-binding protein (bHABP, Calbiochem) at a final concentration of 1 μg/ml. Standards and samples were incubated in the HA-precoated wells at room temperature overnight. Excess HA was removed with 4 washes of PBS, 0.05% Tween 20. The plate was developed using an avidin-biotin HRP system (Vectastain) with 3,3′,5,5′-tetramethylbenzidine as substrate, and absorbance was measured at 650 nm. HA concentration was interpolated from a standard curve and normalized to total protein. Western analysis of whole transfected cell lysates was performed as previously described (24). Primary antibodies were used at a dilution of 1:1,000 for α-UGDH and α-FLAG and 1:750,000 for α-tubulin.
RESULTS
Kinetic Comparison of Wild-type and Mutant UGDH
To determine the role of quaternary structure in the activity and properties of UGDH, we designed, expressed, and purified mutant species of the enzyme that were expected to exhibit disruption from primarily hexameric to exclusively dimeric association. Initially, we selected glutamate 110 and threonine 325 because of their pivotal functions in dimer-dimer association as shown in the ribbon diagram of the ternary complex structure (Fig. 1). The E110A mutant, although dimeric in the apo form, exhibited only ∼50% reduction in Vmax, but was highly unstable in solution and in cultured cells so it could not be evaluated unambiguously. The carboxylate side chain of this residue on one subunit forms salt bridges with two positively charged moieties, one of which is on the adjacent subunit (Lys-329) and one of which stabilizes the same helix that contains Glu-110 (Arg-114, Fig. 1). Therefore, the loss of Glu-110 not only disrupts a direct series of interactions between subunits but also within individual subunits, as this charge likely screens electrostatic repulsion between the two positive charges. This may account for its instability. Instead, we pursued the other highly networked interface residue, Thr-325.
In the apo form Thr-325 directly forms a hydrogen bond with Asp-105 of the opposite subunit (Fig. 1B). A similar view of the interface in the abortive ternary complex structure illustrates a conformational change that reorganizes the hydrogen bond network such that Thr-325 and Asp-105 now coordinate through a highly ordered water molecule (Fig. 1C). Thr-325 residue was altered to an alanine because it would be incapable of hydrogen bonding and to aspartate, which would introduce a new formal negative charge at the interface. Both mutants expressed abundant soluble protein, similar to the wild-type enzyme, and exhibited saturable kinetics. Data were fitted to the Michaelis-Menten equation to calculate steady state kinetic constants and evaluate potential cooperativity. Wild-type UGDH and the T325A mutant exhibited similar Vmax and Km values with respect to both substrate and cofactor when examined under standard assay conditions (Table 1). However, the T325D mutant exhibited an ∼5-fold reduced Vmax relative to the wild-type UGDH. Interestingly, when the wild-type and T325A mutant enzyme were assayed at higher concentrations, previously reported to yield sufficient NADH to inhibit activity (Ki for NADH = 27 μm (21)), the turnover and catalytic efficiency of both were significantly reduced (Fig. 2). In contrast, the efficiency of the T325D mutant was not altered over this enzyme concentration range despite accumulation of NADH to the previously reported inhibitory levels and an unaltered apparent Km for NAD+ relative to the wild-type enzyme. These results suggest the perturbation of the dimer interface by charge introduction eliminates negative cooperativity and product inhibition.
TABLE 1.
Summary of wild type and mutant UGDH kinetic constants
ND, not detectable.
| Enzyme | UDP-glucose |
NAD+ |
||
|---|---|---|---|---|
| Vmax | Km | Vmax | Km | |
| nmol/min/mg | μm | nmol/min/mg | μm | |
| Wild type | 313 ± 8 | 34 ± 3 | 482 ± 15 | 646 ± 73 |
| T325A | 324 ± 8 | 48 ± 4 | 274 ± 9 | 897 ± 99 |
| T325D | 65 ± 1 | 15 ± 1 | 76 ± 1 | 1084 ± 66 |
| Δ132 | ND | ND | ND | ND |
FIGURE 2.
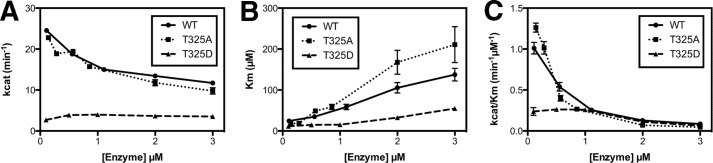
Steady state kinetic analysis reveals selectively reduced catalytic activity and relief from product inhibition by perturbation of threonine 325. Purified recombinant human UGDH wild-type and Thr-325 mutants were assayed for concentration dependence of turnover number (kcat) (A), Km (B), and catalytic efficiency (kcat/Km) (C) with respect to UDP-glucose in the presence of saturating NAD+.
