

Abstract
Amyloid β-peptide (Aβ) plays a central role in pathophysiology of Alzheimer’s disease (AD) through the induction of oxidative stress. This peptide is produced by proteolytic cleavage of amyloid precursor protein (APP) by the action of β- and γ-secretases. Previous studies demonstrated that reduction of Aβ, using an antisense oligonucleotide (AO) directed against the Aβ region of APP, reduced oxidative stress-mediated damage and prevented or reverted cognitive deficits in senescence-accelerated prone mice (SAMP8), a useful animal model to investigate the events related to Aβ pathology and possibly to the early phase of AD.
In the current study, aged SAMP8 were treated by AO directed against PS-1, a component of the γ-secretase complex, and tested for learning and memory in T-maze foot shock avoidance and novel object recognition. Brain tissue was collected to identify the decrease of oxidative stress and to evaluate the proteins that are differently expressed and oxidized after the reduction in free radical levels induced by Aβ. We used both expression proteomics and redox proteomics approaches. In brain of AO-treated mice a decrease of oxidative stress markers was found, and the proteins identified by proteomics as expressed differently or nitrated are involved in processes known to be impaired in AD. Our results suggest that the treatment with AO directed against PS-1 in old SAMP8 mice reverses learning and memory deficits and reduces Aβ-mediated oxidative stress with restoration to the normal condition and identifies possible pharmacological targets to combat this devastating dementing disease.
Keywords: Alzheimer disease, Amyloid β-peptide, Senescence accelerated mouse, Amyloid precursor protein
Introduction
Amyloid β-peptide (Aβ) is the principal component of senile plaques, a pathological hallmark of Alzheimer’s disease (AD). This peptide is processed from amyloid precursor protein (APP) by β- and γ-secretases. Aβ plays a pivotal role in the pathophysiology of AD [1, 2] through its ability to induce free-radical damage to neuronal membrane components [1, 3, 4]. As a small, soluble oligomer, Aβ (1-42) interacts with cell membranes and produces free radicals leading to lipid peroxidation and protein oxidation [5, 6], and to neuronal death as a final consequence of cellular damage [2, 7].
Experimental evidence confirms that amyloid deposition has a critical role in the pathogenesis of AD, and in vivo [8] and in vitro studies [9–12] showed oxidative damage induced by Aβ. Therefore, reduction of Aβ could conceivably reduce oxidative stress-mediated damage [13] and prevent or revert cognitive deficits [14]. To reduce the levels of Aβ, an antisense oligonucleotide (AO) binding a specific sequence in a selected mRNA, with consequent inhibition of the corresponding protein expression, can be used [15]. Several findings indicate that inhibition of the expression of APP or Presenilin-1 (PS-1), a protein member of the γ-secretase complex, can reduce Aβ deposition [16, 17].
The senescence accelerated mouse (SAM) subline known as P8 is a model for studying age-related cognitive impairment. These mice show a natural age-related overexpression of Aβ, [14, 17, 18], and an altered energy metabolism [19]. Plaque formation appears after the manifestation of the cognitive impairment [14]; therefore, the presence of amyloid plaques is not involved in learning and memory defects in SAMP8 mice. These features make SAMP8 mice a good model to investigate the events related to the early phase of AD, and investigation of this model conceivably could lead to a therapy for AD. SAMP8 mice have increased oxidative damage in the brain [20–22].
We earlier demonstrated the beneficial effects of reducing APP levels by AO directed at the Aβ region of the APP gene on the decreased oxidative stress in brain [13, 23]. In the current study, we used SAMP8 mice treated by an intracerebroventricular (ICV) injection of AO directed against the PS-1 gene.
The aim of this study was to examine the effects on oxidative stress in the SAMP8 mice treated by AO against PS-1, how the reduction of Aβ-induced free radical damage affected protein expression levels and their oxidation in hippocampus, and to determine effects on learning and memory deficits in SAMP8 mice. We used proteomics and redox-proteomics approaches and tests of learning and memory.
Materials and methods
A 9-mer phosphorothioate oligodeoxynucleotide antisense directed against PS-1 and random antisense were purchased from Midland Certified (Midland, TX). The sequence for the PS-1 antisense was TCTGTCTCA and for the random was GATCACGTA. The AOs were diluted in water.
All chemicals, proteases, and antibodies used in these studies were purchased from Sigma-Aldrich (St. Louis, MO) with exceptions noted. Criterion precast polyacrylamide gels, TGS and XT MES electrophoresis running buffers, ReadyStrip™ IPG strips, mineral oil, Precision Plus Protein™ All Blue standards, Sypro Ruby® Protein Stain, nitrocellulose membranes, dithiothreitol (DTT), iodoacetamide (IA), Biolytes, and urea were purchased from Bio-Rad (Hercules, CA).
Subjects
Twelve month old SAMP8 mice were from an in-house colony, and the colony derived from stock provided by Dr. Takeda of Kyoto University, Japan. Mice were on a 12-h light: 12-h dark cycle (lights on at 06:00 h) and food (PMI Nutrition LabDiet 5001) and water were available ad libitum. The colony frequently underwent serological testing for viral and bacterial contamination. All procedures, involving rodents were approved by the St. Louis University/VA IACUC. Samples were divided into three groups treated by an ICV injection of water, random AO, and AO direct against PS-1 (Table 1) in the hippocampal region.
Table 1.
Samples of old SAMP8 used in this study
| Old SAMP8 with water, ICV | Old SAMP8 with random AO, ICV | Old SAMP8 with AO against PS-1, ICV |
|---|---|---|
| n=11 | n=12 | n=12 |
Surgery and Drug Administration
Mice were injected with PS-1 antisense, random antisense or water 3 times at one week intervals. Mice were anesthetized with isoflurane, placed in a stereotaxic instrument, the scalp was deflected and a hole drilled through the skull over the injection site. The injection coordinates for the ICV injections were 0.5 mm posterior to the Bregma and 1.0 mm to the right and left of the sagittal suture. The injection depth was 2.0 mm, a 2.0 μl solution of water or AO solution was injected over 60 s through a 30 gauge needle. After ICV injection, the scalp was closed and the mice were returned to their cages.
Sample preparation
Two weeks after the last treatment mice were sacrificed. The hippocampal region of SAMP8 mice were flash frozen in liquid nitrogen. After thawing, samples were homogenized using a Wheaton glass homogenizer (~100 passes) in Media I buffer [0.32 M sucrose, 0.10 mM Tris HCl (pH 8.0), 0.10 mM MgCl2, 0.08 mM EDTA, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, and 11.5 μg/ml aprotinin; pH 8.0]. Homogenates were vortexed and sonicated for 10 s at 20% power with a Fisher 550 Sonic Dismembrator (Pittsburgh, PA). Protein concentrations were determined using the BCA method (Pierce, Rockford, IL).
Validation of PS-1 down-regulation after PS-1 antisense treatment
Hippocampal homogenates (75 μg; n=5) were mixed with sample buffer [0.5 M Tris (pH 6.8), 40% glycerol, 8% sodium dodecyl sulfate (SDS), 20% β-mercaptoethanol, and 0.01% bromophenol blue], heated at 95°C for 5 min and loaded onto a Criterion precast (4–12%) Bis-Tris polyacrylamide gel (Bio-Rad, Hercules, CA). The gel was run in 1x MES running buffer (Bio-Rad) at 80V for 10 min after which the voltage was increased to 120V. Following the gel run, proteins were transferred onto nitrocellulose membrane (0.45 μm) using the Bio-Rad Trans-Blot® Turbo™ transfer system according to the manufacturer’s standard protocol (25V, 1A, 30 min). The membrane was blocked at room temperature (RT) for 1.5 h with 3% bovine serum albumin (BSA) in wash blot [a phosphate-buffered saline (PBS) solution containing 0.04% (v/v) Tween 20 and 0.1 M NaCl] and then incubated with goat anti-PS-1-antibody (Santa Cruz Biotechnology, Dallas, TX) 1:1000 in blocking solution over night at 4°C. After three washes with wash blot the membrane was incubated with rabbit anti-goat IgG antibody conjugated to alkaline phosphatase for 1h at RT. After three washes the membrane was developed colorimetrically using 5-bromo-4-chloro-3-indolyl phosphate dipotassium combined with nitrotetrazolium blue chloride (BCIP/NBT) in ALP buffer [0.1 M Tris, 0.1 M NaCl, 5 mM MgCl2 6 H2O (pH 9.5)]. The membrane was then probed for β-actin as a loading control. The membrane was incubated with mouse anti-β-actin-antibody 1:4000 in blocking for 2h at RT. After washing, sheep anti-mouse IgG antibody conjugated to horseradish peroxidase (HRP) (GE Healthcare, Pittsburgh, PA) was added for 1h at RT. After three washes, the membrane was developed with chemiluminescence (Bio-Rad). Images were acquired on the ChemiDoc™ MP imaging system (Bio-Rad) and image analysis was performed using ImageLab software (Bio-Rad).
