

Abstract
Bone morphogenic protein-4 (BMP4) and fibroblast growth factor-8 (FGF8) are thought to have opposite roles in defining epithelial versus neurogenic fate in the developing olfactory/vomeronasal system. In particular, FGF8 has been implicated in specification of olfactory and gonadotropin releasing hormone-1 (GnRH) neurons, as well as in controlling olfactory stem cell survival. Using different knock-in mouse lines and Cre-lox-mediated lineage tracing, Fgf8 expression and cell lineage was analyzed in the developing nose in relation to the expression of Bmp4 and its antagonist Noggin (Nog). FGF8 is expressed by cells that acquire an epidermal, respiratory cell fate and not by stem cells that acquire neuronal olfactory or vomeronasal cell fate. Ectodermal and mesenchymal sources of BMP4 control the expression of BMP/TGFβ antagonist Nog, whereas mesenchymal sources of Nog define the neurogenic borders of the olfactory pit. Fgf8 hypomorph mouse models, Fgf8neo/neo and Fgf8neo/null, displayed severe craniofacial defects together with overlapping defects in the olfactory pit including (1) lack of neuronal formation ventrally, where GnRH neurons normally form, and (2) altered expression of Bmp4 and Nog, with Nog ectopically expressed in the nasal mesenchyme and no longer defining the GnRH and vomeronasal neurogenic border. Together our data show that (1) FGF8 is not sufficient to induce ectodermal progenitors of the olfactory pit to acquire neural fate and (2) altered neurogenesis and lack of GnRH neuron specification after chronically reduced Fgf8 expression reflected dysgenesis of the nasal region and loss of a specific neurogenic permissive milieu that was defined by mesenchymal signals.
Introduction
Olfactory sensory neurons, pheromone sensory neurons, and gonadotropin releasing hormone-1 (GnRH) neurons originate from heterogeneous progenitors in the olfactory pit (OP; for review, see Forni and Wray, 2012). Although fibroblast growth factor-8 (FGF8) signaling is thought to play a key role, together with bone morphogenic protein (BMP)/TGFβ antagonists, in inducing neuronal cell fate (Wilson and Hemmati-Brivanlou, 1995; Zimmerman et al., 1996; Streit et al., 2000; Chmielnicki et al., 2004; Chiba et al., 2008; Marchal et al., 2009; Tang et al., 2009), the full role played by FGF, BMP, and BMP antagonists in controlling neurogenesis in cranial placodes is not entirely clear (Chung et al., 2008; Maier et al., 2010; Tucker et al., 2010). One example of a cranial placode-derived neuronal population is the GnRH neurons. During embryonic development, GnRH neurons migrate from the olfactory region to the forebrain. In the forebrain, GnRH neurons control reproductive maturation and function (Boehm et al., 2005; Wray, 2009). Developmental pathologies that alter GnRH function, specification, or migration can cause hypogonadotropic hypogonadism (HH; Wray, 2010). Syndromic association of lack or impaired sense of smell and HH is defined as Kallmann syndrome (Kallmann et al., 1944). Forms of HH and Kallmann have been linked to mutations in the FGF8/FgfR1 signaling axis (Ogata et al., 2006; Falardeau et al., 2008; Bajpai et al., 2010; Chung and Tsai, 2010; Trarbach et al., 2010).
FGF8 is essential for correct development of craniofacial mesenchyme. Also, crosstalk between the olfactory placode and craniofacial mesenchyme is crucial to induce OP formation, terminal differentiation, and cell type specification (LaMantia et al., 2000). Defective FGF8 signaling in mice affects progenitor cell identity, olfactory neurogenesis, and GnRH cell fate specification (Riley et al., 2007; Chung et al., 2008; Falardeau et al., 2008; Chung and Tsai, 2010; Sabado et al., 2012). Developmental olfactory defects emerging after reduced FGF8 signal transduction have been previously interpreted as direct result of (1) progressive primordial stem cell death (Kawauchi et al., 2005), (2) changes in precursor cell identity with expansion of uncommitted stem cells and loss of neurogenic progenitors (Tucker et al., 2010), or (3) decreased stem cells that access the neurogenic program with an increase in epidermal cell fate (Maier et al., 2010). However, the nature of these “primordial stem” cells is unclear and no one knows precisely how dysmorphic craniofacial development itself affects development of olfactory/GnRH neurogenesis in Fgf8 mutants.
To address these points we followed Fgf8 expression, cell lineage, and neurogenesis in relation to the expression of craniofacial morphogens Bmp4 and its antagonist Noggin (Nog) in wild-type and hypomorphic Fgf8 mouse models. We observed that reduced Fgf8 levels that affect craniofacial development also altered Bmp expression. Mesenchymal Bmp and Nog expression was found to be crucial in defining neuronal versus epidermal fate in the developing OP. In fact in Fgf8 mutants, altered stem cell markers expression and neurogenic patterns directly reflected changes in Bmp and Nog expression in the nasal mesenchyme. Our data indicate that (1) cell identity, neuralization, and patterning of the OP strictly depend on mesenchymal signals and (2) defects in the olfactory and GnRH systems resulting from altered FGF8 signaling are in large part secondary to craniofacial dysmorphism.