Half-life of UGDH Catalytic Activity in Vitro Is Reduced by Mutations at the Dimer Interface
We previously observed a loss of stability in the catalytic activity of UGDH when quaternary structure was disrupted by mutations that were found in human congenital heart defects. However, we were not able to assert unambiguously that the loss of activity was attributable to the effects of those mutations on UGDH quaternary structure. The Thr-325 mutants afforded this opportunity, as they were rationally designed to disrupt hexamer assembly without directly affecting the integrity of the active site. In addition, we had not tested the effect of substrate and cofactor concentrations that simulated cellular conditions on the overall duration of the enzyme catalytic competence. Purified wild-type or mutant UGDH was incubated with or without NAD+ and UDP-glucuronate, an abortive ternary complex, and activity was monitored over time. Formation of the wild-type ternary complex extended the half-life of enzymatic activity ≈30%, from 59 to 88 h (Fig. 3A). The activity of the T325A mutant was the most labile, with a half-life of only 24 h that was not extended significantly by substrate and cofactor addition (Fig. 3B). Interestingly, UGDH T325D retained its activity similarly to the wild-type enzyme but did not exhibit increased stability in the abortive ternary complex (Fig. 3C).
FIGURE 3.
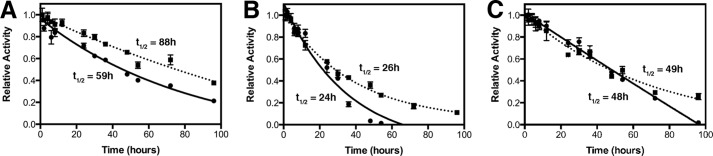
Half-life of wild-type but not mutant UGDH activity in vitro is stabilized by formation of a ternary complex. Purified proteins were placed at 37 °C in the absence (apo, circles, solid line) or presence (holo, squares, dotted line) of UDP-glucuronate and NAD+. At the indicated time points an aliquot was removed, and activity was assayed by the addition of the enzyme to phosphate buffer containing saturating concentrations of NAD+ and UDP-glucose. Values plotted are the mean ± S.D. for triplicate determinations. Absolute turnover was measured as the change in NADH absorbance at 340 nm in a 1-min assay. Absorbance values were normalized to the value at time 0 to illustrate the rate of activity loss. Half-lives were calculated from curve fits of the data from triplicate assays.
UGDH Quaternary Structure Disrupted by Mutagenesis of Thr-325 Is Differentially Restored by Ternary Complex Formation
We directly assessed the quaternary structure of the Thr-325 mutants by size exclusion chromatography. As expected, the wild-type enzyme purified in a hexamer-dimer equilibrium that was stabilized to the hexamer conformation by inclusion of saturating NAD+ cofactor and UDP-glucose substrate in the column running buffer. Moreover, we further found that the presence of either substrate or cofactor alone was also able to shift the eluted peak to the higher mass profile of the hexamer (Fig. 4). As a control, we generated an additional point mutant, Δ132, in which valine 132 was deleted and which had been reported to be exclusively hexameric (25). Consistent with the prior results, this mutant had undetectable activity (Table 1) and eluted at the molecular mass of the hexamer irrespective of substrate or cofactor addition. In contrast, both T325A and T325D mutants were exclusively dimeric in the apo form, indicating the loss of the neutral hydrogen bond network formed by Thr-325 was sufficient to disrupt the dimer-dimer association interface. However, the inclusion of either substrate or cofactor alone with UGDH T325A partially shifted the dimer equilibrium to higher molecular mass complexes, and the addition of both to form the ternary complex fully shifted the peak to the molecular mass of the hexamer. Neither the presence of substrate or cofactor alone nor the inclusion of both together effected any change in the apparent molecular mass of the T325D mutant, which retained its dimeric structure. Thus, we termed the UGDH mutant Δ132 an obligate hexamer, T325A an inducible hexamer, and T325D an obligate dimer.
FIGURE 4.

Quaternary structure of wild-type UGDH and T325A is stabilized from dimeric to hexameric by substrate and cofactor, whereas T325D remains an obligatory dimer. Purified wild-type and mutant UGDH was fractionated by gel filtration FPLC in the absence (dark solid line) or presence of NAD+ alone (gray hatched line), UDP-glucose alone (gray dotted line), or both (dark hatched line). Absorbance was monitored at 280 nm for the apoproteins (left ordinate axis) and 595 nm for the holoproteins (right ordinate axis). Size determination of each peak was made by comparison to molecular weight standards. Representative traces are plotted for wild-type UGDH, Δ132, T325A, and T325D. On each plot the peaks corresponding to hexamer and dimer are labeled H and D, respectively.