Measurement of protein carbonyl, protein-bound 4-hydroxynonenal, and 3-nitrotyrosine
Levels of protein carbonyls, protein-bound 4-hydroxynonenal (HNE) and 3-nitrotyrosine (3-NT) were determined immunochemically. Protein carbonyl levels were detected as adducts of 2,4-dinitrophenylhydrazine (DNPH). Five microliters of the samples were treated with an equal volume of 12% SDS. Samples were then derivatized with 10 μL of 20nM 2,4-DNPH for 20 min, followed by the addition of neutralizing reagent (7.5 μL of 2 M Tris/30% glycerol buffer, pH=8.0). Levels of protein carbonyl were measured by using the slot-blot technique with 250 ng of protein loaded per slot. The 2,4-dinitrophenylhydrazone (DNP) adduct of the carbonyl was detected on the nitrocellulose membrane using a primary rabbit antibody (Millipore) specific for DNP-adducts (1:100) followed by a secondary goat anti-rabbit IgG (Sigma-Aldrich) with conjugated alkaline phosphatase. The reaction product developed colorimetrically using BCIP/NBT in ALP buffer. After developing, blots were allowed to dry overnight, scanned using Adobe Photoshop 6.0 with a Canon CanoScan 8800F scanner, and quantified using ImageQuant TL software (GE Healthcare, Pittsburgh, PA). Previous studies demonstrated that samples treated by NaHB4 resulted in no hydrazone formation nor binding of anti-DNP or secondary antibody, demonstrating specificity for analytical procedures employed [24]. HNE and 3-NT levels were determined in the same manner. Five microliters of the samples were treated with an equal volume of 12% SDS. Samples were then denatured with 10 μL of Laemmli (0.125 M Trizma base, 4% SDS, 20% glycerol) for 20 min. Levels of 3-NT and HNE were measured by slot blot technique using 250 ng of protein per slot for both 3-NT and HNE levels. The HNE levels were detected on the nitrocellulose membrane using a primary antibody (Alpha Diagnostic Intl. Inc.) specific for HNE-modified protein (1:1000). The 3-NT levels were detected by primary rabbit antibody (Sigma-Aldrich) specific for 3-NT (1:1000). The same secondary goat anti-rabbit (Sigma-Aldrich) antibody was then used for detection of each primary antibody. The developing and detection were performed as described above for protein carbonyls.
Two-dimensional electrophoresis
Proteins from hippocampus homogenates (200μg) were precipitated by addition of ice-cold 100% trichloroacetic acid (15% final concentration) and placed on ice for 10 min. Samples were centrifuged at 14,000 rpm (23,700 × g) for 5 min at 4°C. The pellets were washed three times with 0.5 mL of ethanol/ethyl acetate (1:1) and then resuspended in 200 μL of rehydration buffer (8M urea, 2M thiourea, 20mM DTT, 2.0% CHAPS (w/v), 0.2% Biolytes and bromophenol blue) and placed in agitation for 3 hours. Solubilized proteins were finally sonicated twice for 30 sec. For the first-dimension electrophoresis, 200 μL of sample solution were applied to a ReadyStrip™IPG strip pH 3–10 (Bio-Rad). The strips were soaked in the sample solution for 1 h to allow uptake of the proteins. The strips were then actively rehydrated in a Protean IEF Cell Apparatus (Bio-Rad) for 16 h at 50V. The isoelectric focusing was performed at 300V for 2 h linearly; 500V for 2 h linearly; 1000V for 2 h linearly, 8000V for 8 h linearly and 8000V for 10 h rapidly. All the processes above were carried out at room temperature. The focused IEF strips were stored at −80° C until second dimension electrophoresis was performed.
For second dimension electrophoresis, the strips were equilibrated for 10 min in 50 mM Tris-HCl (pH 6.8) containing 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, and 0.5% DTT, and then re-equilibrated for 15 min in the same buffer containing 4.5% IA in place of DTT. Precast criterion gels (8–16 %, Bio-Rad) were used to perform second dimension electrophoresis. Precision ProteinTM Standards (Bio-Rad) were run along with the sample at 200V for 65 min. After electrophoresis, the gels were incubated in fixing solution (7% acetic acid, 10% methanol) for 45 min. Approximately 40 mL of Sypro Ruby Protein Gel Stain (Bio-Rad, Hercules, CA) were used to stain the gels overnight. The gels were placed in deionized water overnight for de-staining.
Gels were scanned into Adobe Photoshop 6.0 with a Molecular Dynamics STORM Phosphoimager (λex/λem: 470/618 nm) and stored in deionized water at 4°C until further use.
Immunochemical detection
For immunoblotting analysis, electrophoresis was carried out in the same way as previously described, and the gels were transferred to a nitrocellulose membrane. The blots were incubated with rabbit anti-3-NT polyclonal antibody (Sigma-Aldrich) diluted 1:1000 in 0.3% BSA in wash blot at RT for 2 h. When the incubation with primary antibody was complete, blots were washed for 10 min in wash blot three times. Secondary antibody (anti-rabbit alkaline phosphatase-conjugated IgG; Sigma-Aldrich, St Louis, MO, USA) diluted 1:2000 in wash blot, was added to membranes for 1 h at room temperature. Membranes were washed three times for 5 min with wash blot and then developed colorimetrically using BCIP/NBT in ALP buffer.
Image Analysis
Differential Protein Levels
Spot intensities from SYPRO Ruby®-stained 2D-gel images of old SAMP8 mice treated with water, old SAMP8 mice treated with random AO, and old SAMP8 mice treated with AO against PS-1 samples were quantified densitometrically according to the total spot density using PDQuest analysis software from Bio-Rad (Hercules, CA). Intensities were normalized to total gel densities and/or densities of all valid spots on the gels. Only spots with a 1.5-fold increase or decrease in normalized spot density in those samples and a statistically significant difference based on a Student’s t-test or Mann-Whitney U test at 95% confidence (i.e., p<0.05) were considered for MS analysis.
Oxidative modification
The analysis of gels and blots to compare protein and 3-NT content between old SAMP8 mice treated with water, old SAMP8 mice treated with random AO, and old SAMP8 mice treated with AO against PS-1 were performed with PDQuest image analysis software (Bio-Rad). Blot immunoreactivity (nitration) was normalized to the total protein content as measured by the intensity of SYPRO Ruby®-stained gels. Only spots with statistically significant increases or decreases in protein-resident 3-NT levels, as calculated by a Student’s t-test and Mann–Whitney U Statistical test at 95% confidence were selected for in-gel trypsin digestion and subsequent MS analysis.
In-gel trypsin digestion
In-gel trypsin digestion of selected gel spots was performed as previously described [25]. Briefly, protein spots identified as significantly altered were excised from 2D-gels with a clean, sterilized blade and transferred to Eppendorf microcentrifuge tubes. Gel plugs were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at RT for 15 min, followed by incubation with 100% acetonitrile at RT for 15 min. After solvent removal, gel plugs were dried in their respective tubes under a flow hood at RT. Plugs were incubated for 45 min in 20 μl of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C. The DTT/NH4HCO3 solution was then removed and replaced with 20 μl of 55 mM IA in 0.1 M NH4HCO3 and incubated with gentle agitation at room temperature in the dark for 30 min. Excess IA solution was removed and plugs incubated for 15 min with 200 μl of 50 mM NH4HCO3 at room temperature. A volume of 200 μl of 100% acetonitrile was added to this solution and incubated for 15 min at room temperature. Solvent was removed and gel plugs were allowed to dry for 30 min at RT under a flow hood. Plugs were rehydrated with 20 ng/μl of modified trypsin (Promega, Madison, WI, USA) in 50 mM NH4HCO3 in a shaking incubator overnight at 37 °C. Enough trypsin solution was added in order to completely submerge the gel plugs.