Materials and Methods
Animals and tissue preparation
Fgf8 hypomorph mouse line Fgf8neo (Meyers et al., 1998) was obtained from Dr. M. Lewandoski [National Cancer Institute (NCI)]. The Fgf8 LacZ knock-in line Fgf8nullLacZ (generated by Drs. D. Brown and G. R. Martin; Grieshammer et al., 2005) was used to follow Fgf8 expression as a null allele (Ilagan et al., 2006) and was obtained from Dr. M. Kelley [National Institute on Deafness and Other Communication Disorders, National Institutes of Health (NIH)]. Fgf8 Cre knock-in line Fgf8Cre has been previously described (Toyoda et al., 2010). NognullLacZ/WT mice (Brunet et al., 1998), also called NogLacZ, were provided by Dr. S. Mackem (NCI). All procedures were approved by National Institute of Neurological Disorders and Stroke Animal Care and Use Committee and performed in accordance with NIH guidelines.
Animals were euthanized in a CO2 chamber. Embryos were removed from the uterus and fixed by immersion in 4% PFA (30 min). Early postnatal mice were decapitated and the head fixed in 4% formaldehyde (1 h). Specimens were cryoprotected in 30% sucrose/PBS at 4°C overnight, embedded in Tissue-Tek OCT compound (Sakura Finetek), frozen in dry ice, and stored at −80°C until sectioning. Frozen samples were cut in serial sections (12–16 μm) using a Leica CM 3050S cryostat (Leica Biosystems) and maintained at −80°C until processing. Parasagittal sections were generated as follows: two serial series were generated for embryonic day E11.5; four serial series were generated for E14.5 embryos; five serial series were generated for E15.5 embryos; and seven serial series were generated for E16.5 and postnatal animals. Coronal sections were generated as follows: six serial series were generated for E11.5 embryos. For all analyzed mouse lines, no male/female distinction was made. All in vivo data were compiled from ≥3 animals of each genotype.
Analysis of BMP4 induction of Noggin in nasal explants
Nasal explants were generated from Noggin-LacZ (Nog-LacZ) timed pregnant mice at E11.5, as previously described (Klenke and Taylor-Burds, 2012). An additional cut was made to remove the rostral part of the nose to facilitate visualization or induced and endogenous Nog signals. Affi-Gel Blue beads (Biorad) were incubated for 1 h at 37°C in 15 μl of either (1) 100 μg/ml recombinant mBMP-4 (R&D Systems) resuspended in 4 mm HCl/0.1% BSA or (2) 4 mm HCl/0.1% BSA storage buffer only. A single bead (control or treated) was placed (Dumont #5 forceps) on an explant immediately after plating. Explants were maintained in serum-free media at 37°C, 5%CO2, for 18–20 h, and washed (PBS). Then the X-gal reaction was performed (3 h at 37°C). After staining, explants were fixed (4% formaldehyde) and coverslipped for imaging.
Immunolabeling
Primary antibodies.
All antibodies used were polyclonal, unless otherwise indicated. Antibodies used were as follows: GnRH-1 (SW-1, 1:3000; Wray et al., 1988), peripherin (peripheral intermediate filament marker; 1:2000; Millipore Bioscience Research Reagents), phosphorylated Sma-related and Mad-related protein (p-SMAD) 1,5,8 (Cell Signaling Technology); Rb anti-SOX2 (1:500; Millipore Bioscience Research Reagents), Rb anti-PAX6 (1:800; Millipore Bioscience Research Reagents), mouse monoclonal biotinylated anti-HuC/D (1:100; Invitrogen), Rb anti ID-3 (1:300; Abcam); mouse monoclonal anti-tubulin III (Tuj-1, 1:700; Sigma-Aldrich); mouse monoclonal anti-ASCL-1/MASH-1 (1:30; BD PharMingen). The mouse monoclonal antibodies anti-AP2α (3B5) and MSX1/2 (4G1) were obtained from Developmental Studies Hybridoma Bank and used at the concentration 1:3 and 1:5 respectively. For AP2α, p-SMAD 1,5,8, MSX1/2, PAX6, SOX2, and ASCL-1/MASH-1, microwave epitope retrieval treatment in citrate buffer, pH ∼6, was performed before immunolabeling.
Chromogen-based reactions.
Sections were first washed in PBS. Then, endogenous peroxidases were inhibited in 0.3% H2O2 (10% methanol in PBS, 15 min). Sections were again washed in PBS and then blocked in 10% normal serum (horse or goat depending on experimental needs)/0.3% Triton X-100, and washed again in PBS. After incubation with primary antibody (at room temperature for 1 h or 4°C overnight) and subsequent PBS washes, sections were placed in either Donkey anti-rabbit (DAR-Bt; Jackson Immuno Research) or donkey anti-mouse (DAM-Bt; Jackson Immuno Research). After PBS washes, the tissue was processed using a standard avidin-biotin-horseradish peroxidase/3′,3-diaminobenzidine (DAB) protocol or nickel-intensified DAB (Kramer et al., 2000).