Intrinsic Thermal Stability and Resistance to Proteolysis Are Reduced by Inability of UGDH to Hexamerize
With these tools in hand, we sought to assess whether the structural properties of the mutants were altered so that we could relate the intrinsic characteristics to the activity of the enzymes in vitro and in vivo and draw conclusions about the importance of hexameric conformation to cellular function. A thermal denaturation assay was previously used to show that the melting temperature (Tm) of the UGDH apoenzyme was increased by 20 °C in the presence of saturating substrate and cofactor to form the holoenzyme (4, 29). We extended our prior characterization of the wild-type enzyme using this assay in the presence of either reduced or oxidized cofactor alone, substrate or product alone, and each permutation of the ternary complex (e.g.; UDP-glucose and NAD+, UDP-glucuronate and NAD+, etc.). As expected, the ternary complexes exhibited ≈20 °C higher Tm values relative to the wild-type UGDH apoenzyme (Fig. 5, solid dark bars compared with the white bars). However, adding either cofactor, substrate, or product alone also increased thermal stability, although to a lesser extent (≈10 °C, Fig. 5, patterned bars). Consistent with the maximal thermal stabilization being attributable to the ability to hexamerize, the UGDH Δ132 mutant had a Tm equivalent to the wild-type ternary complex with or without any additives. The Tm of the UGDH T325A mutant was not altered by the addition of substrate or cofactor alone but was increased by ≈20 °C in each of the ternary complexes, which would be predicted by its apparent hexameric conformation in the gel filtration assay. The T325D mutant, also as predicted, was not stabilized by substrate or cofactor alone in the thermal denaturation assay, but the Tm increased ≈10 °C in the ternary complexes, suggesting there is some intrinsic stability conferred even to the dimeric species that is independent of dimer-dimer association. However, this demonstrates that the hexameric wild-type UGDH enzyme has greater potential to be stabilized in the presence of its metabolites than does the obligate dimer mutant.
FIGURE 5.

UGDH is thermally stabilized by the addition of substrate and cofactor, but thermal stability is compromised by mutations that occur at the dimer interface. Purified wild-type UGDH and point mutants were thermally denatured in the presence of Sypro Orange by increasing temperature from 20 °C to 95 °C in 0.5 °C increments. The change in fluorescence with respect to temperature was used to calculate a melting point (Tm) for each enzyme species in the absence (white bars) or presence of 5 mm NAD+ or NADH, 1 mm UDP-glucose or UDP-glucuronate, and each of three ternary complex combinations, as indicated in the legend. Mean ± S.D. is plotted for the Tm of each enzyme.
As an independent measure of structural integrity in vitro, we evaluated susceptibility of each wild-type and mutant UGDH species to limited trypsin proteolysis. We chose conditions in which the wild-type enzyme was almost completely degraded in the apo form (Fig. 6, top, first two lanes). As observed in the thermal denaturation assay, the addition of substrate, product, reduced or oxidized cofactor, or each permutation of ternary complex components fully protected the wild-type enzyme against proteolysis. Similarly, the obligate hexamer UGDH Δ132 was minimally susceptible to trypsinization in all conditions. The inducible hexamer, T325A, was fully degraded in the apo form and in the presence of substrate alone. The addition of NAD+ stabilized the enzyme fully, and either NADH or UDP-glucuronate partially protected against proteolysis, whereas the ternary complexes were almost completely undigested. These results are consistent with the T325A mutant ability to adopt a hexameric conformation that is thermally and proteolytically stabilized to an increasing degree upon cofactor and substrate addition. Not surprisingly, the T325D obligate dimer mutant was fully degraded by trypsin in the apo form and in the presence of substrate, product, or cofactor individually. Consistent with an increase in its intrinsic subunit stability but without the ability to hexamerize, the T325D mutant experienced only partial protection against trypsin digestion in the ternary complex.
FIGURE 6.