Mass spectrometry (MS)
Salts and contaminants were removed from tryptic peptide solutions using C18 ZipTips (Sigma-Aldrich, St. Louis, MO, USA), reconstituted to a volume of ~ 15 μl in a 50:50 water: acetonitrile solution containing 0.1% formic acid. Tryptic peptides were analyzed with an automated Nanomate electrospray ionization (ESI) [Advion Biosciences, Ithaca, NY, USA] Orbitrap XL MS (Thermo-Scientific, Waltham, MA, USA) platform. The Orbitrap MS was operated in a data-dependent mode whereby the eight most intense parent ions measured in the Fourier Transform (FT) at 60,000 resolution were selected for ion trap fragmentation with the following conditions: injection time 50 ms, 35% collision energy, MS/MS spectra were measured in the FT at 7500 resolution, and dynamic exclusion was set for 120 s. Each sample was acquired for a total of ~ 2.5 min. MS/MS spectra were searched against the International Protein Index (IPI) database using SEQUEST and the following parameters: two trypsin miscleavages, fixed carbamidomethyl modification, variable Methionine oxidation, parent tolerance 10 ppm, and fragment tolerance of 25 mmu or 0.01 Da. Results were filtered with the following criteria: Xcorr1.5, 2.0, 2.5, 3.0 for 1, 2, 3, and 4 charge states, respectively, Delta CN0.1, and P-value (protein and peptide) 0.01. IPI accession numbers were cross-correlated with Swiss Prot accession numbers for final protein identification. Proteins identified with a single peptide were kept for further analyses if multiple spectral counts (SC, number of observed MS/MS spectra) were observed in a single analysis or if the peptide was identified in a separate analysis and workup of the same protein spot.
Immunoprecipitation
Protein A/G–agarose beads (50 μl per sample, i.e., 900 μl for 18 samples) (Calbiochem, La Jolla, CA, USA) were washed with immunoprecipitation (IP) buffer three times for 5 min using a vortex with shaker attachment. IP buffer contained PBS with 0.05% Nonidet P-40 and the protease inhibitors leupeptin (4 μg/ml final concentration), pepstatin (4 μg/ml final concentration), and aprotinin (5 μg/ml final concentration), adjusted to pH 8. Protein samples (250 μg) were first precleared with washed protein A/G–agarose beads (50 μl) for 1 h at 4 °C. Samples were then incubated at RT for 2 h with anti- L-lactate dehydrogenase B chain (LDHB) (ABGENT) antibody, anti-Pin-1 antibody (Santa Cruz Biotechnology), and anti- vesicular-fusion protein (NSF) antibody (BioVision) (5 μg) followed by 1 h incubation with protein A/G–agarose. The antigen–antibody–protein A/G complex was centrifuged at 1000 g for 5 min and the resultant pellet was washed five times with IP buffer (500 μl). The final pellet was suspended in deionized water. Proteins were resolved on SDS–PAGE, followed by immunoblotting on a nitrocellulose membrane (Bio-Rad) as described above, using rabbit polyclonal anti-3-NT (1:2000) primary, and rabbit polyclonal anti-Pin-1 (1:2000) primary (Santa Cruz Biotechnology, Santa Cruz, CA) and Amersham ECL rabbit IgG HRP-linked (1:5000) secondary antibodies. Blots were developed chemifluorescently (λex/λem: 70/618 nm) and quantified using ImageQuant TL software.
Western blotting validation of proteomics-mediated protein identity
1D-PAGE
Hippocampus homogenates (75 μg) were suspended in sample loading buffer, heated at 95 °C for 5 min, and cooled on ice prior to gel loading. Samples and Precision Plus Protein™ All Blue Standards were loaded into a Criterion precast (12%) Bis-Tris polyacrylamide gel and run at RT in a 1x dilution of XT MES running buffer at 80 V for ~10 min to ensure proper protein stacking. The voltage was then increased to 140 V for ~110 min at RT for the duration of the electrophoretic run.
1D & 2D-Western Blotting
Following 1D- or 2D-PAGE, in-gel proteins were transferred using a Trans-Blot Semi-Dry Transfer Cell system at 20 V for 2 h (Bio-Rad, Hercules, CA) into a nitrocellulose membrane (0.45 μm). Post-transfer, membranes were incubated in a blocking solution of 3% BSA in Wash Blot at RT for 2 h. Following blocking, membranes were incubated with rabbit polyclonal antibody anti-Pin-1 (1:2000) (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal antibody anti-isoform M1 pyruvate kinase isozymes M1/M2 (KPYM) (1:500) (ABGENT, USA), and rabbit polyclonal antibody anti-actin (Sigma-Aldrich) at RT on a rocking platform for 2–3 h. Blots were washed three times for 5 min each in wash blot, followed by 1 h incubation with rabbit IgG alkaline phosphatase (Sigma-Aldrich) (1:5000) secondary antibody and Amersham ECL rabbit IgG HRP-linked (1:5000) secondary antibody at RT. Blots were rinsed three times for 5 min each in wash blot and developed colorimetrically using BCIP/NBT in ALP buffer and chemifluorescently (λex/λem: 70/618 nm), respectively. After developing, blots were quantified using ImageQuant TL software (GE Healthcare, Pittsburgh, PA).
Training and Testing
T-Maze training and testing procedures
The T-maze is both a learning task based on working-memory and a reference-memory task. The T-maze consisted of a black plastic alley with a start box at one end and two goal boxes at the other. The start box was separated from the alley by a plastic guillotine door that prevented movement down the alley until raised at the onset of training. An electrifiable floor of stainless steel rods ran throughout the maze to deliver a mild scrambled foot-shock.
Mice were not permitted to explore the maze prior to training. A block of training trials began when a mouse was placed into the start box. The guillotine door was raised and a cue buzzer sounded simultaneously; 5 sec later foot-shock was applied. The arm of the maze entered on the first trial was designated “incorrect” and the mild foot-shock was continued until the mouse entered the other goal box, which in all subsequent trials was designated as “correct” for the particular mouse. At the end of each trial, the mouse was returned to its home cage until the next trial.
Mice were trained until they made 1 avoidance. Training used an intertrial interval of 35 sec, the buzzer was of the door-bell type sounded at 55 dB, and shock was set at 0.35 mA (Coulbourn Instruments scrambled grid floor shocker model E13-08). Retention was tested one week later by continuing training until mice reached the criterion of 5 avoidances in 6 consecutive trials. The results were reported as the number of trials to criterion for the retention test.
Object-Place Recognition
Object-place recognition is a declarative memory task that involves the hippocampus when, as performed here, the retention interval is 24 hours after initial exposure to the objects. Mice were habituated to an empty apparatus for 5 minutes a day for 3 days prior to entry of the objects. During the training session, the mouse was exposed to two similar objects (plastic frogs) which it was allowed to examine for 5 minutes. The apparatus and the objects were cleaned between each mouse. Twenty-four hours later, the mouse was exposed to one of the original objects and a new novel object in a new location and the percent of time spent examining the new object was recorded. The novel object was made out of the same material as the original object and of the same size, but a different shape. This eliminated the possibility of smell associated with a particular object being a factor. The underlying concept of the task is based on the tendency of mice to spend more time exploring novel objects than familiar objects. Thus, the greater the retention/memory at 24 hours, the more time spent with the new object.
Statistical analysis
All data are presented as mean ± S.E.M., as noted, and statistical analyses were performed using a Mann-Whitney U statistical test and a two-tailed Student’s t-test, wherein p<0.05 was considered significant for differential fold-change values. Only proteins with significant P-values from both tests were considered further for MS identification. Protein and peptide identifications obtained with the SEQUEST search algorithm with p<0.01 were considered statistically significant. To further validate SEQUEST identification, the location of protein spots (i.e., molecular weight [MW] and isoelectric point [pI]) on 2D-gels was manually checked based on expected MW and pI values from SwissProt database information. The behavioral tests were analyzed by one way ANOVAs followed by Tukeys post hoc test. The sample size was N = 10 for all behavioral tests.
Results
PS-1 levels
Analysis of PS-1 protein levels showed a significantly decreased PS-1 levels in the hippocampus of SAMP8 mice treated with AO against PS-1 (77.9 ±15.2 (mean ± SEM); p*< 0.03) when compared to SAMP8 mice injected with random AO (Fig. 1).
Figure 1.

Analysis of Western blot for PS-1 levels after treatment with random AO (rAO) or AO directed against PS-1 (anti-PS1). PS-1 levels are normalized to β-actin and resulting values are normalized to (rAO) values. Data is shown as mean ± SEM (n=5).
Protein oxidation
Protein carbonyls level
Analysis of protein carbonyl levels reveals a small, but significant decrease of this oxidative stress marker in hippocampus from old SAMP8 mice treated with AO against PS-1 compared to those in hippocampus from old SAMP8 mice treated with water (p*< 0.008). There are no differences in protein carbonyl levels between old SAMP8 mice treated with water compared to old SAMP8 treated with random AO and between old SAMP8 mice treated with random AO compared to old SAMP8 treated with AO against PS-1. These results are shown in Fig. 2.
Figure 2.