X-gal staining
Cryosections were rinsed in PBS (10 min) then incubated (37°C overnight) in a solution of 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 2 mm MgCl2, 0.1% Tween, 0.1% 5-bromo-4chloro-3-indolyl-b-d-galactoside/dimethylformamide. After enzymatic reaction, slides were either counterstained with Eosin B (Sigma-Aldrich) following standard procedures and coverslipped or washed and immunostained as described above.
In situ hybridization
In situ hybridization was performed as previously described (Bharti et al., 2008). Briefly, 12 μm cryosections were fixed (4% formaldehyde, 5 min), washed, and treated with 1 μg/ml proteinase K (Roche) for 20 s before overnight hybridization with DIG-labeled probe (2 μg/ml) at 60°C. The next morning, excess probe was washed in 0.2× SSC at 60°C. Sections were incubated in anti-DIG alkaline phosphatase conjugated antibody (Roche) overnight at a dilution of 1:3000. Excess antibody was rinsed and the signal was developed using NTMT (NaCl, Tris-HCl, magnesium chloride, Tween; Roche). Fgf8 and Bmp4 probes were purchased from Open Biosystems. Nog probe was obtained from Dr. S. Mackem (NCI).
In vivo analysis and cell scatter plots. Using ImageJ, we analyzed 10× (1360 × 1360 pixel) serial digital images from multiple animals. The OP was defined as region of interest by tracing contours using ImageJ Freehand selection tool. XM and YM coordinates for the center of the OP were defined using Analyze/Measure plugins of ImageJ. X and Y coordinates of cells of interest were recorded as multipoint selections. XM and YM of the OP were subtracted from X and Y position for cells of interest defining XM and YM as coordinates X = 0, Y = 0. X,Y values for different cell type groups were imported and plotted as scatter plots in Microsoft Excel.
Results
Fgf8 is expressed by epidermal cells but not by cells in neurogenic areas
Kawauchi et al. (2005) suggested that an FGF8 autocrine loop might be crucial for sustenance of stem cells of the OP. Tucker et al. (2010) proposed that local FGF8 sources in the developing OP are responsible for specification and transition of primordial stem cells to terminally neurogenic precursors. However, the identity and location of these multipotent stem cells in the OP was still unclear (Kawauchi et al., 2005; Maier et al., 2010; Tucker et al., 2010). To follow neurogenesis and cell fate specification, the expression of neural and epidermal markers was analyzed with respect to expression of Fgf8. SOX2 and PAX6 were used as stem/neural progenitor markers (Kawauchi et al., 2004), HuC/D was used as an early neuronal marker (Fornaro et al., 2003), while AP2α, ID3, and MSX1 were used to identify epidermal cells (Wang et al., 2006; Maier et al., 2010; Forni et al., 2011b). Analysis of knock-in mice expressing β-gal under direct control of Fgf8, Fgf8nullLacZ/WT, confirmed that Fgf8 is expressed in the rostral portion of the invaginating OP [corresponding to the putative respiratory epithelium, (RE)], as well as in the epidermis proximal to the rim of the OP (Fig. 1A,B,D,E). In situ hybridization for Fgf8 confirmed overlapping expression (data not shown). Immunolabeling for HuC/D on Fgf8nullLacZ/WT (Fig. 1B) highlighted that Fgf8 expression is confined to the non-neurogenic portions of the OP (HuC/D negative). Notably, the neuroepithelial stem/progenitor markers PAX6 and SOX2 had similar expression patterns (Fig. 1F,G), which were also similar to those of HuC/D (Fig. 1E), with higher levels of PAX6, SOX2, and HuC/D expression distal to Fgf8 sources (Fig. 1B,C,E–G). Epithelial cells expressing Fgf8 were positive for the epidermal markers ID3 (data not shown) and AP2α (Fig. 1H). Thus, the transition from Fgf8-expressing epithelium to Fgf8-negative epithelium delineated a border between non-neuronal regions of the OP and highly neurogenic vomeronasal organ (VNO) epithelium and olfactory epithelium (OE; Fig. 1B,C,E–H, dotted line).
Figure 1.

