

Abstract
The precise mechanisms underlying contrast-induced acute kidney injury (CI-AKI) are not well understood. Intracellular Ca2+ overload is considered to be a key factor in CI-AKI. Voltage-dependent Ca2+ channel (VDC) and Na+/Ca2+ exchanger (NCX) system are the main pathways of intracellular Ca2+ overload in pathological conditions. Here, we review the potential underlying mechanisms involved in CI-AKI and discuss the role of NCX-mediated intracellular Ca2+ overload in the contrast media-induced renal tubular cell injury and renal hemodynamic disorder.
1. Pathogenesis of CI-AKI
Contrast-induced acute kidney injury (CI-AKI) is the third leading cause of hospital-acquired acute renal failure accounting for 10–12% of all causes of hospital-acquired renal failure [1]. In general population, the incidence is 1–6%. In some special populations, such as patients with underlying hypertension, cardiovascular diseases, diabetes mellitus, or preexisting renal insufficiency, the incidence is higher and may be as high as 20–50% [2–4]. In patients undergoing coronary angiography in China, the incidence of CI-AKI is 8.7%–23.5% [5, 6]. The precise mechanisms underlying CI-AKI are not fully understood, especially its cellular and molecular mechanism. But, it is clear that disturbance of renal hemodynamics and direct toxic action on renal tubular cells are main factors responsible for CI-AKI. Previous investigations [7, 8] have shown that contrast media administration can result in initial renal vasodilatation (about 20 minutes), followed by prolonged vasoconstriction (about 20 minutes to several hours). Subsequent studies [9, 10] demonstrated that there were regional differences in the vascular response to contrast media, with a greater reduction in flow to the outer medulla. And now, it has been verified that contrast-induced selective reduction in renal medullary blood flow and the secondary hypoxia in this region is a major underlying cause of CI-AKI [10]. It has been reported that calcium channel blockers (CCB) can reverse the acute hemodynamic alterations induced by contrast administration and alleviated CI-AKI [11–13]. Furthermore, our experimental animal investigation [14] also verified that tail vein injection of an inhibitor of reverse mode of Na+/Ca2+ exchanger (NCX) can suppress the contrast-induced ET-1 overproduction and renal vasoconstriction. These findings suggested that intracellular Ca2+ overload plays an important role in contrast-induced renal hemodynamic disorder. Besides changes in calcium physiology, contrast-induced vasoconstriction might also be a result of a direct effect on vascular smooth muscle [15] or from a local increase in adenosine [16] and endothelin [17] production.
It must be pointed out that, under normal circumstances, the contrast-induced renal hemodynamic disorder was not enough to induce CI-AKI based on the facts that humans as well as experimental animals without risk factors do not usually exhibit CI-AKI following contrast media injection. This is because, under physiological state, the renal circulation is subjected to autoregulation which is associated with neural, hormonal, paracrine, and autocrine influences. Injured autoregulation of microcirculation might be the cause that all kinds of risk factors such as preexisting renal impairment, diabetes mellitus, and hypercholesterolemia, make the kidney vulnerable to iodinated contrast media.
Renal tubular cells apoptosis is a key mechanism of CI-AKI. Studies have shown that contrast media can induce renal tubular epithelial cell apoptosis via ROS (reactive oxygen species) pathway, JNK/p38 stress kinase pathway, and intrinsic apoptotic pathways [18–20] and can also result in renal tubular epithelial cell injury by dephosphorylation (inactivation) of the kinase Akt [21]. But it is still unclear why contrast media can cause ROS overproduction and why contrast media can activate p38 Mitogen-Activated Protein Kinases (MAPK). Our recent studies showed that contrast-induced ROS overproduction, p38 activation, and tubular cell apoptosis might be associated with intracellular calcium overload [19, 22, 23].