Data show the average level of protein carbonyl, 3-NT and HNE of hippocampus from old SAMP8 mice previously treated with water (V), old SAMP8 mice treated with random AO (rAO) and old SAMP8 mice treated by AO directed against PS-1 (anti-PS1). Error bars indicate SEM for ten animals in each group. Measured values are normalized to the (V) values.
3-Nitrotyrosine (3-NT)
3-NT levels of brain from old SAMP8 mice after injection of the AO against PS-1 are significantly decreased compared to those in brain from old SAMP8 mice treated with water or with random AO (p*<0.03; p**<0.0003, respectively). These results are shown in Figure 2.
Lipid peroxidation
Protein-bound HNE
HNE is a product of lipid peroxidation [26] and its levels have been found elevated in AD brain [5, 6]. The protein-bound HNE levels, as Figure 2 shows, were not affected greatly by AO treatment against PS-1 (Fig. 2).
Proteomics and redox proteomics
To identify proteins of differential amounts, proteomics analysis using 2-DE and Sypro Ruby staining was performed by comparing the differential patterns of protein expression between hippocampus from old SAMP8 mice treated with water, old SAMP8 mice after injection of the random AO and old SAMP8 mice after injection of the AO against PS-1. Fig. 3 shows 2-D gel images related to the matching between hippocampal proteins from old SAMP8 mice treated by water vs. old SAMP8 mice treated by random AO, old SAMP8 mice treated by AO against PS-1 vs. old SAMP8 mice treated by water, and old SAMP8 mice treated by AO against PS-1 vs. old SAMP8 mice treated by random AO, in which proteins of different amounts expressed differently are highlighted by circles. Eighteen proteins expressed differently were found, and, interestingly, seventeen of these were significantly up-regulated in the hippocampus of old SAMP8 mice treated by AO against PS-1. These proteins were excised from the gels, digested with trypsin and analyzed by MS/MS analyses. Table 2 lists the proteins identified with the number of peptide sequences, the score, the coverage, MW, pI, fold-change levels, and p-value. All protein identifications were consistent with comparison of protein positions on the gel with MW and pI from databases.
Figure 3.

Proteomic profile of representative 2D-gels with proteins differentially expressed in the three groups of matching. (A) old SAMP8 mice previously treated with water vs. old SAMP8 mice treated by random AO; (B) old SAMP8 mice treated by water vs. old SAMP8 mice treated by AO against PS-1; (C) old SAMP8 mice treated by random AO vs. old SAMP8 mice treated by AO against PS-1.
Table 2.
Proteins Expressed Differently when Old SAMP8 Mice are Injected with AO against PS-1 (C) Compared to Old SAMP8 Treated with Water (A) or Random AO (B)
| Spot | Protein Identified | Accession # | Coverage | Number of identified peptides (a) | Score | MW (kDa) | pI | Groups | P value (c) | Fold (b) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Guanine nucleotide-binding protein G (0) subunit alpha (GNAO) | P18872 | 17.80 | 5 | 29.01 | 40.1 | 5.53 | A-B | 0.034 | 3.21 ↑B |
| 2 | Isocitrate dehydrogenase (NAD) subunit alpha, mitochondrial (IDH3A) | Q9D6R2 | 21.04 | 7 | 35.33 | 39.6 | 6.73 | B-C | 0.021 | 2.27 ↑C |
| 3 | Isocitrate dehydrogenase (NAD) subunit alpha, mitochondrial (IDH3A) | Q9D6R2 | 16.94 | 6 | 23.67 | 39.6 | 6.73 | A-C B-C |
0.035 0.022 |
1.62 ↑C 1.79 ↑C |
| 4 | Actr3 protein | Q3ULF7 | 16.75 | 7 | 25.72 | 47.3 | 5.88 | A-C B-C |
0.0041 0.0037 |
2.1 ↑C 2.71 ↑C |
| 5 | Actr3 protein | Q3ULF7 | 14.35 | 6 | 24.53 | 47.3 | 5.88 | A-C B-C |
0.0081 0.018 |
2.86 ↑C 2.63 ↑C |
| 6 | V-type proton ATPase subunit B, brain isoform (V-ATPase) | P62814 | 4.11 | 2 | 32.46 | 56.5 | 5.81 | A-C B-C |
0.031 0.036 |
1.7 ↑C 1.9 ↑C |
| 7 | Dihydropyrimidinase-related protein 2 (CRMP-2) | O08553 | 18.01 | 8 | 48.51 | 62.2 | 6.38 | A-C B-C |
0.0084 0.019 |
3.26 ↑C 2.93 ↑C |
| 8 | Dihydropyrimidinase-related protein 2 (CRMP-2) | O08553 | 8.39 | 4 | 15.38 | 62.2 | 6.38 | A-C | 0.021 | 1.9 ↑C |
| 9 | Dihydropyrimidinase-related protein 2 (CRMP-2) | O08553 | 15.03 | 7 | 40.68 | 62.2 | 6.38 | B-C | 0.029 | 2.95 ↑C |
| 10 | Alpha-enolase (ENO) | P17182 | 38.02 | 15 | 74.44 | 47.1 | 6.80 | A-C | 0.041 | 2.05 ↑C |
| 11 | Isoform M1 Pyruvate kinase isozymes M1/M2 (KPYM) | O08749 | 7.07 | 8 | 27.41 | 57.9 | 7.14 | A-C | 0.038 | 1.69 ↑C |
| 12 | Pyruvate dehydrogenase E1 component subunit alpha (PDHA1) | P35486 | 11.54 | 4 | 22.97 | 43.2 | 8.19 | B-C | 0.0002 | 2.02 ↑C |
| 13 | Aspartate aminotransferase, cytoplasmic (AATC) | P05201 | 26.88 | 10 | 52.62 | 46.2 | 7.14 | B-C | 0.011 | 1.38 ↑C |
| 14 | Isoform A of Cytosolic acyl coenzyme A thoiester hydrolase (BACH) | Q91V12-2 | 20.71 | 5 | 29.01 | 37.5 | 7.52 | B-C | 0.035 | 1.75 ↑C |
| 15 | Isoform A of Cytosolic acyl coenzyme A thioester hydrolase (BACH) | Q91V12-2 | 10.36 | 3 | 15.87 | 37.5 | 7.52 | B-C | 0.013 | 1.43 ↑C |
| 16 | ATPsynthase subunit alpha, mitochondrial (ATPase) | Q03265 | 23.51 | 11 | 53.66 | 59.7 | 9.19 | B-C | 0.026 | 1.98 ↑C |
| 17 | ES1 protein homolog, mitochondrial (ES1) | Q9D172 | 6.77 | 2 | 4.35 | 28.1 | 8.78 | A-C B-C |
0.025 0.076 |
1.58 ↑C 2.03 ↑C |
| 18 | Peptidyl-prolyl cis-trans isomerase A (PIN-1) | P17742 | 17.68 | 3 | 17.02 | 18.0 | 7.90 | A-C | 0.049 | 1.72 ↑C |
| 19 | Triosephosphate isomerase (TPI) | P17751 | 16.47 | 3 | 16.47 | 26.7 | 7.30 | A-C | 0.038 | 2.4 ↑C |
| 20 | Protein DJ-1 (PARK7) | Q99LX0 | 42.33 | 6 | 22.84 | 20.0 | 6.77 | B-C | 0.044 | 1.67 ↑C |
| 21 | Prohibitin (Fragment) (PHB) | Q5SQG5 | 10.63 | 3 | 9.57 | 23.0 | 6.37 | B-C | 0.01 | 1.59 ↑C |
| 22 | N-ethylmaleimide sensitive fusion protein attachment protein beta (SNAPs) | A2APW8 | 18.12 | 4 | 28.01 | 33.5 | 5.47 | A-B B-C |
0.01 0.031 |
1.8 ↑A 1.98 ↑C |
| 23 | Pyruvate dehydrogenase E1 component subunit beta, mitochondrial (PDH) | Q9d051 | 19.50 | 5 | 31.84 | 38.9 | 6.87 | B-C | 0.044 | 1.6 ↑C |
The number of peptide sequences identified by nanospray ESI-MS/MS of tryptic peptides.
The fold-change in spot density from (C) compared with (A) and (B). The arrow indicates the direction of change.
The p-value associated with fold-change calculated using a Student’s t-test.
Specific nitrated proteins were detected immunochemically using 2D-gels and 2D-western blots. 2D-gels and 2D-blots probed with 3-NT antibody were matched using the PDQuest software and the specific 3-NT levels were obtained by dividing the 3-NT level of a single spot on the blot by the protein level of its corresponding protein spot on the gel. Fig. 4 shows 2D-blots related to the matching between hippocampus from old SAMP8 mice treated by water vs. old SAMP8 mice treated by random AO; old SAMP8 mice treated by AO against PS-1 vs. old SAMP8 mice treated by water; and old SAMP8 mice treated by AO against PS-1 vs. old SAMP8 mice treated by random AO, in which proteins differently nitrated are highlighted by circles.