Fgf8 is expressed in the non-neurogenic portion of the olfactory pit. A–H, E11.5 Fgf8nullLacz/WT. A D, Head of embryo, as reference for parasagittal (B, C) and coronal sections (E–H), showing Fgf8 expression (X-gal enzymatic reaction, blue) at the rim of the OP, nasal process (NP), and maxillary process (MP). D, Dorsal; V, ventral; R, rostral; C, caudal; L, lateral; M, medial. B, E, X-gal enzymatic reaction (blue, arrows) and HuC/D immunostaining (brown) highlights the occurrence of neuron formation (brown cells) out of Fgf8 (β-gal/blue)-expressing RE and epidermis (Ep). Stem cell transcription factors SOX2 (F) and PAX6 (C, G) were also not detected in Fgf8-positive areas (arrows; compare with E), with higher expression levels coinciding with the neurogenic areas (compare area dorsal to dashed line in E–G). H, The transcription factor Ap2α was expressed along and proximal to the Fgf8-expressing epithelia. Scale bars: (in B) B, C, 100 μm; (in E) E–H, 100 μm.
Bmp4 is expressed in the mesenchyme proximal to Fgf8 sources
BMP4 has been shown to suppress neurogenesis and to initiate an epidermal/RE differentiation program in progenitor cells in the nasal pit (Wilson and Hemmati-Brivanlou, 1995; Hu et al., 2008; Maier et al., 2011). Though opposite roles for FGF8 and BMP in controlling neurogenesis have been proposed (Maier et al., 2010), the spatial relation between the sources of these signals and their direct effect on neural induction in the OP is unclear (Liu et al., 2005). Thus, analysis of Bmp4 expression at E11.5 by in situ hybridization was performed. Bmp4 was expressed by mesenchymal cells in the nasal process and in the rostral aspect of the RE (Fig. 2A, white and black arrows respectively). Nasal mesenchymal cells positive for Bmp4 expression were juxtaposed to Fgf8-expressing RE, which also expressed low levels of Bmp4 (Fig. 2, compare A, B). We analyzed the activation of BMP downstream intracellular signaling molecules SMAD 1,5,8, and MSX1/2, as they had been previously associated with epidermal cell fate (Marchal et al., 2009; Maier et al., 2010, 2011). The pattern of p-SMAD 1,5,8 and MSX1/2 reflected Bmp4 expression (Fig. 2, compare A, C, D), though the MSX1/2 staining defined sharper borders than p-SMADs. The MSX1/2 staining was stronger within and proximal to Bmp4 sources, consistent with high BMP signal thresholds inducing MSX1/2 and with MSX1 controlling BMP expression (Zhang et al., 2002; Hu et al., 2008).
Figure 2.
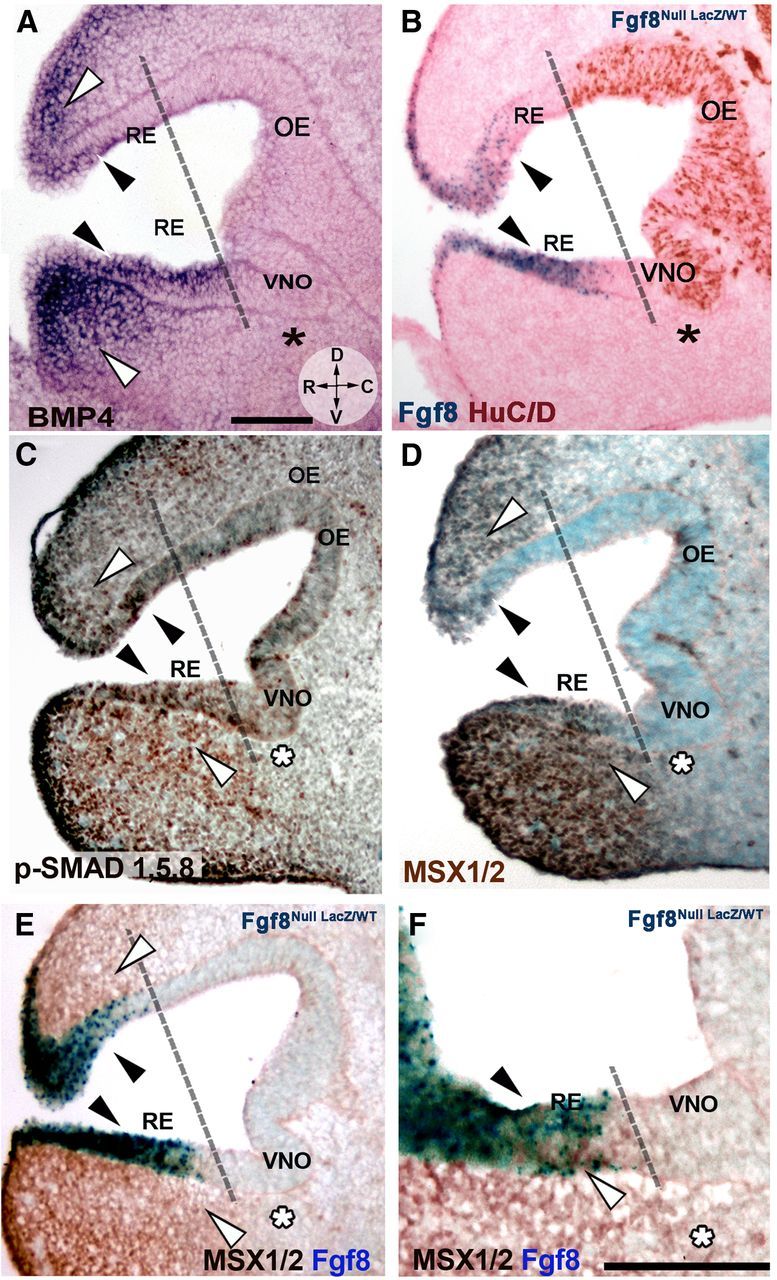
Bmp4 is expressed in the developing nasal mesenchyme and is coexpressed with FGF8 in the RE. A–D, E11.5, parasagittal sections, orientation indicated in A. R, Rostral; D, dorsal; V, ventral; C, caudal. Use dashed line as reference to separate non-neurogenic and neurogenic OP areas. A, Bmp4 in situ hybridization showed expression along the RE (black arrowheads) and in rostral, dorsal, and ventral nasal mesenchyme (white arrowheads). Bmp4 expression was not detected in the caudal nasal mesenchyme (asterisk) facing the neurogenic VNO (compare to B). B, Fgf8nullLacz/WT; X-gal enzymatic reaction (blue) and HuC/D immunostaining (brown) highlights the occurrence of neurogenesis mainly caudal to Fgf8-expressing areas (blue). Fgf8 