

Abstract
Effectively determining masses of proteins is critical to many biological studies (e.g. for structural biology investigations). Accurate mass determination allows one to evaluate the correctness of protein primary sequences, the presence of mutations and/or post-translational modifications, the possible protein degradation, the sample homogeneity, and the degree of isotope incorporation in case of labelling (e.g. 13C labelling).
Electrospray ionisation (ESI) mass spectrometry (MS) is widely used for mass determination of denatured proteins, but its efficiency is affected by the composition of the sample buffer. In particular, the presence of salts, detergents, and contaminants severely undermines the effectiveness of protein analysis by ESI-MS. Matrix-assisted laser desorption/ionization (MALDI) MS is an attractive alternative, due to its salt tolerance and the simplicity of data acquisition and interpretation. Moreover, the mass determination of large heterogeneous proteins (bigger than 100 kDa) is easier by MALDI-MS due to the absence of overlapping high charge state distributions which are present in ESI spectra.
Here we present an accessible approach for analysing proteins larger than 100 kDa by MALDI-time of flight (TOF). We illustrate the advantages of using a mixture of two matrices (i.e. 2,5-dihydroxybenzoic acid and α-cyano-4-hydroxycinnamic acid) and the utility of the thin layer method as approach for sample deposition. We also discuss the critical role of the matrix and solvent purity, of the standards used for calibration, of the laser energy, and of the acquisition time. Overall, we provide information necessary to a novice for analysing intact proteins larger than 100 kDa by MALDI-MS.
Keywords: Chemistry, Issue 79, Chemistry Techniques, Analytical, Mass Spectrometry, Analytic Sample Preparation Methods, biochemistry, Analysis of intact proteins, mass spectrometry, matrix-assisted laser desorption ionization, time of flight, sample preparation
Introduction
Structural biology relies on the production of high quality proteins 1 and therefore needs to be coupled with efficient and reliable techniques for protein analysis 2,3. In our mass spectrometry (MS) laboratory within an institute of structural biology we need to confirm primary sequence of proteins, evaluate the presence of mutations and posttranslational modifications, the degradation of proteins, the sample homogeneity, and the quality of isotopic labelling (e.g. deuterated proteins for nuclear magnetic resonance studies4). Since structural biologists use limited proteolysis to distinguish structurally rigid domains from flexible parts, we need to reliably characterise such truncated proteins using MS.
When biomolecules are analysed by MS, two possible approaches are utilized to softly ionise such heavy and labile molecules. Electrospray ionisation (ESI) ionises molecules directly from the liquid phase 5; matrix-assisted laser desorption ionisation (MALDI) requires that the biomolecules are co-crystallized with ultraviolet-absorbing organic molecules (i.e. matrix molecules) 6.
ESI time-of-flight (TOF) MS coupled to liquid chromatography has become a routine technique for the analysis of intact proteins, because it allows mass determination with high accuracy (≤ 50 ppm). However, it is quite susceptible to the composition of the sample buffer (in particular to salts and detergents) and to contaminants (i.e. polymers) which are sometimes difficult to eliminate, causing the suppression of the analyte signal.
MALDI-TOF MS represents an effective alternative to ESI-MS because its performance is less affected by buffer components, detergents and contaminants, and allows intact protein mass determination with sufficient accuracy (≤ 500 ppm) for sequence validation. After protein digestion, MALDI-TOF MS can be also utilized to analyse the obtained peptides for further primary sequence confirmation by the so-called "peptide mass fingerprinting".
In our hands, the mass determination of intact proteins, which are larger than 100 kDa and heterogeneous (due to modifications or truncations), is easier by MALDI-MS than by ESI-MS. This is due to the lack of overlapping charge state distributions present in ESI spectra. Moreover, since the MALDI process is not dependent of the analyte size7, such method yields high sensitivity when a biomolecule mass is above 100 kDa. Remarkable analyses of intact proteins were carried out using home-made instruments between the end of the 1980's and the beginning of the 1990's8-11.