Figure 4.

Proteomic profile of representative 2D-blots with proteins differentially nitrated in the three groups of matching. (A) old SAMP8 mice treated previously with water vs. old SAMP8 mice treated previously with random AO; (B) old SAMP8 mice treated previously with water vs. old SAMP8 mice treated by AO against PS-1; (C) old SAMP8 mice treated previously with random AO vs. old SAMP8 mice treated previously with AO against PS-1.
Table 3 lists the nitrated proteins identified by MS/MS and interrogation of databases. Interestingly, most of these proteins are excessively nitrated in old SAMP8 mice treated with random AO compared to either old SAMP8 mice treated by water or old SAMP8 mice treated by AO against PS-1. This result confirms our data presented above about the measure of total levels of nitrated proteins.
Table 3.
Level of Nitrated Proteins from Old SAMP8 Mice Injected with AO against PS-1 (C) Compared to Old SAMP8 Mice Treated with Water (A) or Random AO(B)
| Spot | Protein Identified | Accession # | Coverage | Number of identified peptides (a) | Score | MW (kDa) | pI | Groups | P value (c) | Fold (b) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Isoform 2 of Glial fibrillary acidic protein (GFAP) | P03995-2 | 30.84 | 14 | 67.67 | 49.3 | 5.50 | A-C | 0.027 | 15.4 ↑A |
| 2 | Peptidase (Mitochondrial processing) alpha (DPL2) | A2AIW8 | 8.02 | 4 | 13.37 | 58.2 | 6.83 | B-C | 0.037 | 3.79 ↑C |
| 3 | Dihydropyrimidinase-related protein 2 | O08553 | 32.52 | 14 | 103.38 | 62.2 | 6.38 | B-C | 0.047 | 9.5 ↑C |
| 4 | Isoform 5 of Dynamin-1 (DYN1) | P39053-5 | 22.91 | 18 | 91.49 | 96.0 | 7.01 | B-C | 0.04 | 2.79 ↑B |
| 5 | Vesicle-fusing ATPase/Vesicular-fusion protein NSF (NSF) | P46460 | 26.08 | 18 | 86.01 | 82.6 | 6.95 | A-B | 0.048 | 3.85 ↑B |
| 6 | Dihydropyrimidinase-related protein 1 (CRMP-1) | P97427 | 7.17 | 3 | 9.26 | 62.1 | 7.12 | B-C | 0.018 | 3.37 ↑B |
| 7 | Isoform 3 of Septin-11 (SEP11) | Q8C1B7-3 | 22.35 | 9 | 48.34 | 48.9 | 6.81 | B-C | 0.0072 | 5.24 ↑B |
| 8 | Alpha-enolase (ENO) | P17182 | 21.43 | 7 | 28.71 | 47.1 | 6.8 | A-B | 0.04 | 7.14 ↑B |
| 9 | Actin-related protein 2 (Actr2) | P61161 | 27.41 | 9 | 47.52 | 44.7 | 6.74 | B-C | 0.047 | 1.88 ↑B |
| 10 | L-lactate dehydrogenase B chain (LDHB) | P16125 | 8.08 | 2 | 9.00 | 36.5 | 6.05 | B-C | 0.047 | 3.79 ↑B |
| 11 | Isoform 2 of Isocitrate dehydrogenase (NAD) subunit alpha, mitochondrial (IDH3A) | Q9D6R2-2 | 7.64 | 2 | 4.96 | 31.4 | 6.04 | A-C | 0.036 | 42.52 ↑A |
The number of peptide sequences identified by nanospray ESI-MS/MS of tryptic peptides.
The fold-change in spot density of (C) compared with (A) and (B). The arrow indicates the direction of change.
The p-value associated with fold-change calculated using a Student’s t-test.
To confirm the protein expression burden obtained by proteomics data, we carried out western blot analysis of KPYM and immunoprecipitation followed by western blot analysis of Pin1. The results shown in Fig. 5 demonstrate an increase of KPYM in hippocampus from old SAMP 8 mice treated by AO against PS-1 vs. old SAMP8 mice treated by water, and an increase of Pin1 in old SAMP 8 mice treated by AO against PS-1 vs. old SAMP 8 mice treated by water, validating the trend in levels obtained by proteomics.
Figure 5.

(A) Western blot to confirm the expression level of isoform M1 pyruvate kinase isozymes M1/M2 (KPYM) of old SAMP8 mice treated by water (V) and old SAMP 8 mice treated by AO against PS-1 (anti-PS1). Error bars indicate SEM for four animals in each group and all values were normalized to β-actin. (B) Immunoprecipitation followed by western blot analysis was performed to confirm the expression level of Pin-1 between old SAMP8 mice treated by water (V) and old SAMP 8 mice treated by AO against PS-1(anti-PS1). Pin-1 protein was immunoprecipitated using anti-Pin-1 antibody and then probed for Pin-1 level. Error bars indicate SEM for two animals in each group.
Further, to validate nitration levels achieved by proteomics data, immunoprecipitation analysis of LDH and NSF were performed. Fig. 6 shows an increase of LDH in hippocampus from old SAMP8 treated by random AO compared to old SAMP8 mice treated by AO against PS-1, and an increase of NSF in old SAMP8 treated by water compared to old SAMP8 mice treated by random AO, validating data from proteomics. In addition, we confirmed the correct identification of two of the proteins, KPYM and Pin-1, by performing 2D-western blot. The blots were probed with anti-KPYM and anti-Pin-1 antibodies, respectively (Fig. 7). The protein spots of interest are circumscribed by a circle.
Figure 6.

Immunoprecipitation followed by western blot analysis was performed to confirm the nitration levels of (A) L-lactate dehydrogenase B chain (LDHB) of and (B) vesicular-fusion protein (NSF) of old SAMP8 mice treated by random AO (rAO) vs. old SAMP8 mice treated by AO against PS-1 (anti-PS1) and old SAMP8 mice treated by water (V) vs. old SAMP8 mice treated by random AO (rAO), respectively. The proteins first were immunoprecipitated using anti-LDH or anti-NSF antibody and then probed with 3-NT antibody. Representative blots of LDH and NSF probed with 3-NT antibody were shown. Error bars indicate SEM for two animals in each group.
Figure 7.

Validation of protein identified by MS using Western blot analysis: (A) 2D-blot probed with anti- isoform M1 pyruvate kinase isozymes M1/M2 (KPYM) antibody. (B) 2D-blot probed with anti-Pin-1 antibody. A circle is drawn around the protein spot of interest.
Effect of PS-1 antisense on memory in 12 month old SAMP8 mice
For the acquisition test, the one-way ANOVA with trials to criterion as the independent variable showed a significant treatment effect. Dunnett’s post hoc analysis indicated that the mice that received AO against PS-1 took significantly fewer trials to reach criterion than the mice that received water and the mice that received random AO. There was no difference between the mice that received random AO and the mice that received water (see Figure 8A). A one-way ANOVA for trials to criterion on the retention test showed a significant treatment effect. As above, Dunnett’s post hoc analysis indicated that the mice that received AO against PS-1 took significantly fewer trials to reach criterion than the mice that received water and the mice that received random antisense. There was no difference between the mice that received water and the mice that received random antisense (See Figure 8B). The ANOVA for percent time exploring the novel object showed a significant treatment effect. The mice that received AO against PS-1 spent a greater amount of time exploring the novel object than the mice that received water and the mice that received random antisense. There was no difference between the mice that received water and the mice that received random antisense (Figure 8C).
Figure 8.

The mice that received PS-1 antisense took significantly fewer trials to make one avoidance than the mice that received water or random antisense (8A), on the one–week retention test the mice that received PS-1 antisense took significantly fewer trials to reach criterion than the mice that received water or random antisense (8B) and in object recognition the mice that received PS-1 antisense spent significantly more time exploring the novel object than the mice that received water or random antisense (8C). The * indicates p<0.05 and ** indicates p<0.01.