was expressed in areas where Bmp4 expression was detected (compare black arrows in A and B). C, Immunostaining for p-SMAD 1,5,8 (brown) detected active BMP4 downstream signaling in areas of Bmp4 expression (A, C, white arrowheads) and along the RE (black arrowheads). Decreased p-SMAD 1,5,8 immunoreactivity was detected in area facing the VNO (asterisk). D, MSX1/2 immunolabeling (dark brown) revealed a pattern similar to p-SMAD (compare with C) with high MSX1/2 levels in the RE (black arrowheads) and mesenchyme proximal to Bmp4 sources (white arrowheads; compare with A). No expression was found in the mesenchyme facing the neurogenic VNO (asterisk). E, F, MSX1/2 immunolabeling (brown) and Fgf8 expression (X-gal enzymatic reaction, blue) revealed co-localization in the RE (black arrowheads). Scale bars: (in A) A–D, 100 μm; F, 100 μm.
Previous data in chick suggested a direct role for FGF8 in antagonizing BMP4 intracellular signaling (Maier et al., 2010). The expression pattern in mouse does not support this idea. Notably we (1) found Bmp4 expression and active downstream signaling within the non-neurogenic Fgf8-expressing RE and in the nasal mesenchyme adjacent to the Fgf8 source (Fig. 2C–F); and (2) observed a decrease in Bmp4 expression and immune reactivity for BMP downstream molecules only distant from the Fgf8 sources in both nasal mesenchyme and in the neurogenic areas of the VNO and OE (Fig. 2A–F; use dashed line as reference in B).
Fgf8 genetic lineage tracing shows Fgf8 is expressed by epidermal cells and not by neurogenic progenitor/stem cells
“Primordial” stem cells were thought to form the olfactory and vomeronasal neuroepithelium after migrating from the Fgf8-expressing domains of the OP (Kawauchi et al., 2005; Maier et al., 2010; Tucker et al., 2010). Our expression data are inconsistent with this idea (Figs. 1, 2), and indicate instead that Fgf8 was specifically expressed by the non-neurogenic epithelium and that immunoreactivity for the neuronal stem cell markers (SOX2, PAX6) increased distally from Fgf8 sources. Thus, Fgf8 genetic lineage cell fate tracing (Fig. 3) was performed to further clarify this issue. Heterozygous Fgf8Cre/WT knock-in mice (Toyoda et al., 2010) were crossed with RosaYFP (Srinivas et al., 2001) reporters. Fgf8Cre/RosaYFP embryos were collected for analysis at E11.5 (n = 3) and at E15.5 (n = 4) and immunostained for yellow fluorescent protein and the neuronal markers HuC/D and tubulin-III. Analysis at E11.5 (Fig. 3A–E) and E15.5 (Fig. 3F–I) confirmed that the epidermis, rostral RE, and a large part of the oral mucosa (Tucker et al., 1999) originated from Fgf8-expressing cells, while the neurons of the OE and VNO did not. Parasagittal and coronal cuts showed sharp transitions between the non-neurogenic Fgf8-expressing domain and the neurogenic areas of the OP (Fig. 3A–D). These data show that the Fgf8-expressing cells and the neurogenic progenitor cells belong to distinct borders and that the neurons forming in the OP are not derived from Fgf8-expressing progenitors. At both stages (E11.5 and E15.5), Fgf8 was expressed by epidermal cells and not by the stem cell reservoir responsible for formation of the olfactory and vomeronasal neuroepithelium.
Figure 3.