MALDI-TOF can be also used as screening tool to evaluate the quality of protein samples because it requires less time for sample preparation and is less susceptible to interferences due to common impurities (e.g. salts). After a first, quick evaluation by MALDI-MS, a sample can be further analysed by ESI-TOF to determine its mass with higher accuracy. Furthermore, MALDI generates ions containing fewer charges than ESI and therefore acquiring and interpreting MALDI data is more straightforward. This allows students working in structural biology to analyse their recombinant proteins just after a brief training.
Two key factors influence the quality of the MALDI spectra: the matrix and the technique used for the matrix deposition (e.g. dried droplet 8 and thin layer 12-16,17,18). A single organic matrix [e.g. sinapinic acid (SA) 19-21 or α-cyano-4-hydroxycinnamic acid (α-CHCA) 14,22,23] is often used for the MS examination of intact proteins and cross-linked protein complexes. Using α-CHCA, Chait group previously presented a detailed protocol for preparing an ultra thin layer prior to MALDI analysis of soluble and membrane proteins14,15. Recently, Gorka et al. illustrated the graphite-based target coating to improve the MALDI analysis of peptides and proteins using α-CHCA as matrix24.
Here, we present a simple protocol for the analysis of intact proteins by MALDI-TOF MS, utilising a mixture of two matrices: 2,5-dihydroxybenzoic acid (DHB) and α-CHCA 25. We systematically evaluated the performance of the DHB-CHCA mix compared to the SA and α-CHCA matrices, for the control of intact proteins. The matrix mixture allows a better resolution (i.e. the protein peaks are much sharper). Moreover, the presence of intense multiple charges ions (i.e. M+2H2+, M+3H3+, etc.) enables a more accurate mass determination because the resolution of axial MALDI-TOF instruments is higher at lower mass over charge (m/z) 26. This is particularly useful for the molecular weight determination of proteins larger than 100 kDa.
Higher sensitivity is also reached using the DHB-CHCA mixture (0.5 pmoles of protein spotted on the MALDI target).
As we mentioned above, an important factor that should be considered when using MALDI instrument is the matrix deposition. Laugesen et al. proposed the use of DHB-CHCA mixture for the first time 25, utilising the dried droplet deposition. However, we observed better results (e.g. much higher sensitivity) when we utilized the thin layer method where the first layer is formed by the α-CHCA dissolved in acetone. The thin layer method 12,27 implies the formation of a homogeneous substratum of matrix crystals on the MALDI target, which was described in a Jove video previously 15. Then, the sample is deposited on this substratum, and finally additional matrix is deposited (see below). In this article, we also illustrate how to deposit the sample on the MALDI target, but also how to clean the target15, to recrystallize, and prepare the matrices.
To conclude we aim to provide all the necessary information for analysing intact proteins to scientists (in particular, structural biologists) who need to evaluate the quality of produced proteins in a rapid and simple way and who are not so familiar with MALDI-TOF MS. As predicted in mid-1990s28, MS has had an increased impact on biological research, as its accessibility to biologists has increased. We hope that the information we provided will be useful to make MALDI-TOF accessible to biologists and scientists who would like to start using mass spectrometry.
Protocol
1. Protein Sample Preparation: Buffer Exchange (Optional)
5-25 μl of micromolar concentrations of protein (1 to 20 μM) are necessary. Buffer exchange can be carried out using centrifugal ultrafiltration devices (e.g. Vivaspin, Sartorius) or microcentrifuge gel filtration columns (e.g., Micro Bio-Spin 6 chromatography columns, Bio-Rad) 29,30. Buffer exchange steps can be repeated 2-3x . Detailed description of buffer exchange was previously presented 29,30. For example, we utilize 20 mM tris(hydroxymethyl)aminomethane (i.e. Tris) pH 8 as final buffer.