Discussion
Much evidence supports a central role of Aβ as a causative agent of AD [2, 4, 27]. Inhibition of APP or γ-secretase can decrease Aβ formation [28] with consequent reversal of memory impairment [14]. γ-secretase is a complex that consists of PS-1, nicastrin, anterior pharynx homolog 1 and presenilin enhancer protein 2 among other moieties [29]. PS-1 has an essential role in the γ-secretase complex and in the processing of APP. Mutations in the PS-1 gene are causative of FAD and are responsible for an increase in Aβ42 in humans [30, 31]. PS-1 and PS-2 knock-out mice have no γ-secretase [32], demonstrating the critical role of PS-1 for γ-secretase activity. For this reason PS-1 could be a promising therapeutic target for AD. Several studies demonstrated that the reduction of PS-1 leads to a downregulation of amyloid β protein [16, 33]. Kumar et al. noticed that an increased expression or mutation of PS-1 may modulate regulation of the γ-secretase activity, with consequent increased Aβ accumulation and early impairment in SAMP8 mice [34].
We previously demonstrated that the AO treatment against APP in 12-month-old SAMP8 mice is due to a decrease in Aβ-mediated oxidative stress [13]. Hence, antisense therapy conceivably could be curative for AD through reduction of Aβ levels and reversal of deficits in learning and memory [17].
This study was designed to determine whether there was a decrease of oxidative stress in hippocampus from old SAMP8 mice after treatment with AO against PS-1, and to identify the hippocampal proteins that were less oxidized or expressed differently after AO-mediated reduction of oxidative-nitrosative stress induced by Aβ. Our results show that the amounts of protein oxidation (protein carbonyl and 3-NT) are reduced in old SAMP8 mice after injection of AO against PS-1, consistent with previous data [13, 35]. In contrast, treatment with AO against PS-1 did not exert the same protective effect on protein-bound HNE levels.
We determined specific nitrated proteins, as well as the proteins that are expressed differently, to gain insight into mechanisms that are affected by reduction of oxidative stress in brain secondary to the decrease of Aβ levels. The proteins identified as expressed differently or nitrated are grouped in Tables 4 and 5 correlated with their functions.
Table 4.
Functionalities of Identified Proteins Differently Expressed
| Functions | Proteins involved |
|---|---|
| Energy or mitochondrial dysfunction | Isocitrate dehydrogenase (NAD) subunit α (IDH3A) Isoform M1 Pyruvate kinase isozymes M1/M2 (KPYM) Pyruvate dehydrogenase E1 component subunit alpha (PDHA1) Aspartate aminotransferase (AATC) ATP synthase (ATPase) Pyruvate dehydrogenase component subunit beta, mitochondrial (PDH) Prohibitin (Fragment) (PHB) Triosephosphate isomerase (protect against neuronal apoptosis) (TPI) ES1 protein homolog, mitochondrial (ES1) Alpha-enolase (ENO) |
| Neuritic abnormalities | Actin-related protein 3 (Actr3) Dihydropyrimidinase-related protein 2 (CRMP-2) |
| Synaptic abnormalities and LTP | N-ethylmaleimide sensitive fusion protein attachment protein beta (SNAPs) Guanine nucleotide protein G (0) subunit alpha (GNAO) |
| Cell cycle; tau phosphorylation; and Abeta production | Pin-1 |
| Lipid abnormalities | Isoform A of Cytosolic acyl coenzyme A thioester hydrolase (BACH) |
| Lysosomal dysfunction | V-type proton ATPase (V-ATPase) |
| Antioxidant defense | Protein DJ-1 (PARK7) |
Table 5.
Functionalities of Identified Nitrated Proteins
| Functions | Proteins involved |
|---|---|
| Energy or mitochondrial dysfunction | α-enolase (ENO) L-lactate dehydrogenase (LDH) Isoform 2 of isocitrate dehydrogenase (NAD) subunit α Peptidase α (DPL2) Vesicle-fusing ATPase (NSF) |
| Neuritic abnormalities | Dihydropyrimidinase-related protein 2 (CRMP-2) Dihydropyrimidinase-related protein 1(CRMP-1) Actin-related protein 2 (Actr2) Septin-11 (SEP11) |
| Synaptic abnormalities | Isoform 5of Dynamin-1 (DYN1) |
| Marker of inflammation | Isoform 2 of Glial fibrillary acidic protein (GFAP) |
Energy dysfunction or mitochondrial dysfunction
Several studies showed aberrant mitochondrial morphology and reduced rate of cerebral metabolism in AD brain [36–38]. Given that glucose is the principal source for the production of ATP in healthy brain, diminished glucose utilization leads to less production of ATP with loss of synapses and synaptic function producing memory impairment. Further, decreased levels of ATP would disturb ion homeostasis, impairing ion motive ATPases, glucose and glutamate transporters and membrane asymmetry, rendering neurons susceptible to excitotoxicity and apoptosis [39].
We identified IDH3A, KPYM, PDHA1 and PDH, TPI, ATP synthase, AATC, prohibitin, ES1 and α-enolase with higher levels in brain from old SAMP8 mice treated by AO against PS-1 compared to old SAMP8 treated with random AO and old SAMP8 treated with water. We also found LDH and isoform 2 of IDH3A less nitrated in old SAMP8 mice treated by AO against PS-1 compared to old SAMP8 treated with random AO and old SAMP8 treated with water. These proteins are related to the glycolytic and TCA pathways.
Mitochondrial-resident ATP synthase α chain plays a crucial role in production and release of ATP. As a component of complex V this protein transforms the mitochondrial proton gradient into chemical energy. Previous studies showed decrease levels of complex V [40] and its oxidative modification in AD [41]. Here, we showed an increased expression of ATP synthase α chain in brains of old SAMP8 mice treated by AO against PS-1, and, together with other identified proteins noted above, this suggests that energy metabolism is improved by AO treatment against PS-1, which significantly improves learning and memory in SAMP8 mice and is connected with the behavioral measures shown in this study.
LDH is an important enzyme that, during anaerobic conditions, converts pyruvate to lactate, an alternative source for neuronal energy production. We found LDH nitrated in AD brain, and this modification decreased the enzymatic activity and compromised the additional use of lactate for energy production [42]. Further, in aged SAMP8 mouse brain, LDH was identified as oxidized and with low expression levels, suggesting an impairment of LDH activity even in aged SAMP8 mice [43]. In our study, LDH was found less nitrated in old SAMP8 after injection with AO against PS-1. These data suggest a reduction of oxidative stress and a recovery of LDH activity with consequent increased energy production.
α-Enolase, a subunit of enolase, is involved in energy metabolism but also has roles in neuronal outgrowth and cellular differentiation, cell survival, and Aβ degradation [44]. Enolase is the only protein with increased amounts of protein carbonyls, protein-bound HNE, and 3-NT in common in brain from subjects with AD and mild cognitive impairment (MCI) [44–46]. Enolase also was found oxidized in aged SAMP8 [13], but its level was not altered in aged brains [47] and in AD brains [47]. We found an increased expression of α-enolase in aged SAMP8 mice after injection with AO against PS-1 that may help to reverse the reduced glucose metabolism and abnormal neuronal growth observed in aged SAMP8 mice, as well as activate pro-survival pathways, and increase Aβ clearance [44]. Taken together, our proteomics results confirm an interaction between PS-1 and cellular metabolism, and suggest that PS-1 mediated γ-secretase activity may be an important therapeutic target for AD.
Neuritic abnormality
CRMP-1 and CRMP-2, actin-related protein 2 and 3, and septin-11 are involved in axonal outgrowth, neuronal repair and they have a crucial role for neuronal connections preserving learning and memory processes. CRMP-2 was found down regulated and oxidatively modified in AD [45], adult Down syndrome (DS) [48] and fetal DS [49], indicating that the decreased activity of CRMP-2 has a role in these diseases. Previously [13] we found an increased specific carbonyl level of CRMP-2 in aged SAMP8 brains. This oxidative modification may be associated with the impairment in learning and memory in aged SAMP8 mice. We show here an increased expression and a decreased oxidation of CRMP-1 and actin-related proteins 2 and 3 in old SAMP8 mice after treatment with AO against PS-1 and this is consistent with the notion of restored normal extention of neurites, thereby improving interneuronal communication.
Cell cycle; tau phosphorylation; Abeta production
Pin-1 plays an important role in cell cycle regulation, recently seen to be abnormal in AD and MCI [50], and modulates the phosphorylation of tau protein, acting both on kinases and phosphatates. Pin-1 was found to be down-regulated and also oxidatively modified in AD brain [51, 52] and MCI brain[50, 53]. These alterations cause a significantly diminished activity of Pin1 that may lead to hyperphosphorylation of tau [54]. Hyperphosphorylation of tau causes the formation of neurofibrillary tangles, which damage and kill neurons. We found an increased expression level of Pin-1 in brain in aged SAMP8 treated by AO. This increased could restore the activity of Pin-1 crucial to counteract the neurofibrillary tangle formation in AD.