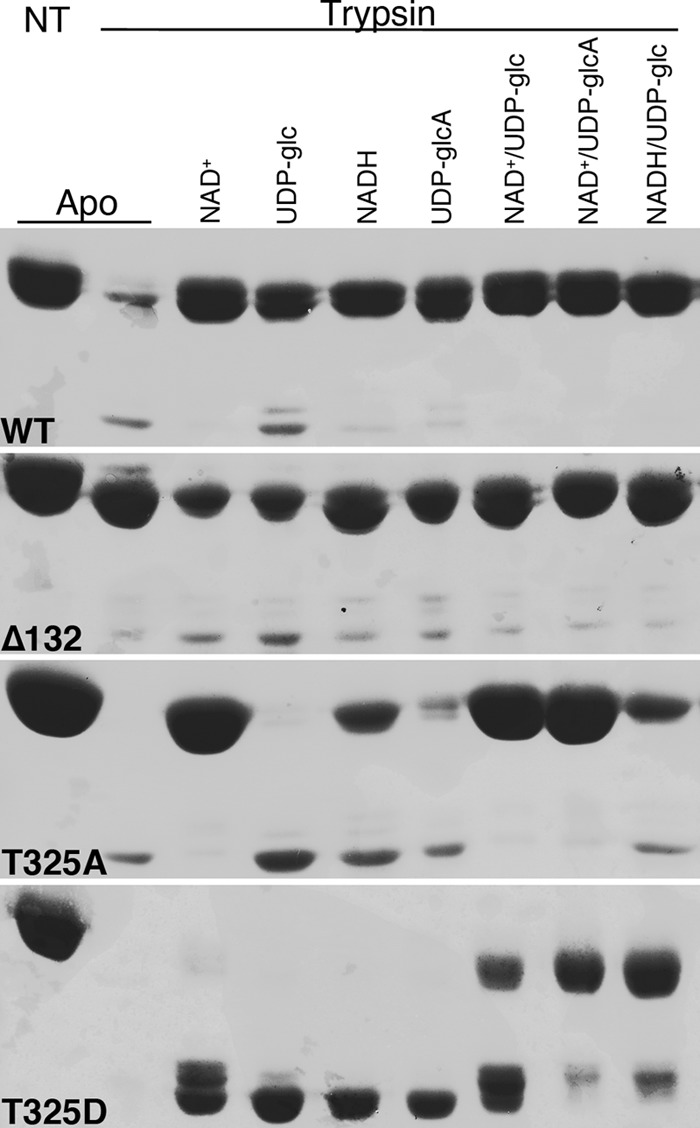
Differential sensitivity to limited proteolysis reveals altered conformation conferred to the mutant UGDH enzymes by impaired ability to assemble in hexamers. Purified wild-type and mutant UGDH was prepared in the absence or presence of the indicated substrate and/or cofactor combinations and then treated with trypsin and analyzed by SDS-PAGE. Results are representative of at least three different experimental replicates. NT, no trypsin control.
Cellular Function of UGDH Is Dependent on Hexameric Stability
Though cellular ratios of NAD+/NADH and UDP-glucose/UDP-glucuronate are likely to undergo considerable flux depending upon local or systemic nutrient and oxygen status, the pool of NAD cofactors in a given cell type is relatively fixed. Therefore, we compared the effects of obligate and inducible quaternary structure on the ability to provide precursors for HA synthesis in a cellular context. HEK293 cells, which express very low endogenous UGDH, and HAS, the enzyme that uses UDP-glucuronate to produce HA, were transfected with eukaryotic expression constructs for wild-type or mutant UGDH with or without HAS3. Equivalent steady state expression of all UGDH species was observed in each transfection condition (Fig. 7, upper panel). HA was quantified in the conditioned media of each transfectant and normalized to total protein in the transfected cell lysate analyzed by Western blots in the upper panels. Concurrent expression of wild-type UGDH and HAS3 yielded ≈3-fold more HA production than transfection with HAS3 alone, as we have previously observed (12). Strikingly, expression of the T325A mutant with HAS3 was not fully effective in promoting HA synthesis, yielding ∼60% less HA than the wild-type cotransfection. The UGDH T325D mutant did not support HA production in excess of HAS3 alone, and coexpression of HAS3 with the obligate hexamer Δ132 modestly but significantly suppressed HA production relative to HAS3 alone. Thus, the cellular activity of UGDH is maximized when its conformational sampling statistically favors the hexameric species but permits dynamic movements in quaternary structure. These results support a model in which the hexamer-dimer ratio of UGDH senses flux in the presence of cellular cofactor, substrate, and product.
FIGURE 7.