Note: This step could be omitted in many cases. It should be carried out when a protein sample contains molecules or buffers that could strongly interfere with MS detection [e.g. glycerol; 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, HEPES]. It is also useful to improve the quality of the spectra.
2. Recrystallization of Matrices in Order to Improve Their Purity (Optional)
Pour 10 ml of 40% ethanol (EtOH) into a Pyrex flask.
Add 600 mg of a matrix.
Using a water bath and a resistance heater, warm the matrix solution and stir it until the matrix is completely dissolved using a glass rod or a magnetic stirrer.
Repeat step 2 until you have a saturated solution.
Note: Using a too large volume of solvent or dissolving the matrix far below the boiling point of the solution will cause a poor yield of crystals or no crystals at all.
Allow the solution to cool slowly. You can leave it at room temperature for several hours and then at 4 °C overnight. Note: if crystals are not formed, crystallization can be induced by scratching the inside of the beaker (just below the solution surface) using a glass stirring rod.
Collect the matrix crystals by filtration.
Rinse the crystals with a minimum amount of ice-cold solvent and filtrate them.
Allow the crystals to dry. You can use vacuum for this step.
3. Cleaning of MALDI Stainless Steel Target
Rinse the MALDI plate with methanol (MeOH) and wipe gently with a cleaning tissue specially made for laboratory applications (e.g. Kimwipe).
Rinse the target with H2O and wipe it with a cleaning tissue.
Insert the MALDI plate in a 600 ml beaker and cover it with 50% EtOH.
Sonicate the target in the EtOH solution for 10 min in an ultrasonic bath.
If there are residues remaining on the plate, repeat the steps 1-4 once again.
Finally, rinse the target with MeOH (or with water, see the note below), tilt it in a way that all the liquid is collected on a cleaning tissue. Let the target dry at room temperature or using a nitrogen-gas flow.
Note: A similar procedure was previously described 15.
Note: The solvent (MeOH or water) used for the last rinsing of the target determines some target features. If the target is rinsed with MeOH, the sample spreads on the target much more than when using water.
4. α-CHCA Thin Layer Solution: Preparation and Deposition on the MALDI Target
Dissolve α-CHCA in acetone in order to make a saturated solution.
By hand (without any pipette) dip a 10 μl tip (e.g. GELoader Tip, Eppendorf) in α-CHCA saturated solution: a small amount of the solution will flow in the tip (due to capillarity).
Touch very rapidly (i.e. 1 sec) the MALDI target with pipette tip and deposit the α-CHCA acetone solution on the MALDI target. This will constitute the matrix thin layer, where the protein sample will be deposited. Try to generate a thin layer spot as small as possible.
Note: When using SA as matrix, we prepare a thin layer using a saturated solution of SA in acetone.
5. Preparation of Natrix Solutions: SA, α-CHCA, DHB and CHCA_DHB Mixture 25
Prepare a 20 mg/ml SA solution in ACN, 0.1% TFA (70:30, vol/vol).
Prepare 20 mg/ml α-CHCA solution in ACN and 5% formic acid (70:30, vol/vol) [named as "α-CHCA solution"].
Prepare a 20 mg/ml DHB solution in ACN and 0.1% trifluoroacetic acid (TFA) (70:30, vol/vol) [named as "DHB solution"].
Mix "α-CHCA solution" and "DHB solution" in 1:1 ratio (vol/vol), to obtain the "CHCA_DHB solution".
Note: One can mix "α-CHCA solution" and "DHB solution" in different ratios, when analysing proteins with mass less than 100 kDa. For example a ratio of 40:60 between α-CHCA and DHB (vol/vol) yields a better resolution, but less sensitivity (see discussion).
6. Sample Deposition on the MALDI Target
Deposit 0.5 μl of the protein sample on the previously prepared α-CHCA thin layer. Immediately after that, add 0.5 μl of the matrix solution (i.e. the SA solution or the α-CHCA solution or the "CHCA_DHB mix"). This implies mixing the sample and the matrix on the target.