2. The Role of Intracellular Ca2+ in the Pathogenesis of Contrast-Induced Acute Kidney Injury
Intracellular calcium overload is considered to be a key factor in ischemic cell injury and CI-AKI [12]. Studies have shown that both renal vasoconstriction and renal tubular apoptosis induced by contrast media are associated with changes in calcium physiology [11, 13, 22, 23]. Although physiological and pathophysiological mechanisms of Ca2+ overload in ischemic kidney and CI-AKI have not been fully elucidated, there is evidence indicating that increased cytosolic Ca2+ may be an important mediator of epithelial cell apoptosis and necrosis [24]. So, theoretically, CCB would have protective effects on CI-AKI. In clinical practice, CCB can reverse the acute hemodynamic alterations induced by radiocontrast administration and alleviated CI-AKI [11–13]. However, acute administration of CCB before contrast media administration is not enough to prevent CI-AKI [25]. Only one small trial demonstrated any value with CCB [13] whereas other studies showed no beneficial effects [26, 27]. The fact that acute administration of CCB before contrast media administration was not enough to prevent CI-AKI suggested that the intracellular Ca2+ overload induced by contrast media might not be completely suppressed by CCB. So we cannot conclude based on these clinical data that intracellular calcium overload was not associated with CI-AKI because VDC is not the only pathway that induces Ca2+ influx. There is the possibility that other channels besides VDC may also be involved in the contrast-induced intracellular calcium overload. Our recent study [22] has shown that contrast media resulted in NRK-52E cell apoptosis via the induction of an increase in intracellular Ca2+ and reactive oxygen species and KB-R7943, inhibitor of the reverse mode of NCX, attenuated the contrast media-induced renal tubular epithelial cell apoptosis by suppressing the intracellular Ca2+ overload and reducing oxidative stress, which suggested that intracellular Ca2+ overload via the NCX system is also involved in contrast-induced renal tubular apoptosis.
3. The Role of Na+/Ca2+ Exchanger System in the Pathogenesis of CI-AKI
NCX is a bidirectional plasma membrane transporter that catalyzes the exchange of 3 or 4 Na+ for 1 Ca2+, depending on the electrochemical gradients of the substrate ions [28, 29] and is encoded by a multigene family comprising 3 NCX isoforms: NCX1, which is expressed in various organs including the kidney [30]; and NCX2 and NCX3, which are expressed mainly in the brain and skeletal muscle [31, 32]. Under physiological conditions, NCX can pump the Ca2+ outside the cell using the Na+ concentration gradient across the cell membrane to keep a low intracellular Ca2+ level, which is referred to as the forward-mode operation of the exchanger. In pathological conditions, NCX can reversely extrude Na+ for Ca2+ influx and result in intracellular Ca2+ overload, which is referred to as the reverse mode or calcium influx mode of NCX. In the normal kidney, NCX plays an important role in the active calcium transport in distal convoluted tubules [33]. In the ischemia-reperfusion kidney and in the hypoxia-reoxygenation renal tubular epithelial cells, NCX reversely extrudes Na+ for Ca2+ influx and results in intracellular Ca2+ overload and tubular epithelial cell injury [34, 35].
It has been verified that contrast media can induce renal tubular epithelial cell apoptosis via ROS pathway, JNK/p38 pathway, and intrinsic apoptosis pathway [18, 20]. Our recent in vitro studies [19, 36] demonstrated that contrast-induced ROS overproduction, p38 activation, and apoptosis in renal tubular cell were associated with the increase of intracellular Ca2+. The inhibitor of reverse mode of NCX, KB-R7943, can alleviate contrast-induced renal tubular apoptosis through suppressing the increase of intracellular Ca2+ and subsequent ROS overproduction and p38 activation. These data demonstrate that intracellular Ca2+ overload via the reverse mode of NCX system is involved in contrast-induced renal tubular epithelial cell apoptosis.
Recent animal model experiments [14, 37] also showed that pretreatment with tail vein injection of KB-R7943 markedly and dose-dependently suppressed the increase in renal ET-1 production and the reduction in renal blood flow induced by contrast medium administration and prevented contrast-induced acute renal failure, which suggested that Ca2+ overload via the reverse mode of NCX, followed by renal ET-1 overproduction and renal vasoconstriction, plays an important role in the pathogenesis of CI-AKI.