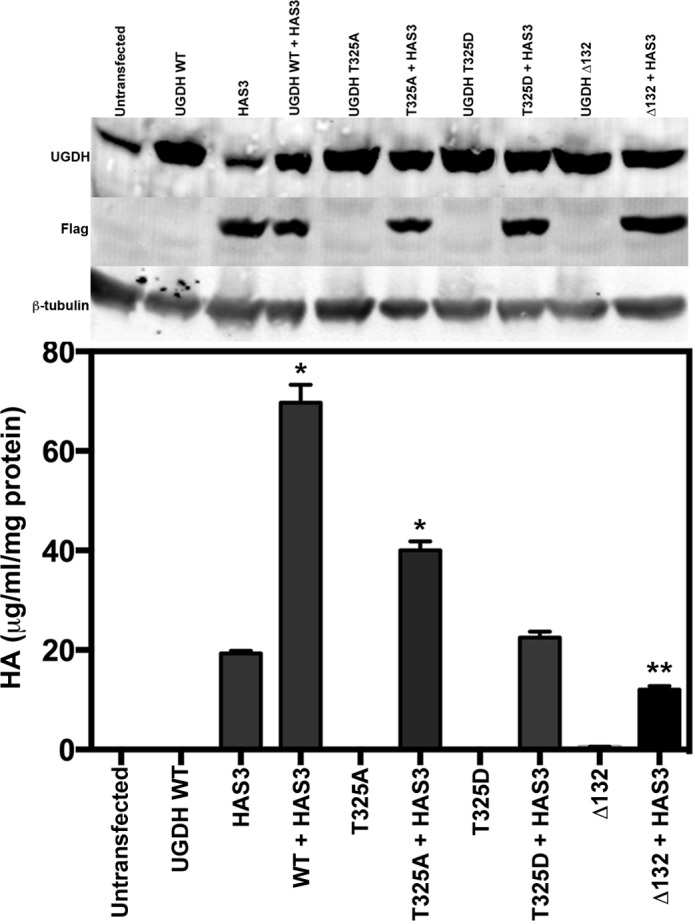
Loss of UGDH intrinsic activity impacts cellular HA production. HEK293 cells were cotransfected with constructs encoding GFP (vector control) and human HAS3, or GFP and wild-type or mutant UGDH, or with HAS3 and each UGDH construct. After 48 h, conditioned media were analyzed for HA content, and cell lysates were analyzed by Western blot for UGDH, HAS3 (FLAG), or tubulin (control). HA was calculated in units of mass per volume of conditioned medium and normalized to protein content (in mg) of the whole cell lysates. The mean ± S.E. is plotted for quadruplicate measurements repeated three times. Statistical significance was evaluated by Student's t test; *, p < 0.01; **, p < 0.05, relative to HAS3 only. Western data are shown for the relevant lysates.
DISCUSSION
UGDH targeted disruption and loss of function polymorphisms that interfere with the enzyme quaternary structure and cellular stability have been implicated in cardiac malformation in multiple organisms (4, 5, 33, 34). Moreover, the androgen-stimulated expression of UGDH is important for prostate epithelial androgen solubilization and elimination. Our goal was to engineer obligatory forms of the enzyme that represented its two most stable sampled oligomeric states while minimally disrupting overall or active site stability to determine the mechanistic function of this hexamer-dimer equilibrium in vitro and in a cellular context. The engineered UGDH mutants allowed us to determine that the hexameric enzyme has only modestly greater maximal catalytic activity than the dimer but is significantly more responsive to structural stabilization and activation by substrate and cofactor and capable of supporting greater output of the extracellular matrix product HA. Importantly, however, the dimeric mutant retains significant activity and is considerably less sensitive to product inhibition by reduced cofactor.
Examination of the dimer-dimer subunit interface revealed an extensive network of charge interactions and hydrogen bonding that coordinately stabilize the hexamer in the presence and absence of its cofactor or substrate. Besides Thr-325, we had also identified a glutamate residue, Glu-110, as a critical contributor to non-covalent interactions. However, the intrinsic instability of the E110A mutant made it unsuitable for subsequent cellular studies. Thr-325 is ideally positioned to accommodate reorganization of the interface hydrogen bonds that occurs as the active site shifts to form binary and ternary complexes and exchange cofactors during the catalytic cycle. Although elimination of the hydroxyl by substituting alanine for threonine was expected to prevent dimer-dimer association, stability of the hexamer in the ternary complex indicates that there was sufficient redundancy and flexibility in the network to prevent loss of activity. Placement of a negative charge was necessary and sufficient to prevent subunit association, as the new negative charge was proximal to both the Asp-105 and Glu-110 side chains of the adjacent subunit, which probably was electrostatically repulsive.
It was important to characterize the mutants sufficiently to ascertain that an obligate dimer had been achieved, as we had previous mutants that purified in a dimeric form as an apoprotein, and later we discovered that although they had activity, they also had restored hexameric structure in the active ternary complex. K339A purified as a dimer but had been designed as a substrate binding mutant. Consistent with this, its Vmax was identical to the wild-type enzyme, but the Km for UDP-glucose was increased several hundred-fold. We interpreted this to mean that the dimeric form of the enzyme was equally active, but we did not characterize the solution oligomerization state of the ternary complex. In contrast, a previously reported mutant, K94E, was described as a dimer with 100-fold reduced activity and was used to argue that the dimeric species was inactive (26). The Km for both substrate and cofactor was also elevated 1–2 orders of magnitude, so the mutant was severely compromised, but the conformation of the ternary complex was also not verified. Inspection of the positioning of Lys-94 at the dimer-dimer interface reveals that its side chain directly forms hydrogen bonds with the Glu-360 side chain and the Met-98 backbone carbonyl of the opposite subunit and that, within its own subunit, its backbone amide and carbonyl hydrogen bond with the backbone carbonyl of Ala-103 and the backbone amide of Gly-101, respectively. Significantly increased disorder is evident for residues 88–110. Thus, residue Lys-94 is critically involved in stabilization of a major element of secondary structure that directly communicates with the active site. For these reasons the K94E dimer did not unambiguously address the relation of UGDH quaternary structure to its function in vitro and is also not suitable for studies in vivo. Our finding that substrate, product, and/or cofactor addition to either the wild-type or T325A mutant UGDH could partially or completely shift the structural equilibrium to the hexameric species led us to examine multiple features of the enzyme that could report on its structural integrity in reaction conditions. In particular, we wanted to identify a mutant that was minimally perturbed on an overall structural level other than being unable to oligomerize. In doing so we affirmed that the dimeric species had reduced catalytic activity but not as a consequence of altered substrate or cofactor affinity in the context of the reaction.