Fgf8 genetic lineage cell fate tracing. A–E, Parasagittal (A–C) and coronal (D, E) sections of E11.5 Fgf8Cre/RosaYFP embryos. A, Yellow fluorescent protein (YFP) immunostaining (brown) reveals Fgf8 lineage (YFP expression) in the facial epidermis (Ep) and along the rostral RE. YFP expression was not detected in the OE and VNO, confirming distinct genetic lineage for the Fgf8-expressing epidermis and these structures. B–C, YFP (green) and tubulin+HuCD immunostaining (red). Neurons (red) of the OE (B) and VNO (C) derive from progenitors negative for Fgf8 expression (green). D, Section shows the distinct lineage for neuronal cells of OE, VNO, and migratory mass (MM; red only) and the Fgf8 (YFP+)-expressing epidermal cells (green). Fgf8 lineage was found for the superficial ectoderm forming the epidermis (Ep) along the lateral (LNP) and medial nasal process (MNP). Note that neurons positive for Fgf8 lineage were detected in the developing forebrain (arrowheads, Br). E, Immunostaining against YFP (brown) and tubulin III (gray-blue); use boxed area in F as reference. No neuronal Fgf8 lineage was detected in the lateral or medial neuroepithelium. F–I, E15.5 Fgf8Cre/RosaYFP embryo. F, YFP expression (brown) was detected in the facial epidermis (Ep), rostral RE, and oral mucosa (OM), but not in the OE and VNO. YFP (green) and tubulin+HuCD immunostaining (red) confirmed lack of Fgf8 lineage in the OE (G) and VNO (H). I, YFP expression (brown) was detected in the oral mucosa (OM) and dental enamel (TE), but not the tongue (Tng). Scale bars: A, D, F, I, 100 μm; B, C, E, G, H, 50 μm.
Nog defines neurogenic permissive areas under the control of BMP4
Active BMP signaling can trigger expression of BMP antagonists, such as Nog Chordin, Follisatin (Stottmann et al., 2001), redundant molecules with a key role in controlling olfactory neurogenesis (Kawauchi et al., 2009; Maier et al., 2010). Using a NogLacZ knock-in mouse (NognullLacZ/WT, Fig. 4), the physiological expression pattern Nog was assessed at E11.5. Nog-driven β-gal expression was found in epidermal cells, and in the dorsal and ventral non-neurogenic RE, areas also positive for Bmp4 expression and downstream signals (Figs. 2, 4A,B). In addition, strong Nog expression was found in a distinct group of mesenchymal cells, devoid of Bmp expression, adjacent to the developing VNO (Fig. 4A, asterisk). A Nog-positive border was created by ventral RE cells and mesenchymal cells that delineated the caudal boundary of the MSX1-positive epithelium and mesenchyme (Fig. 4A,B). Molecules able to silence BMP, according to the default program (Wilson and Hemmati-Brivanlou, 1995), allow ectodermal progenitors to access neuronal cell fate. Neuronal formation in the ventral OP, as assayed via expression of HuC/D and the transit-amplifying neural progenitor marker ASCL1, occurred caudal to the RE Nog source and proximal to the mesenchymal Nog source (Fig. 4C,D). Neurons were not detected in the Nog-expressing areas of the OP unless proximal to Nog-expressing mesenchyme. These data suggest that Bmp4 might play a “positive” role in defining the neurogenic permissive area by driving the expression pattern the BMP/Tgfβ antagonist Nog in the mesenchyme.
Figure 4.
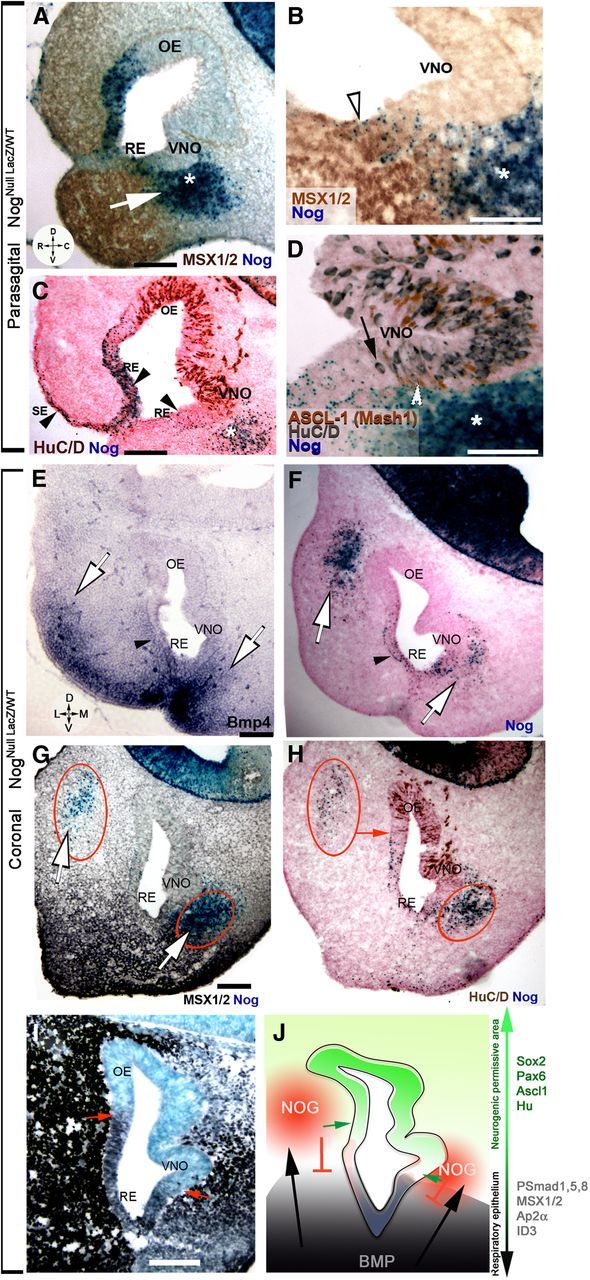