Synaptic abnormalities
SNAPs are highly conserved proteins that exist in three isoforms: α-, β-, and γ-SNAP, and exert a central role in intracellular membrane fusion and vesicular trafficking, crucial for the function of the synapse. γ-SNAP was found oxidized in AD brain [51]. The oxidation of γ-SNAP may alter neurotransmitter systems, which is correlated with altered memory and cognition in AD. Here, the expression levels of β-SNAP are increased in aged SAMP8 treated by AO against PS-1 compared to aged SAMP8 treated by random AO, which is consistent with improved learning and memory following this treatment that are observed in this study.
Isoform 5 of Dyn-1, another protein involved in synaptic vesicle recycling and related to memory formation, has a central role in mitochondrial dynamics, and malfunction of this mechanism can contribute to cell injury during neurodegenerative diseases like AD. Nitric oxide can lead to S-nitrosylation of Dyn-1 with activation of its fission activity and consequent mitochondrial fragmentation, synaptic damage and neuronal apoptosis [55]. We found Dyn-1 is less oxidized in old SAMP8 mice treated by AO against PS-1 compared to old SAMP8 treated by random AO. Therefore, the treatment with AO against PS-1 might lead to reduced Aβ levels and consequently nitric oxide levels with consequent improved mitochondrial dynamics.
Lipid abnormalities
Isoform Acyl-CoA thioesterases are a group of enzymes that maintain lipid homeostasis, a pathway compromised in AD [56]. We found an increase in the expression levels of this protein that if produced in AD could modulate loss of lipid asymmetry reported in AD brain [57, 58] as well as affect beta-oxidation of fatty acids, improving the decreased energy metabolism in AD.
Lysosomal Dysfunction
Lysosomes exert an important role in the degradation of old and damaged proteins and organelles through two pathways: autophagic and endocytic pathways [59] known to be impaired in AD [60]. A recent study reports that PS-1 mutations involved in FAD compromised these pathways [61]. PS-1 is important for lysosomal acidification since it is responsible for the maturation of V-ATPase, a proton pump crucial to set lysosomal pH. PS-1 mutations cause incorrect localization of V-ATPase with consequent autophagic and lysosomal impairment.
In our study, V-ATPase is up-regulated in old SAMP8 mice after injection with AO against PS-1 compared to aged SAMP8 treated by random AO and aged SAMP8 treated with water. This increase conceivably could contribute to reversing the dysfunction of autophagic and endocytic pathways [62].
Figure 9.
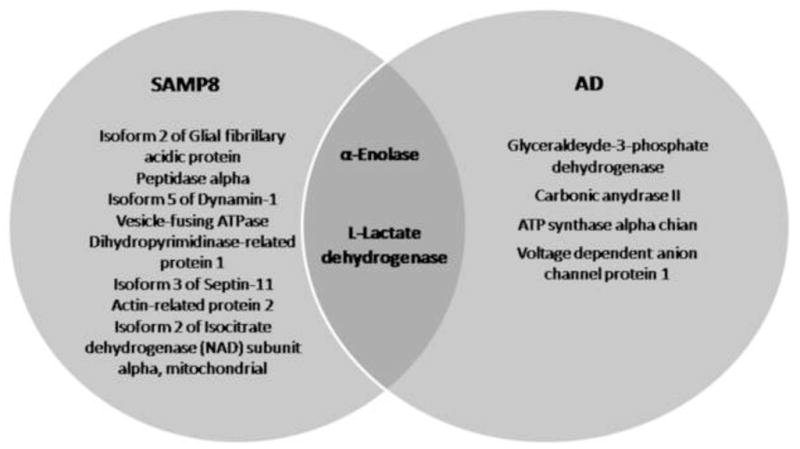
Venn diagram of relationship between nitrated brain proteins in subjects with AD and SAMP8 mice.
Summary of findings.
In our study, a decreased oxidative stress markers (protein carbonyls and 3-NT) were found, and all of the brain proteins identified as differently expressed and oxidized appear over expressed (except guanine nucleotide-binding protein G) and less nitrated (except CRMP-2 and peptidase α) in old SAMP8 mice treated by AO direct against PS-1. These proteins are involved in processes related to: energy metabolism, neuritic growth, lipid abnormalities, cell cycle, synaptic abnormalities, tau function, and lysosomal function, which are all impaired in AD (Fig. 9). That is, AO treatment against PS-1 leads to elevated levels or less nitration of proteins known to be altered in AD. Therefore, the treatment with AO directed against PS-1 in old SAMP8 mice, can reduce Aβ-mediated oxidative stress and increase expression of proteins known to be altered in AD and may support a possible pharmacologic strategy to combat this devastating dementing disease.
Highlights.
The senescence accelerate mouse P8 (SAMP8) is a model for cognitive impairment and amyloidosis.
SAMP8 mice were treated by an antisense directed against presenilin-1.
Inhibition of expression of presenilin-1 can reduce amyloid-beta deposition.
AO treatment reversed oxidative damage and cognitive deficits in brain.
Antisense against PS-1 represents a potential therapy for Alzheimer disease.
Acknowledgments
This work was supported in part by a NIH grant to DAB [AG-05119].
Abbreviations
- AD
Alzheimer’s Disease
- MCI
Mild Cognitive Impairment
- Aβ
Amyloid β-peptide
- FAD
Familial Alzheimer’s Disease
- NFT
Intracellular neurofibrillary tangles
- APP
Amyloid precursor protein
- PS-1
Presenilin-1
- PS-2
Presenilin-2
- AO
Antisense oligonucleotide
- SAM
Senescence accelerated mouse
- BCIP/NBT
5-Bromo-4-chloro-3-indolyl phosphate/Nitrotetrazolium Blue chloride
- NAD
Isocitrate dehydrogenase
- IDH3A
subunit alpha
- KPYM
Isoform M1 pyruvate kinase isozymes M1/M2
- PDHA1 and PDH
Pyruvate dehydrogenase E1 component subunit alpha and beta
- LDH
L-lactate dehydrogenase
- AATC
Aspartate aminotransferase
- ES1
ES1 protein homolog
- NSF
Vesicle-fusing ATPase/Vesicular-fusion protein NSF
- CRMP-1
Dihydropyrimidinase-related protein 1
- CRMP-2
Dihydropyrimidinase-related protein 2
- SNAPs
Soluble N-ethylmaleimide-sensitive factor attachment proteins
- V-ATPase
vacuolar-type H+-ATPase
- TPI
Triosephosphate isomerase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Butterfield DA. β-Amyloid-Associated Free Radical Oxidative Stress and Neurotoxicity: Implications for Alzheimer’s Disease. Chem Res Toxicol. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid β-peptide. Trends in Molecular Medicine. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 3.Butterfield DA, Hensley K, Harris M, Mattson M, Carney J. β-Amyloid Peptide Free Radical Fragments Initiate Synaptosomal Lipoperoxidation in a Sequence-Specific Fashion: Implications to Alzheimer’s Disease. Biochemical and Biophysical Research Communications. 1994;200:710–715. doi: 10.1006/bbrc.1994.1508. [DOI] [PubMed] [Google Scholar]
- 4.Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer’s β-Amyloid peptide (1–40) in cultured hippocampal neurons. Experimental Neurology. 1995;131:193–202. doi: 10.1016/0014-4886(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 5.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radical Biology and Medicine. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 6.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Aβ1–42. Journal of Neurochemistry. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 7.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yatin S, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1-42) Neurobiology of Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. [DOI] [PubMed] [Google Scholar]
- 9.Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, Banks WA, Butterfield DA. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: Implications for Aβ accumulation in AD brain. Free Radical Biology and Medicine. 2010;49:1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiology of Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 11.Kanski J, Aksenova M, Butterfield D. The hydrophobic environment of Met35 of Alzheimer’s Aβ(1–42) is important for the neurotoxic and oxidative properties of the peptide. Neurotoxicity Research. 2002;4:219–223. doi: 10.1080/10298420290023945. [DOI] [PubMed] [Google Scholar]
- 12.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 13.Poon HF, Joshi G, Sultana R, Farr SA, Banks WA, Morley JE, Calabrese V, Butterfield DA. Antisense directed at the Aβ region of APP decreases brain oxidative markers in aged senescence accelerated mice. Brain Research. 2004;1018:86–96. doi: 10.1016/j.brainres.2004.05.048. [DOI] [PubMed] [Google Scholar]
- 14.Morley JE, Kumar VB, Bernardo AE, Farr SA, Uezu K, Tumosa N, Flood JF. Beta-amyloid precursor polypeptide in SAMP8 mice affects learning and memory. Peptides. 2000;21:1761–1767. doi: 10.1016/s0196-9781(00)00342-9. [DOI] [PubMed] [Google Scholar]
- 15.Gewirtz AM, Sokol DL, Ratajczak MZ. Nucleic Acid Therapeutics: State of the Art and Future Prospects. 1998;92:3. [PubMed] [Google Scholar]
- 16.Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neuroscience. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA, Morley JE. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21:1769–1775. doi: 10.1016/s0196-9781(00)00339-9. [DOI] [PubMed] [Google Scholar]
- 18.Morley JE, Farr SA, Kumar VB, Armbrecht HJ. The SAMP8 Mouse: A Model to Develop Therapeutic Interventions for Alzheimer’s Disease. Curr Pharm Des. 2012;18:1123–1130. doi: 10.2174/138161212799315795. [DOI] [PubMed] [Google Scholar]