Note: Usually one mixes the protein sample with the matrix in a 1:1 ratio. This ratio is critical for the good quality of the MALDI spectra. If you would like to test different sample concentrations, you can use the ACN_formic acid solution (described above) to dilute the sample.
Note: If you prefer, you can mix the sample and the matrix in a tube and then deposit the mixed "sample_matrix solution" on the thin layer.
Note: We suggest you to test different sample concentrations because diluting the sample allows you to reduce the interference of contaminants. To dilute the sample, you can use the ACN_formic acid solution (described above) to dilute the sample.
7. Calibrant Deposition
Deposit 0.5 μl of the calibrant standard (e.g. "protein standard II", Bruker Daltonics, Bremen) then add 0.5 μl of the matrix solution.
Note: Load the calibrant on the MALDI plate next to the protein sample spots (see discussion).
8. MALDI Spectra Acquisition of Calibrant and Protein Samples
Once the samples and the calibrant are dried, you can observe the "spots" under a microscope. This observation is optional and may be interesting mainly to novices. To acquire the sample spectra, insert the target in the MALDI-TOF instrument and choose the appropriate instrumental parameters which are different for each type of equipment. To get the most appropriate set-up one should follow the manufacturer recommendations. As general considerations when analysing intact proteins, one should use the instrument in linear mode (not in reflector mode, which is appropriate for peptides analyses). Normally we utilize our instrument in positive-ion mode. Moreover, a key parameter is the "pulsed ion extraction" which improves the instrumental resolution 31. In simple terms, the "pulsed ion extraction" can be defined as a pause between the generation of the sample ions and the time when the ions are accelerated toward the detector. When analysing intact proteins, one should utilized a "pulsed ion extraction" (e.g. 500 nsec) larger than when observing peptides (e.g. 80 nsec).
Choose the appropriate m/z range, acquire the spectra of the calibrant, and calibrate the instrument. When you acquire the spectra of your samples, use the appropriate laser intensity (see discussion).
Representative Results
We analyzed an intact protein (Chromosome region maintenance 1 protein, Crm1; molecular weight: 123386 Da) using two different matrices, two deposition methods, and the same laser intensity (Figure 1). SA and a CHCA_DHB mixture were utilized as matrices. The mixture yielded mass spectra of higher quality in terms of signal to noise ratio and of sensitivity (Figures 1A and B). In particular, we could detect 0.5 pmoles of protein deposited on the MALDI target (Figure 1C). The same amount of protein was barely detectable using SA (Figure 1D). Moreover, using the matrices mixture, the method of choice for the matrix deposition was the "thin layer" approach (Figure 1A). Using the "dried droplet" method, we did not detect any protein with a mass higher than 100 kDa (data not shown). Utilizing the CHCA_DHB mixture (Figures 1A and 1C), multiply charged ion of the protein were observed, allowing the mass determination with higher accuracy because the peak resolution in axial MALDI-TOF instruments is inversely proportional to the m/z26.
We also analyzed monomeric beta-galactosidase (116,300 Da) (Figure 2). The CHCA_DHB spectra were better than the SA spectra in terms of sensitivity and resolution. Furthermore, the presence of multiply charged ions (M2+, M3+ and M4+) allowed us to confirm the mass of the protein with higher accuracy (Figures 2A and 2C). Using the thin layer approach, we compared the performance of the CHCA_DHB mixture, SA and α-CHCA alone for the analysis of an immunoglobulin, IgG (148500 Da) (Figure 3). When we correlate the different data, protein signals were higher and with better resolution in the CHCA_DHB spectra. To conclude, the use of the CHCA_DHB mixture and the thin layer deposition method demonstrated significant improvements in the quality of large intact protein mass spectra acquired using a MALDI TOF instrument.