4. Hypothesis about the Molecular Mechanism of CI-AKI
Based on the findings [19, 22, 36] that inhibition of the reverse mode of NCX alleviated contrast-induced renal tubular cell apoptosis through suppressing the increase of intracellular Ca2+, ROS overproduction, p38 MAPK activation, and Caspase-3 overexpression and the findings [14, 37] that tail vein injection of inhibitor of reverse mode of NCX can exert protective effects on CI-AKI in rats through suppressing contrast-induced renal ET-1 overproduction and renal vasoconstriction, we propose the following hypothesis regarding the molecular mechanism of CI-AKI. Contrast medium exposure activates the reverse mode of NCX1 expressed in renal tubular epithelial cells; NCX reversely extrudes Na+ for Ca2+ influx and results in increased intracellular Ca2+. The increased intracellular Ca2+ can stimulate Ca2+ release from the mitochondrial and endoplasmic reticulum and result in intracellular Ca2+ overload [38]. The intracellular Ca2+ overload via the reverse mode of NCX and VDC induced by contrast media in the renal tubular epithelial cell can result in ROS overproduction and oxidative stress. Increased ROS and intracellular Ca2+ can induce upregulation of p38 and p-p38 MAPK expression [36] and subsequently activate intrinsic apoptotic pathways such as bcl-2, bax, and caspase-3 and result in renal tubular epithelial cell apoptosis, which is the underlying cause of contrast-induced direct renal tubular toxicity. p38 MAPK activation via the reverse mode of NCX and VDC could also result in renal ET-1 overproduction, followed by renal vasoconstriction and renal ischemia, which is one of the underlying causes of contrast-induced renal hemodynamic abnormalities. ET-1 overproduction and renal ischemia can cause depletion of adenosine triphosphate (ATP) and development of intracellular acidosis. The accumulation of intracellular Na+, which is caused by inhibition of Na+/K+-ATPase activity because of decreased ATP production [39] and activation of the Na+/H+ exchange because of intracellular acidosis [40], can also activate the reversion of the mode of NCX and subsequently cause calcium overload and ET-1 overproduction, forming a vicious cycle. The diagram of the hypothesis about the molecular mechanism of CI-AKI is seen in Figure 1. Contrast media exposure activates VDC and the reverse mode of NCX expressed in the renal tubular epithelial cell and induces Ca2+ influx. The increased intracellular Ca2+ stimulates Ca2+ release from the mitochondrial and endoplasmic reticulum and results in intracellular Ca2+ overload, which induced ROS overproduction and oxidative stress. Increased ROS and intracellular Ca2+ activate p38 MAPK. On one hand, p38 MAPK activates intrinsic apoptotic pathways such as bcl-2, bax, and caspase-3 and induces renal tubular epithelial cell apoptosis, which is the underlying cause of contrast-induced direct renal tubular toxicity. On the other hand, activated p38 MAPK also results in renal ET-1 overproduction, followed by renal vasoconstriction and renal ischemia, which is one of the underlying causes of contrast-induced renal hemodynamic abnormalities. ET-1 overproduction and renal ischemia can cause depletion of ATP and development of intracellular acidosis, which can result in accumulation of intracellular Na+ and further activate the reversion of the mode of NCX and subsequently cause Ca2+ influx and ET-1 overproduction, forming a vicious cycle.
Figure 1.
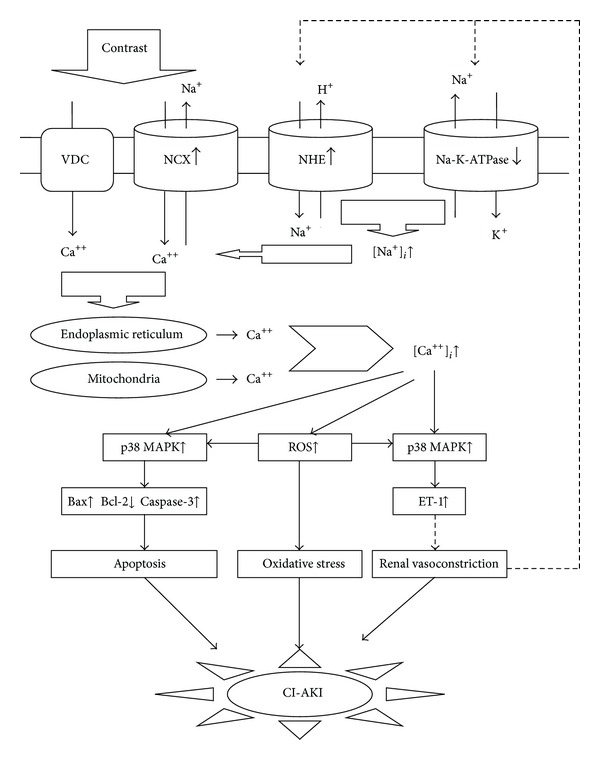
The diagram shows the proposed molecular mechanism of CI-AKI. CI-AKI, contrast-induced acute renal injury; NCX, Na+/Ca2+ exchanger; VDC, the voltage-dependent Ca2+ channel; NHE, Na+/H+ exchange; [Ca++]i, intracellular Ca2+ concentration; [Na+]i, intracellular Na+ concentration; p38 MAPK (p38 Mitogen-Activated Protein Kinases); ROS, reactive oxygen species. ET-1, endothelin-1; ATP, adenosine triphosphate.