Gel filtration shows UDP-glucose alone and NAD+ alone stabilize the wild-type hexamer. However, the T325A mutant was only induced to form a hexamer if both substrate and cofactor were present. In addition, induction of T325A hexamer formation required concentrations of substrate and cofactor that were saturating for wild-type UGDH and for maximal catalytic rate in the T325A mutant. In these conditions, the hexameric conformation of T325A is then fully stabilized. In contrast, the obligate dimer remains dimeric in all conditions. When we compare with thermal stability, there is a strong increase in melting temperature of the wild type when substrate, product, or cofactor are added alone and an additional increase when either a productive or an abortive ternary complex is formed. An interpretation of increased stability in the hexameric form is further supported by the observed protection from proteolysis, with all of the binary and ternary permutations tested as well as in the obligate hexameric mutant. Results of proteolysis and thermal denaturation further support only a partial restoration in complex formation and its resultant increase in stability for the T325A mutant as indicated in gel filtration and suggest greater intrinsic structural stability is conferred by cofactor binding than by UDP-glucose binding. Also consistently, the ternary complexes of T325A and wild-type UGDH are equally stable when assessed both by thermal denaturation and limited proteolysis. The obligate dimer shows no intrinsic increase in thermal or proteolytic resistance except in the ternary complex, which suggests that approximately half the increase in quantified stability parameters is due to stability of the dimer and half is the result of further ability to hexamerize. Moreover, because the UDP-glucose substrate and UDP-glucuronate product minimally protected the T325A mutant against proteolysis, the stability conferred in the dimeric ternary complex likely derives from the binding of cofactor after substrate association.
Previous kinetic data have shown that substrate binds first, with the cofactor more surface-exposed to facilitate exchange during catalysis (30). This is consistent with the above proteolytic results and together with the observed product inhibition in the hexameric form suggests that the availability of the substrate may be a key regulatory condition. The cooperative rate increase of the enzyme in the presence of increasing substrate is evidence that a conformational shift may precede achievement of full catalytic potential. In addition, it has been suggested that the dynamic hexamer is the relevant catalytic species on the basis of relatively low activity in both the K94E dimer and the Δ132 obligate hexamer. Our results with a single substitution at position Thr-325 are consistent with lower activity of the obligate dimer but with a more modest reduction in turnover than in K94E. This retention of significant catalytic activity in the less perturbed T325D dimeric mutant suggests the dimeric species may also be catalytically relevant in some cellular conditions, particularly in the presence of product inhibition, but does not negate the more significant role of the hexamer during maximum cellular demand.
The wild-type enzyme overexpressed in cells transfected with HA synthase, which exhibit a high demand for HA production, generates a 3-fold increase in achievable steady state HA levels. In contrast, the T325D obligate dimeric and Δ132 obligate hexameric enzyme species were unable to support the acute increase in cellular demand for HA precursors to any detectable extent, which suggests that in the cellular context neither a dimeric nor a hexameric conformation can be exclusively considered the catalytically relevant species. This is not surprising, as the transiently dissociable interface of the wild-type enzyme is likely essential for cofactor exchange between the two oxidation events that occur in a single turnover. However, it is interesting that the T325A mutant has equivalent steady state kinetic parameters to the wild-type UGDH enzyme and a comparable ternary complex stability but demonstrates reduced support for HA production. We attribute this to the increased lability of the dimer-dimer interface in cellular conditions that normally support activity, as the individual substrate/cofactor dissociation events leave the subunits more susceptible to loss of maximally active hexameric conformation. The equilibrium between hexamer and dimer presumably does not imply full dissociation in the cell but rather involves a spectrum of enzyme species with transiently exposed regions of the dimer-dimer interface. At cellular levels of substrate, cofactor, and product, the exposure of the wild-type enzyme interface is likely to be low frequency, since these individual components stabilize all properties we measured for the hexameric conformation. Because the T325A mutant is not fully stabilized to a hexamer when only one of these components is present and is minimally stabilized if any component is subsaturating, this mutant may be considerably less functional in the cell as a result of increased dissociation. One additional explanation may be that the subunit interfaces of the intact hexamer versus the hexamer with a transiently dissociated but catalytically engaged dimer interface may interact differently with other cellular regulatory factors. Such interactions could covalently or non-covalently modify the subunit interface to favor or discourage subunit association and thereby alter activity of the enzyme.