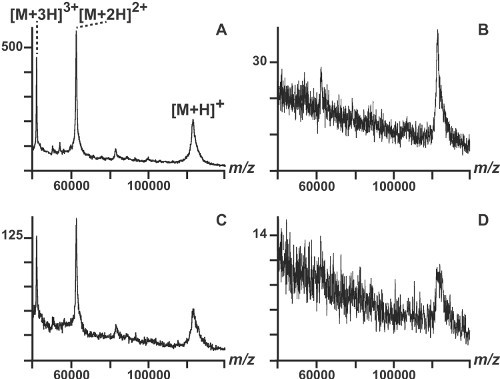 Figure 1. MALDI-TOF analysis of the intact Chromosome region maintenance 1 protein, (Crm1), molecular weight: 123,386 Da. A) 1 pmole of Crm1 was analysed using the DHB_CHCA mix as matrix. B) amount: 1 pmole; matrix: SA. C) amount: 0.5 pmole; matrix: DHB_CHCA. D) amount: 0.5 pmole; matrix: SA. The signal of the peaks, expressed in an arbitrary intensity unit scale, is normalized to the maximum value present in each spectrum.
Figure 1. MALDI-TOF analysis of the intact Chromosome region maintenance 1 protein, (Crm1), molecular weight: 123,386 Da. A) 1 pmole of Crm1 was analysed using the DHB_CHCA mix as matrix. B) amount: 1 pmole; matrix: SA. C) amount: 0.5 pmole; matrix: DHB_CHCA. D) amount: 0.5 pmole; matrix: SA. The signal of the peaks, expressed in an arbitrary intensity unit scale, is normalized to the maximum value present in each spectrum.
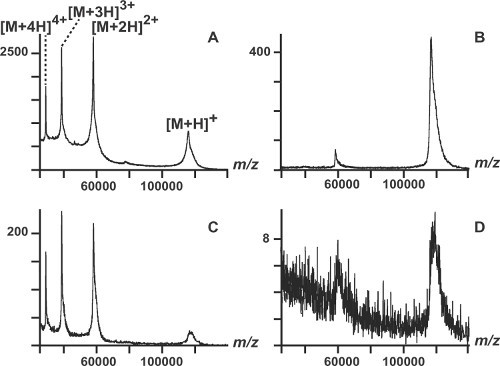 Figure 2. MALDI-TOF analysis of intact beta-galactosidase (116,300 Da). A) amount: 20 pmoles; matrix DHB_CHCA. B) amount: 20 pmoles; matrix: SA. C) amount: 5 pmoles; matrix: DHB_CHCA. D) amount: 5 pmoles; matrix: SA.
Figure 2. MALDI-TOF analysis of intact beta-galactosidase (116,300 Da). A) amount: 20 pmoles; matrix DHB_CHCA. B) amount: 20 pmoles; matrix: SA. C) amount: 5 pmoles; matrix: DHB_CHCA. D) amount: 5 pmoles; matrix: SA.
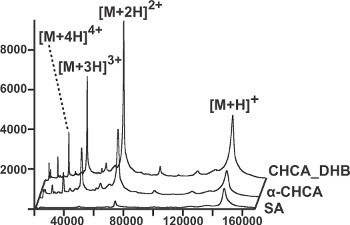 Figure 3. MALDI-TOF analysis of an intact immunoglobulin, IgG (148,500 Da); amount: 1.7 pmoles. The spectra obtained using the CHCA_DHB mixture were much more intense (maximum: 8200 arbitrary unit, au) than the SA spectra (maximum: 1,200 au) and α-CHCA spectra (maximum: 4500 au)
Figure 3. MALDI-TOF analysis of an intact immunoglobulin, IgG (148,500 Da); amount: 1.7 pmoles. The spectra obtained using the CHCA_DHB mixture were much more intense (maximum: 8200 arbitrary unit, au) than the SA spectra (maximum: 1,200 au) and α-CHCA spectra (maximum: 4500 au)
Discussion
We presented a detail protocol to acquire high quality mass spectra of large intact proteins with molecular weights larger than 100 kDa, using a MALDI-TOF instrument. A number of critical aspects concerning the sample preparation should be carefully considered in order to improve the quality of the mass spectra. Matrices and solvents of high purity are of critical importance because all the contaminants present in the matrices and solvents are concentrated on the MALDI target after solvent evaporation. In order to enhance the purity of the MALDI matrices it is possible to purify them using classical recrystallization methods (see above). We also recommend to freshly preparing the matrices solutions (at least once a week) to ameliorate the matrix crystallization and to avoid matrix degradation.