5. Conclusion
In summary, Ca2+ overload via the reverse mode of NCX1 and VDC, followed by ROS overproduction, p38 MAPK activation, and ET-1 overproduction, plays an important role in the contrast-induced renal hemodynamic disorder and renal tubular epithelial cell apoptosis, which suggests that, in clinical practice, CCB should be recommended to patients with hypertension who are undergoing radiographic examination or therapy requiring contrast media and that selective inhibitors of NCX1 may be beneficial in the prevention and treatment of CI-AKI in humans.
Disclosure
This paper was not published or submitted elsewhere.
Conflict of Interests
There does not exist any financial or other conflict of interests.
Acknowledgments
This study was supported by research Grants from the National Natural Science Funds of China (81370841) and the Natural Science Fund of Hubei province (2012FFB04426).
References
- 1.Hou SH, Bushinsky DA, Wish JB. Hospital-acquired renal insufficiency: a prospective study. American Journal of Medicine. 1983;74(2):243–248. doi: 10.1016/0002-9343(83)90618-6. [DOI] [PubMed] [Google Scholar]
- 2.Goldenberg I, Matetzky S. Nephropathy induced by contrast media: pathogenesis, risk factors and preventive strategies. Canadian Medical Association Journal. 2005;172(11):1461–1471. doi: 10.1503/cmaj.1040847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh Y, Yano T, Sendo T, Oishi R. Clinical and experimental evidence for prevention of acute renal failure induced by radiographic contrast media. Journal of Pharmacological Sciences. 2005;97(4):473–488. doi: 10.1254/jphs.crj05002x. [DOI] [PubMed] [Google Scholar]
- 4.Ledneva E, Karie S, Launay-Vacher V, Janus N, Deray G. Renal safety of gadolinium-based contrast media in patients with chronic renal insufficiency. Radiology. 2009;250(3):618–628. doi: 10.1148/radiol.2503080253. [DOI] [PubMed] [Google Scholar]
- 5.Gao F, Zhou YJ, Zhu X, Wang ZJ, Yang SW, Shen H. C-reactive protein and the risk of contrast-induced acute kidney injury in patients undergoing percutaneous coronary intervention. American Journal of Nephrology. 2011;34(3):203–210. doi: 10.1159/000329534. [DOI] [PubMed] [Google Scholar]
- 6.Ling W, Zhaohui N, Ben H, et al. Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron Clinical Practice. 2008;108(3):176–181. doi: 10.1159/000117814. [DOI] [PubMed] [Google Scholar]
- 7.Bakris GL, Burnett JC., Jr. A role for calcium in radiocontrast-induced reductions in renal hemodynamics. Kidney International. 1985;27(2):465–468. doi: 10.1038/ki.1985.32. [DOI] [PubMed] [Google Scholar]
- 8.Katzberg RW, Morris TW, Burgener FA. Renal renin and hemodynamic responses to selective renal artery catheterization and angiography. Investigative Radiology. 1977;12(5):381–388. doi: 10.1097/00004424-197709000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Nygren A. Contrast media and regional renal blood flow. A study of the effects of ionic and non-ionic monomeric and dimeric contrast media in the rat. Acta Radiologica. 1992;378(part 3):123–135. [PubMed] [Google Scholar]
- 10.Liss P, Nygren A, Olsson U, Ulfendahl HR, Erikson U. Effects of contrast media and mannitol on renal medullary blood flow and red cell aggregation in the rat kidney. Kidney International. 1996;49(5):1268–1275. doi: 10.1038/ki.1996.181. [DOI] [PubMed] [Google Scholar]
- 11.Russo D, Testa A, Della Volpe L, Sansone G. Randomised prospective study on renal effects of two different contrast media in humans: protective role of a calcium channel blocker. Nephron. 1990;55(3):254–257. doi: 10.1159/000185971. [DOI] [PubMed] [Google Scholar]