Acknowledgments
Chimera was supported by NIGMS, National Institutes of Health Grant P41-GM103311 (to the University of California at San Francisco).
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM077289 (to J. J. B.) and R01 CA165574 and P20 GM103489 (to M. A. S.).
- HA
- hyaluronan
- UGDH
- UDP-glucose dehydrogenase
- HAS3
- hyaluronan synthase 3.
REFERENCES
- 1. Tukey R. H., Strassburg C. P. (2000) Human UDP-glucuronosyltransferases. Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 40, 581–616 [DOI] [PubMed] [Google Scholar]
- 2. Fraser J. R., Laurent T. C., Laurent U. B. (1997) Hyaluronan. Its nature, distribution, functions, and turnover. J. Intern. Med. 242, 27–33 [DOI] [PubMed] [Google Scholar]
- 3. Prydz K., Dalen K. T. (2000) Synthesis and sorting of proteoglycans. J. Cell Sci. 113, 193–205 [DOI] [PubMed] [Google Scholar]
- 4. Hyde A. S., Farmer E. L., Easley K. E., van Lammeren K., Christoffels V. M., Barycki J. J., Bakkers J., Simpson M. A. (2012) UDP-glucose dehydrogenase polymorphisms from patients with congenital heart valve defects disrupt enzyme stability and quaternary assembly. J. Biol. Chem. 287, 32708–32716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walsh E. C., Stainier D. Y. (2001) UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science 293, 1670–1673 [DOI] [PubMed] [Google Scholar]
- 6. Campbell R. E., Sala R. F., van de Rijn I., Tanner M. E. (1997) Properties and kinetic analysis of UDP-glucose dehydrogenase from group A streptococci. Irreversible inhibition by UDP-chloroacetol. J. Biol. Chem. 272, 3416–3422 [DOI] [PubMed] [Google Scholar]
- 7. Ge X., Penney L. C., van de Rijn I., Tanner M. E. (2004) Active site residues and mechanism of UDP-glucose dehydrogenase. Eur. J. Biochem. 271, 14–22 [DOI] [PubMed] [Google Scholar]
- 8. Stewart D. C., Copeland L. (1998) Uridine 5′-diphosphate-glucose dehydrogenase from soybean nodules. Plant Physiol. 116, 349–355 [Google Scholar]
- 9. Turner W., Botha F. C. (2002) Purification and kinetic properties of UDP-glucose dehydrogenase from sugarcane. Arch. Biochem. Biophys. 407, 209–216 [DOI] [PubMed] [Google Scholar]
- 10. Schiller J. G., Bowser A. M., Feingold D. S. (1972) Studies on the mechanism of action of UDP-d-glucose dehydrogenase from beef liver. II. Carbohydr. Res. 25, 403–410 [DOI] [PubMed] [Google Scholar]
- 11. Sommer B. J., Barycki J. J., Simpson M. A. (2004) Characterization of human UDP-glucose dehydrogenase. CYS-276 is required for the second of two successive oxidations. J. Biol. Chem. 279, 23590–23596 [DOI] [PubMed] [Google Scholar]
- 12. Wei Q., Galbenus R., Raza A., Cerny R. L., Simpson M. A. (2009) Androgen-stimulated UDP-glucose dehydrogenase expression limits prostate androgen availability without impacting hyaluronan levels. Cancer Res. 69, 2332–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Campbell R. E., Tanner M. E. (1999) UDP-glucose analogues as inhibitors and mechanistic probes of UDP-glucose dehydrogenase. J. Org. Chem. 64, 9487–9492 [Google Scholar]
- 14. Easley K. E., Sommer B. J., Boanca G., Barycki J. J., Simpson M. A. (2007) Characterization of human UDP-glucose dehydrogenase reveals critical catalytic roles for lysine 220 and aspartate 280. Biochemistry 46, 369–378 [DOI] [PubMed] [Google Scholar]
- 15. Eccleston E. D., Thayer M. L., Kirkwood S. (1979) Mechanisms of action of histidinol dehydrogenase and UDP-Glc dehydrogenase. Evidence that the half-reactions proceed on separate subunits. J. Biol. Chem. 254, 11399–11404 [PubMed] [Google Scholar]
- 16. Franzen J. S., Ashcom J., Marchetti P., Cardamone J. J., Jr., Feingold D. S. (1980) Induced versus pre-existing asymmetry models for the half-of-the-sites reactivity effect in bovine liver uridine diphosphoglucose dehydrogenase. Biochim. Biophys. Acta 614, 242–255 [DOI] [PubMed] [Google Scholar]