The matrix deposition approach is very critical for the final quality of the spectra. As described above, the thin layer method is our method of choice 12. Moreover, we optimized the approach for depositing the first layer. When the matrix is dissolved in acetone to obtain a saturated solution, the propanone allows fast evaporation of the solvent, but also makes the size of the first layer very difficult to control. We suggest using a GELoader tip as little brush that you dip in the matrix_acetone solution to get a minimum volume which you quickly deposit on the target. The size of the first layer somehow controls the final size of the MALDI spot: a small spot (i.e. 0.5-0.75 mm of diameter) is preferable because in this case the sample concentration is higher than in the case of a large spot. Obtaining a small spot differs from the ultra-thin layer approach previously proposed in a JOVE video 15. In Fenyo et al. the thin layer solution is spread all over the MALDI target using the side of a pipette tip. This approach requires testing the thin layer which should satisfy specific criteria (e.g. speed of solvent evaporation). If the criterion is not met, the MALDI target should be washed and a new thin layer should be prepared. This makes the preparation quite laborious for a novice. Furthermore, the deposition of sample/matrix mixture on the plate requires the aspiration of the excess solvent using a vacuum line and a washing step using TFA15 is necessary, making the Fenyo et al. preparation a bit more difficult than our approach.
The volume ratio between the matrix and the sample is quite important for a successful analysis of intact proteins by MALDI-TOF. After testing different ratios we suggest to use a 1:1 ratio. A different matrix:sample proportion could compromise the quality of the spectra, probably due to inhomogeneous crystallization. The order in which matrix and sample are deposited is also critical. After depositing the matrix thin layer, the sample is deposited and immediately after (before the sample spot becomes dry) the CHCA_DHB mixture is added. This procedure has been defined as "sandwich method" 27, where the sample alone is deposited between two matrix layers. As an alternative, the CHCA_DHB solution and the sample can be mixed in 0.5 ml tube and then spotted on the CHCA thin layer 12. This yields a spot with a homogenous layer of crystals resulting in high quality spectra. When analysing proteins with a mass lower than 100 kDa, the concentration of DHB can be increased (e.g. 60:40 DHB:CHCA); this can produce sharper peak, but could compromise the measurement sensitivity (i.e. less intense peaks).
For a correct instrument calibration, it is essential to deposit the calibrant standards very close to the sample spots, allowing one to obtain a better mass accuracy. A "pseudo-internal calibration procedure" was previously suggested 15: after the acquisition of a sample spectrum, the calibrant spot is laser shot and signal of the calibrant are added to the sample spectrum. This allows you to internally calibrate the sample spectrum using the calibrant peaks.
The laser energy should be also carefully controlled during the spectra acquisition. In most cases, higher energy increases the sensitivity, but lowers the resolution. We suggest using the minimum possible laser energy without compromising the sensitivity of the acquisition. Moreover, to improve spectral quality, one can acquire more shots on each sample spot. Normally the more shots you acquire (1,000-2,000 shots), the higher the intensity of the signal. When hitting the spot with the laser, one needs to find the right position (i.e. "sweet spot"), since the sample is not homogenously distributed. You can shoot on the same area until the signal is increasing, and then try to find another area containing the sample.
In conclusion, high purity of matrices and solvents, the methods used for sample and matrices deposition on the target, the position of the standards used for calibration, the intensity of laser energy, the time of acquisition (number of shots/spot) are critical factors that one should take into account when analysing intact proteins by MALDI-TOF MS.