- 19.Shimano Y. [Studies on aging through analysis of the glucose metabolism related to the ATP--production of the senescence accelerated mouse (SAM)] Hokkaido Igaku Zasshi. 1998;73:557–569. [PubMed] [Google Scholar]
- 20.Butterfield DA, Poon HF. The senescence-accelerated prone mouse (SAMP8): A model of age-related cognitive decline with relevance to alterations of the gene expression and protein abnormalities in Alzheimer’s disease. Experimental Gerontology. 2005;40:774–783. doi: 10.1016/j.exger.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. Journal of Neurochemistry. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 22.Morley JE, Armbrecht HJ, Farr SA, Kumar VB. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim Biophys Acta. 2011;1822:650–656. doi: 10.1016/j.bbadis.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 23.Poon HF, Farr SA, Banks WA, Pierce WM, Klein JB, Morley JE, Butterfield DA. Proteomic identification of less oxidized brain proteins in aged senescence-accelerated mice following administration of antisense oligonucleotide directed at the Aβ region of amyloid precursor protein. Brain Res Mol Brain Res. 2005;138:8–16. doi: 10.1016/j.molbrainres.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 25.Thongboonkerd V, Luengpailin J, Cao J, Pierce WM, Cai J, Klein JB, Doyle RJ. Fluoride Exposure Attenuates Expression of Streptococcus pyogenes virulence factors. J Biol Chem. 2002;277:16599–16605. doi: 10.1074/jbc.M200746200. [DOI] [PubMed] [Google Scholar]
- 26.Butterfield DA. Protein oxidation processes in aging brain. Cell aging gerontol. 1997;2:161–191. [Google Scholar]
- 27.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radical Biology and Medicine. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomita T, Iwatsubo T. The inhibition of gamma-secretase as a therapeutic approach to Alzheimer’s disease. Drug News Perspect. 2004;17:321–325. doi: 10.1358/dnp.2004.17.5.829036. [DOI] [PubMed] [Google Scholar]
- 29.Morohashi Y, Kan T, Tominari Y, Fuwa H, Okamura Y, Watanabe N, Sato C, Natsugari H, Fukuyama T, Iwatsubo T, Tomita T. C-terminal Fragment of Presenilin Is the Molecular Target of a Dipeptidic γ-Secretase-specific Inhibitor DAPT (N-[N- (3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-Butyl Ester) J Biol Chem. 2006;281:14670–14676. doi: 10.1074/jbc.M513012200. [DOI] [PubMed] [Google Scholar]
- 30.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 31.Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ, Arango JC. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med. 1996;2:1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 32.Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibitearlyembryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE 1, the Alzheimer’s disease beta-secretase, have normal phenotype and abolish beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- 34.Kumar VB, Franko M, Banks WA, Kasinadhuni P, Farr SA, Vyas K, Choudhuri V, Morley JE. Increase in presenilin 1 (PS1) levels in senescence-accelerated mice (SAMP8) may indirectly impair memory by affecting amyloid precursor protein (APP) processing. J Exp Biol. 2009;212:494–498. doi: 10.1242/jeb.022780. [DOI] [PubMed] [Google Scholar]
- 35.Butterfield DA, Howard BJ, LaFontaine MA. Brain oxidative stress in animal models of accelerated aging and the age-related neurodegenerative disorders, Alzheimer’s disease and Huntington’s disease. Curr Med Chem. 2001;8:815–828. doi: 10.2174/0929867013373048. [DOI] [PubMed] [Google Scholar]
- 36.Atamna H, Frey WHn. Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion. 2007;7:297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 38.Crouch PJ, Cimidins K, Duce JA, Bush AI, Trounce IA. Mitochondria in aging and Alzheimer’s disease. Rejuvenation Res. 2007;10:349–357. doi: 10.1089/rej.2007.0592. [DOI] [PubMed] [Google Scholar]
- 39.Sultana R, Reed T, Perluigi M, Coccia R, Pierce WM, Butterfield DA. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med. 2007;11:839–851. doi: 10.1111/j.1582-4934.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schägger H, Ohm TG. Human Diseases with Defects in Oxidative Phosphorylation. European Journal of Biochemistry. 1995;227:916–921. doi: 10.1111/j.1432-1033.1995.tb20219.x. [DOI] [PubMed] [Google Scholar]
- 41.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiology of Disease. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. Journal of Neurochemistry. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 43.Poon HF, Castegna A, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience. 2004;126:915–926. doi: 10.1016/j.neuroscience.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 44.Butterfield DA, Lange MLB. Multifunctional roles of enolase in Alzheimer’s disease brain: beyond altered glucose metabolism. Journal of Neurochemistry. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, α-enolase and heat shock cognate 71. Journal of Neurochemistry. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 46.Schonberger SJ, Edgar PF, Kydd R, Faull RLM, Cooper GJS. Proteomic analysis of the brain in Alzheimer’s disease: Molecular phenotype of a complex disease process. PROTEOMICS. 2001;1:1519–1528. doi: 10.1002/1615-9861(200111)1:12<1519::aid-prot1519>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 47.Kato K, Suzuki F, Morishita R, Asano T, Sato T. Selective increase in S-100 beta protein by aging in rat cerebral cortex. J Neurochem. 1990;54:1269–1274. doi: 10.1111/j.1471-4159.1990.tb01958.x. [DOI] [PubMed] [Google Scholar]
- 48.Lubec G, Nonaka M, Krapfenbauer K, Gratzer M, Cairns N, Fountoulakis M. Expression of the dihydropyrimidinase related protein 2 (DRP-2) in Down syndrome and Alzheimer’s disease brain is downregulated at the mRNA and dysregulated at the protein level. J Neural Transm Suppl. 1999;57:161–177. doi: 10.1007/978-3-7091-6380-1_10. [DOI] [PubMed] [Google Scholar]
- 49.Weitzdoerfer R, Fountoulakis M, Lubec G. Aberrant expression of dihydropyrimidinase related proteins-2, -3 and -4 in fetal Down syndrome brain. J Neural Transm Suppl. 2001:95–107. doi: 10.1007/978-3-7091-6262-0_8. [DOI] [PubMed] [Google Scholar]
- 50.Keeney JR, Swomley A, Harris J, Fiorini A, Mitov M, Perluigi M, Sultana R, Butterfield DA. Cell Cycle Proteins in Brain in Mild Cognitive Impairment: Insights into Progression to Alzheimer Disease. Neurotoxicity Research. 2012;22:220–230. doi: 10.1007/s12640-011-9287-2. [DOI] [PubMed] [Google Scholar]
- 51.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiology of Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 53.Butterfield DA, Poon HF, StClair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiology of Disease. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 54.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiology of Aging. 2006;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Nakamura T, Cieplak P, Cho D-H, Godzik A, Lipton SA. S-Nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion. 2010;10:573–578. doi: 10.1016/j.mito.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional Membrane Phospholipid Alterations in Alzheimer’s Disease. Neurochemical Research. 1998;23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- 57.Bader Lange ML, Cenini G, Piroddi M, Mohmmad Abdul H, Sultana R, Galli F, Memo M, Butterfield DA. Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiology of Disease. 2008;29:456–464. doi: 10.1016/j.nbd.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Culter RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lynch G, Bi X. Lysosomes and brain aging in mammals. Neurochem Res. 2003;28:1725–1734. doi: 10.1023/a:1026069223763. [DOI] [PubMed] [Google Scholar]
- 60.Cataldo AM, Paskevich PA, Kominami E, Nixon RA. Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc Natl Acad Sci USA. 1991;88:10998–11002. doi: 10.1073/pnas.88.24.10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Domenico FSR, Ferree A, Smith K, Barone E, Perluigi M, Coccia R, Pierce W, Cai J, Mancuso C, Squillace R, Wiengele M, Dalle-Donne I, Wolozin B, Butterfield DA. Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: relevance to Parkinson disease. Antioxid Redox Signal. 2012;17:1490–1506. doi: 10.1089/ars.2011.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]