- 12.Duan SB, Liu FY, Luo JA, et al. Nephrotoxicity of high- and low-osmolar contrast media: the protective role of amlodipine in a rat model. Acta Radiologica. 2000;41(5):503–507. doi: 10.1080/028418500127345794. [DOI] [PubMed] [Google Scholar]
- 13.Neumayer HH, Junge W, Kufner A, Wenning A. Prevention of radiocontrast-media-induced nephrotoxicity by the calcium channel blocker nitrendipine: a prospective randomised clinical trial. Nephrology Dialysis Transplantation. 1989;4(12):1030–1036. [PubMed] [Google Scholar]
- 14.Yang DW, Yang DP, Jia RH, Tan J. Na+/Ca2+ exchange inhibitor, KB-R7943, attenuates contrast-induced acute kidney injury. Journal of Nephrology. 2013;26:877–885. doi: 10.5301/jn.5000259. [DOI] [PubMed] [Google Scholar]
- 15.Karstoft J, Bååth L, Jansen I, Edvinsson L. Vasoconstriction of isolated arteries induced by angiographic contrast media. A comparison of ionic and non-ionic contrast media iso-osmolar with plasma. Acta Radiologica. 1995;36(3):312–316. [PubMed] [Google Scholar]
- 16.Pflueger A, Larson TS, Nath KA, King BF, Gross JM, Knox FG. Role of adenosine in contrast media-induced acute renal failure in diabetes mellitus. Mayo Clinic Proceedings. 2000;75(12):1275–1283. doi: 10.4065/75.12.1275. [DOI] [PubMed] [Google Scholar]
- 17.Bagnis C, Idee JM, Dubois M, et al. Role of endothelium-derived nitric oxide-endothelin balance in contrast medium-induced acute renal vasoconstriction in dogs. Academic Radiology. 1997;4(5):343–348. doi: 10.1016/s1076-6332(97)80115-8. [DOI] [PubMed] [Google Scholar]
- 18.Quintavalle C, Brenca M, De Micco F, et al. In vivo and in vitro assessment of pathways involved in contrast media-induced renal cells apoptosis. Cell Death and Disease. 2011;2(5, article e155) doi: 10.1038/cddis.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang DP, Jia RH, Ding GH, Zhang J. KB-R7943 decreased renal tubular cell apoptosis and p38 expression induced by contrast media. Chinese Journal of Experimental Surgery. 2008;5:641–643. [Google Scholar]
- 20.Gong X, Celsi G, Carlsson K, Norgren S, Chen M. N-acetylcysteine amide protects renal proximal tubular epithelial cells against iohexol-induced apoptosis by blocking p38 MAPK and iNOS signaling. American Journal of Nephrology. 2010;31(2):178–188. doi: 10.1159/000268161. [DOI] [PubMed] [Google Scholar]
- 21.Andreucci M, Fuiano G, Presta P, et al. Radiocontrast media cause dephosphorylation of Akt and downstream signaling targets in human renal proximal tubular cells. Biochemical Pharmacology. 2006;72(10):1334–1342. doi: 10.1016/j.bcp.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Yang DP, Yang DW, Jia RH, Ding GH. Selective inhibition of reverse mode of Na+/Ca2+ exchanger attenuates contrast-induced cell injury. American Journal of Nephrology. 2013;37:264–273. doi: 10.1159/000348526. [DOI] [PubMed] [Google Scholar]
- 23.Yang DP, Jia RH, Ding GH, Yang DW. Felodipine attenuates NRK52E cell injury induced by contrast media. Herald of Medicine. 2008;27(10):1153–1156. [Google Scholar]
- 24.Wilson DR, Arnold PE, Burke TJ, Schrier RW. Mitochondrial calcium accumulation and respiration in ischemic acute renal failure in the rat. Kidney International. 1984;25(3):519–526. doi: 10.1038/ki.1984.48. [DOI] [PubMed] [Google Scholar]
- 25.Carraro M, Mancini W, Artero M, et al. Dose effect of nitrendipine on urinary enzymes and microproteins following non-ionic radiocontrast administration. Nephrology Dialysis Transplantation. 1996;11(3):444–448. [PubMed] [Google Scholar]
- 26.Khoury Z, Schlicht JR, Como J, et al. The effect of prophylactic nifedipine on renal function in patients administered contrast media. Pharmacotherapy. 1995;15(1 I):59–65. [PubMed] [Google Scholar]