- 17. Franzen J. S., Ishman R., Feingold D. S. (1976) Half-of-the-sites reactivity of bovine liver uridine diphosphoglucose dehydrogenase toward iodoacetate and iodoacetamide. Biochemistry 15, 5665–5671 [DOI] [PubMed] [Google Scholar]
- 18. Franzen J. S., Kuo I., Eichler A. J., Feingold D. S. (1973) UDP-glucose dehydrogenase. Substrate binding stoichiometry and affinity. Biochem. Biophys. Res. Commun. 50, 517–523 [DOI] [PubMed] [Google Scholar]
- 19. Franzen J. S., Marchetti P. S., Feingold D. S. (1980) Resonance energy transfer between catalytic sites of bovine liver uridine diphosphoglucose dehydrogenase. Biochemistry 19, 6080–6089 [DOI] [PubMed] [Google Scholar]
- 20. Franzen J. S., Marchetti P. S., Lockhart A. H., Feingold D. S. (1983) Special effects of UDP-sugar binding to bovine liver uridine diphosphoglucose dehydrogenase. Biochim. Biophys. Acta 746, 146–153 [DOI] [PubMed] [Google Scholar]
- 21. Kadirvelraj R., Sennett N. C., Custer G. S., Phillips R. S., Wood Z. A. (2013) Hysteresis and negative cooperativity in human UDP-glucose dehydrogenase. Biochemistry 52, 1456–1465 [DOI] [PubMed] [Google Scholar]
- 22. Kadirvelraj R., Sennett N. C., Polizzi S. J., Weitzel S., Wood Z. A. (2011) Role of packing defects in the evolution of allostery and induced fit in human UDP-glucose dehydrogenase. Biochemistry 50, 5780–5789 [DOI] [PubMed] [Google Scholar]
- 23. Nelsestuen G. L., Kirkwood S. (1971) The mechanism of action of uridine diphosphoglucose dehydrogenase. Uridine diphosphohexodialdoses as intermediates. J. Biol. Chem. 246, 3824–3834 [PubMed] [Google Scholar]
- 24. Ordman A. B., Kirkwood S. (1977) Mechanism of action of uridine diphoglucose dehydrogenase. Evidence for an essential lysine residue at the active site. J. Biol. Chem. 252, 1320–1326 [PubMed] [Google Scholar]
- 25. Sennett N. C., Kadirvelraj R., Wood Z. A. (2011) Conformational flexibility in the allosteric regulation of human UDP-α-d-glucose 6-dehydrogenase. Biochemistry 50, 9651–9663 [DOI] [PubMed] [Google Scholar]
- 26. Sennett N. C., Kadirvelraj R., Wood Z. A. (2012) Cofactor binding triggers a molecular switch to allosterically activate human UDP-α-d-glucose 6-dehydrogenase. Biochemistry 51, 9364–9374 [DOI] [PubMed] [Google Scholar]
- 27. Campbell R. E., Mosimann S. C., van De Rijn I., Tanner M. E., Strynadka N. C. (2000) The first structure of UDP-glucose dehydrogenase reveals the catalytic residues necessary for the two-fold oxidation. Biochemistry 39, 7012–7023 [PubMed] [Google Scholar]
- 28. Spicer A. P., Kaback L. A., Smith T. J., Seldin M. F. (1998) Molecular cloning and characterization of the human and mouse UDP-glucose dehydrogenase genes. J. Biol. Chem. 273, 25117–25124 [DOI] [PubMed] [Google Scholar]
- 29. Egger S., Chaikuad A., Kavanagh K. L., Oppermann U., Nidetzky B. (2011) Structure and mechanism of human UDP-glucose 6-dehydrogenase. J. Biol. Chem. 286, 23877–23887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Egger S., Chaikuad A., Klimacek M., Kavanagh K. L., Oppermann U., Nidetzky B. (2012) Structural and kinetic evidence that catalytic reaction of human UDP-glucose 6-dehydrogenase involves covalent thiohemiacetal and thioester enzyme intermediates. J. Biol. Chem. 287, 2119–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 32. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357, 289–298 [DOI] [PubMed] [Google Scholar]
- 33. Häcker U., Lin X., Perrimon N. (1997) The Drosophila sugarless gene modulates Wingless signaling and encodes an enzyme involved in polysaccharide biosynthesis. Development 124, 3565–3573 [DOI] [PubMed] [Google Scholar]
- 34. Hwang H. Y., Horvitz H. R. (2002) The Caenorhabditis elegans vulval morphogenesis gene sqv-4 encodes a UDP-glucose dehydrogenase that is temporally and spatially regulated. Proc. Natl. Acad. Sci. U.S.A. 99, 14224–14229 [DOI] [PMC free article] [PubMed] [Google Scholar]