Mesenchymal sources of Nog define neurogenic permissive borders. A–F, X-gal enzymatic reaction (blue) on NognullLacz/WT. A, Msx1/2 expression (brown) decreased as Nog (blue) expression increased along the RE and in the nasal mesenchyme. White arrow depicts potential Bmp signal in inducing Nog expression. B, Nog (blue) was expressed by MSX1-positive cells (brown) along the RE while mesenchymal Nog expression defined MSX1-negative cells in the VNO. C, HuC/D immunostaining (brown) confirmed that Nog was in cells along the superficial epidermis (SE, arrowhead), non-neuronal RE (arrowheads) and in the ventral mesenchyme (asterisk). D, Vomeronasal area; immunostaining against transit amplifying progenitor cell marker ASCL1 (brown) and HuC/D (gray) showed similar expression patterns with respect to Nog sources (blue); HuC/D (black arrow) and ASCL1 (white arrowhead) were rarely in Nog-expressing areas, but were proximal to the Nog-expressing mesenchyme (asterisk). E–I, E11.5 coronal sections, orientation indicated in E. D, Dorsal; V, ventral; L, lateral; M, medial. E, In situ hybridization against Bmp4 highlighted Bmp4 sources in lateral, ventrolateral, and ventromedial mesenchyme (white arrows). Bmp4 was also expressed by mesenchyme proximal to the RE and by the RE (black arrowhead). F, X-gal staining (blue) revealed Nog expression in proximity to the lateral and ventromedial sources of Bmp4 (compare with E) as well as along the Bmp4-expressing RE (black arrowhead). G, Immunostaining highlighting MSX1 expression (black) in response to Bmp signaling in the mesenchyme and rostral RE. Nog expression (blue; red circles) was found in the dorsolateral and ventromedial nasal mesenchyme. H, HuC/D immunostaining (brown) indicated that neuron formation in the dorsolateral OE coincided with Nog expression in the mesenchyme (red arrow), while neuron formation in the VNO occurred proximal to the medioventral source of Nog (lower red circle). I, Immunostaining for the transcription factor AP2α revealed expression mainly in the non-neurogenic epithelium (use F as reference). AP2α levels decreased in proximity of the dorsolateral and ventromedial source of Nog (compare with G, H; lower red circle). J, Schematic representing relation between Bmp4 expression that induces Nog expression (red) in the nasal mesenchyme. By silencing Bmp signaling, Nog expression defines the neurogenic permissive areas of the OP. Markers related to neurogenic (green arrow) or epithelial fate (black arrow) are indicated. Scale bars, 100 μm.
Previous work by Tucker et al. (2010) indicated that subsets of embryonic progenitor/stem cells responsible for OE formation were located in the lateral portion of the OP. To determine the full extent of Nog sources and confirm the role played by mesenchymal sources of BMP inhibitors in controlling neurogenesis, coronal sections of E11.5 NogLacZ embryos were examined (Fig. 4E–I). In situ hybridization against Bmp4 revealed the existence of lateral/ventrolateral and ventromedial sources of Bmp4 (Fig. 4E). X-gal reaction alone (Fig. 4F) or followed by MSX1 immunostaining (Fig. 4G) confirmed that both medial and lateral BMP sources (Liu et al., 2005) correlated with Nog expression in a dorsolateral and ventromedial fashion. Staining for Nog:β-gal together with HuC/D (Fig. 4H) or AP2α (Fig. 4I) showed that the transition from non-neurogenic RE (AP2α+; Fig. 4I, arrow) 128 128 neurogenic epithelium (HuC/D+; Fig. 4H, arrow) coincided with decreased BMP4 signals and was defined by mesenchymal sources of Nog (Fig. 4G,H, red circles; summarized in Fig. 4J, scheme). In summary, in apparent reaction to BMP, Nog is expressed by epithelial cells of the OP and by distinct clusters of mesenchymal cells, the latter delineating a transition zone from epidermal to neurogenic precursors.