Author Contributions
L.S. and E.B.E. designed the experiments, performed the mass spectrometry experiments, analysed the data, and wrote the manuscript.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Dr. Christophe Masselon (iRTV, CEA, Grenoble) and members of the Viral Infection and Cancer Group at the IBS, for their critical evaluation of the manuscript and for useful discussions. We are grateful to Cyril Dian for the kind gift of the protein Crm1. This scientific work took place in the mass spectrometry facility of Grenoble Instruct Centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL). It was financially supported by the French Infrastructure for Integrated Structural Biology Initiative (FRISBI, ANR-10-INSB-05-02), GRAL (ANR-10-LABX-49-01) [within the Grenoble Partnership for Structural Biology] and by the French National Centre for Scientific Research (CNRS).
References
- Rupp B. Biomolecular Crystallography : Principles, Practice, and Application to Structural Biology. Garland Science - Taylor & Francis Group; 2010. [Google Scholar]
- Cohen SL, Chait BT. Mass spectrometry as a tool for protein crystallography. Annual review of biophysics and biomolecular structure. 2001;30:67–85. doi: 10.1146/annurev.biophys.30.1.67. [DOI] [PubMed] [Google Scholar]
- Chait BT. Mass spectrometry--a useful tool for the protein X-ray crystallographer and NMR spectroscopist. Structure. 1994;2:465–467. doi: 10.1016/s0969-2126(00)00047-2. [DOI] [PubMed] [Google Scholar]
- Gans P, et al. Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins. Angew Chem Int Ed Engl. 2010;49:1958–1962. doi: 10.1002/anie.200905660. [DOI] [PubMed] [Google Scholar]
- Wilm M. Principles of electrospray ionization. Molecular & cellular proteomics : MCP. 2011;10:M111 009407. doi: 10.1074/mcp.M111.009407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban PL, Amantonico A, Zenobi R. Lab-on-a-plate: extending the functionality of MALDI-MS and LDI-MS targets. Mass spectrometry reviews. 2011;30:435–478. doi: 10.1002/mas.20288. [DOI] [PubMed] [Google Scholar]
- De Hoffmann E, Stroobant V. Mass Spectrometry: Principles and Applications. Wiley; 2007. [Google Scholar]
- Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Analytical chemistry. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- Spengler B, Cotter RJ. Ultraviolet laser desorption/ionization mass spectrometry of proteins above 100,000 daltons by pulsed ion extraction time-of-flight analysis. Analytical chemistry. 1990;62:793–796. doi: 10.1021/ac00207a004. [DOI] [PubMed] [Google Scholar]
- Beavis RC, Chait BT. High-accuracy molecular mass determination of proteins using matrix-assisted laser desorption mass spectrometry. Analytical chemistry. 1990;62:1836–1840. doi: 10.1021/ac00216a020. [DOI] [PubMed] [Google Scholar]
- Hillenkamp F, Karas M, Beavis RC, Chait BT. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Analytical chemistry. 1991;63:1193A–1203A. doi: 10.1021/ac00024a002. [DOI] [PubMed] [Google Scholar]
- Beavis RC, Chait BT. Matrix-assisted laser desorption ionization mass-spectrometry of proteins. Methods in enzymology. 1996;270:519–551. doi: 10.1016/s0076-6879(96)70024-1. [DOI] [PubMed] [Google Scholar]
- Xiang F, Beavis RC. A method to increase contaminant tolerance in protein matrix-assisted laser desorption/ionization by the fabrication of thin protein-doped polycrystalline films. Rapid Communications in Mass Spectrometry. 1994;8:199–204. [Google Scholar]
- Cadene M, Chait BT. A robust, detergent-friendly method for mass spectrometric analysis of integral membrane proteins. Analytical chemistry. 2000;72:5655–5658. doi: 10.1021/ac000811l. [DOI] [PubMed] [Google Scholar]