- 27.Spangberg-Viklund B, Berglund J, Nikonoff T, Nyberg P, Skau T, Larsson R. Does prophylactic treatment with felodipine, a calcium antagonist, prevent low-osmolar contrast-induced renal dysfunction in hydrated diabetic and nondiabetic patients with normal or moderately reduced renal function? Scandinavian Journal of Urology and Nephrology. 1996;30(1):63–68. doi: 10.3109/00365599609182351. [DOI] [PubMed] [Google Scholar]
- 28.Philipson KD, Nicoll DA, Ottolia M, et al. The Na+/Ca2+ exchange molecule: an overview. Annals of the New York Academy of Sciences. 2002;976:1–10. doi: 10.1111/j.1749-6632.2002.tb04708.x. [DOI] [PubMed] [Google Scholar]
- 29.Condrescu M, Opuni K, Hantash BM, Reeves JP. Cellular regulation of sodium-calcium exchange. Annals of the New York Academy of Sciences. 2002;976:214–223. doi: 10.1111/j.1749-6632.2002.tb04744.x. [DOI] [PubMed] [Google Scholar]
- 30.Nicoll DA, Longoni S, Philipson KD. Molecular cloning and functional expression of the cardiac sarcolemmal Na+-Ca2+ exchanger. Science. 1990;250(4980):562–565. doi: 10.1126/science.1700476. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Matsuoka S, Hryshko LV, et al. Cloning of the NCX2 isoform of the plasma membrane Na+-Ca2+ exchanger. Journal of Biological Chemistry. 1994;269(26):17434–17439. [PubMed] [Google Scholar]
- 32.Nicoll DA, Quednau BD, Qui Z, Xia YR, Lusis AJ, Philipson KD. Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. Journal of Biological Chemistry. 1996;271(40):24914–24921. doi: 10.1074/jbc.271.40.24914. [DOI] [PubMed] [Google Scholar]
- 33.Magyar CE, White KE, Rojas R, Apodaca G, Friedman PA. Plasma membrane Ca2+-ATPase and NCX1 Na+/Ca2+ exchanger expression in distal convoluted tubule cells. American Journal of Physiology—Renal Physiology. 2002;283(1):F29–F40. doi: 10.1152/ajprenal.00252.2000. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita J, Itoh M, Kuro T, et al. Pre- or post-ischemic treatment with a novel Na+/Ca2+ exchange inhibitor, KB-R7943, shows renal protective effects in rats with ischemic acute renal failure. Journal of Pharmacology and Experimental Therapeutics. 2001;296(2):412–419. [PubMed] [Google Scholar]
- 35.Yamashita J, Kita S, Iwamoto T, et al. Attenuation of ischemia/reperfusion-induced renal injury in mice deficient in Na+/Ca2+ exchanger. Journal of Pharmacology and Experimental Therapeutics. 2003;304(1):284–293. doi: 10.1124/jpet.102.039024. [DOI] [PubMed] [Google Scholar]
- 36.Yang DP, Jia RH, Ding GH, Yang DW, Xiong XL. Reverse mode of Na+Ca2+ exchange inhibitor, KB-r7943 attenuates tubular epithelial cell apoptosis induced by contrast media. Chinese Journal of Emergency Medicine. 2008;17(7):713–716. [Google Scholar]
- 37.Yang DW, Yang DP, Jia RH, Lin S. Effects of selective inhibition of reverse mode of Na+/Ca2+ exchanger on rats with contrast-induced acute kidney injury. National Medical Journal of China. 2013;93(22):1750–1754. [PubMed] [Google Scholar]
- 38.Yu L, Netticadan T, Xu YJ, Panagia V, Dhalla NS. Mechanisms of lysophosphatidylcholine-induced increase in intracellular calcium in rat cardiomyocytes. Journal of Pharmacology and Experimental Therapeutics. 1998;286(1):1–8. [PubMed] [Google Scholar]
- 39.Cross HR, Radda GK, Clarke K. The role of Na+/K+ ATPase activity during low flow ischemia in preventing myocardial injury: a 31P, 23Na and 87Rb NMR spectroscopic study. Magnetic Resonance in Medicine. 1995;34(5):673–685. doi: 10.1002/mrm.1910340505. [DOI] [PubMed] [Google Scholar]
- 40.Scholz W, Albus U, Lang HJ, et al. Hoe 694, a new Na+/H+ exchange inhibitor and its effects in cardiac ischaemia. British Journal of Pharmacology. 1993;109(2):562–568. doi: 10.1111/j.1476-5381.1993.tb13607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]