- Fenyo D, et al. MALDI sample preparation: the ultra thin layer method. J. Vis. Exp. 2007. p. e192. [DOI] [PMC free article] [PubMed]
- Gabant G, Cadene M. Mass spectrometry of full-length integral membrane proteins to define functionally relevant structural features. Methods. 2008;46:54–61. doi: 10.1016/j.ymeth.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Hook P, et al. Long range allosteric control of cytoplasmic dynein ATPase activity by the stalk and C-terminal domains. The Journal of biological chemistry. 2005;280:33045–33054. doi: 10.1074/jbc.M504693200. [DOI] [PubMed] [Google Scholar]
- Vorm O, Roepstorff P. Peptide sequence information derived by partial acid hydrolysis and matrix-assisted laser desorption/ionization mass spectrometry. Biological mass spectrometry. 1994;23:734–740. doi: 10.1002/bms.1200231204. [DOI] [PubMed] [Google Scholar]
- Beavis RC, Chait BT. Cinnamic acid derivatives as matrices for ultraviolet laser desorption mass spectrometry of proteins. Rapid Communications in Mass Spectrometry. 1989;3:432–435. doi: 10.1002/rcm.1290031207. [DOI] [PubMed] [Google Scholar]
- Madler S, Boeri Erba , E , Zenobi R. MALDI-ToF Mass Spectrometry for Studying Noncovalent Complexes of Biomolecules. Topics in current chemistry. 2012. [DOI] [PubMed]
- Wortmann A, Pimenova T, Alves S, Zenobi R. Investigation of the first shot phenomenon in MALDI mass spectrometry of protein complexes. The Analyst. 2007;132:199–207. doi: 10.1039/b615411e. [DOI] [PubMed] [Google Scholar]
- Beavis RC, Chaudhary T, Chait BT. Alpha-Cyano-4-hydroxycinnamic Acid as a Matrix for Matrix-assisted Laser Desorption Mass Spectrometry. Organic Mass Spectrometry. 1992;27:156–158. [Google Scholar]
- Liu Z, Schey KL. Optimization of a MALDI TOF-TOF mass spectrometer for intact protein analysis. Journal of the American Society for Mass Spectrometry. 2005;16:482–490. doi: 10.1016/j.jasms.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Gorka J, Bahr U, Karas M. Graphite supported preparation (GSP) of alpha-cyano-4-hydroxycinnamic acid (CHCA) for matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) for peptides and proteins. Journal of the American Society for Mass Spectrometry. 2012;23:1949–1954. doi: 10.1007/s13361-012-0478-8. [DOI] [PubMed] [Google Scholar]
- Laugesen S, Roepstorff P. Combination of two matrices results in improved performance of MALDI MS for peptide mass mapping and protein analysis. Journal of the American Society for Mass Spectrometry. 2003;14:992–1002. doi: 10.1016/S1044-0305(03)00262-9. [DOI] [PubMed] [Google Scholar]
- Sparkman DO. Mass Spectrometry Desk Reference. Global View Publishing; 2000. [Google Scholar]
- Kussmann M, Roepstorff P. Sample preparation techniques for peptides and proteins analyzed by MALDI-MS. Methods Mol Biol. 2000;146:405–424. doi: 10.1385/1-59259-045-4:405. [DOI] [PubMed] [Google Scholar]
- Wang R, Chait BT. High-accuracy mass measurement as a tool for studying proteins. Current opinion in biotechnology. 1994;5:77–84. doi: 10.1016/s0958-1669(05)80074-6. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum N, Michaelevski I, Sharon M. Analyzing large protein complexes by structural mass spectrometry. J. Vis. Exp. 2010. p. e1954. [DOI] [PMC free article] [PubMed]
- Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nature protocols. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Vestal ML, Juhasz P, Martin SA. Delayed extraction matrix-assisted laser desorption time-of-flight massspectrometry. Rapid Communications in Mass Spectrometry. 1995;9:1044–1050. [Google Scholar]