Sources of BMP4 in the nasal area directly control Nog expression pattern
Morphological observations suggested a potential cause–effect relation between BMP4 sources in the nasal area and Nog expression. To test whether this biochemical relation existed, beads soaked in 0.1% BSA (Fig. 5A,B) or BMP4 (Fig. 5C–H) were placed on nasal explants generated from Nog-LacZ and WT embryos (Fig. 5C–H). X-gal reaction was performed 18 h after bead placement. A BMP4-soaked bead placed on either the mesenchyme (Fig. 5C,D) or along the midline (Fig. 5E,F) of nasal explants generated from Nog-LacZ mice induced Nog expression (Fig. 5C–F, arrowheads). This induction was not seen with BSA-soaked beads implanted on Nog-LacZ tissue (Fig. 5A,B) or BMP4-soaked beads on WT tissue (Fig. 5G,H). These data are consistent with previous studies performed in different embryonic regions (Gazzerro et al., 1998; Merino et al., 1998; Stottmann et al., 2001) and confirm our hypothesis that sources of BMP4 directly induce Nog expression in the nasal area.
Figure 5.
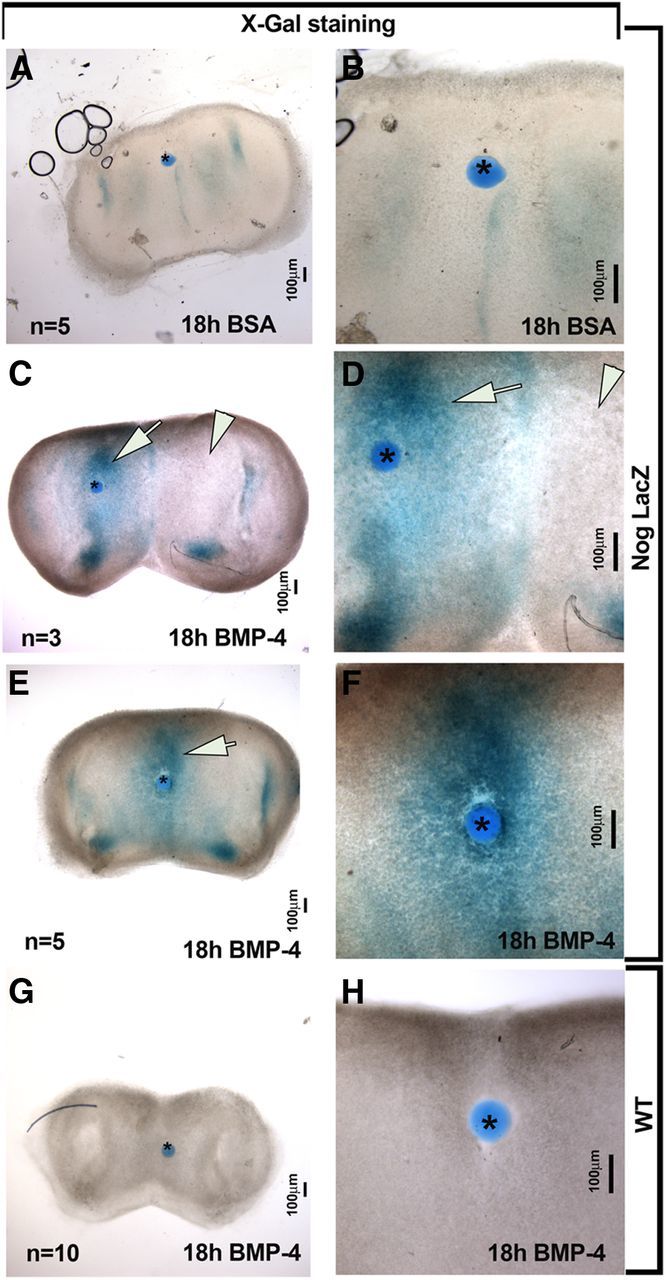
BMP4 directly induced Nog expression in developing nasal mesenchyme. A–F, Coronal nasal explant from NognullLacz/WT. A, B, Control bead soaked in BSA did not induce Nog expression in the surrounding tissue. C–F, BMP4-soaked bead placed on lateral (C, D) or midline (E, F) mesenchymal tissue of explants from Nog-LacZ mice induced Nog expression. Evidence of BMP4-induced expression of Nog was present regardless of bead position (see arrows in C, D, E vs internal negative control arrowheads in C, D). Endogenous levels of Nog served as internal controls in these experiments. BSA-soaked beads placed on Nog-LacZ explant tissue (A, B) as well as BMP4-soaked beads placed on explants from WT mice (G, H) served as negative controls and showed no induction of Nog:β-gal after X-gal staining. Bead is marked by asterisk in all images. High-magnification images are shown in B–H.
Mesenchymal sources of Nog define the GnRH neurogenic niche
GnRH neurons were analyzed to confirm the relationship between mesenchymal Nog and neuronal formation of this specific cell type (Fig. 6). GnRH neurons originate in a spatially limited area in the ventral portion of the OP (Tucker et al., 2010; Forni et al., 2011a). Analyzing NogLacZ E11.5 embryonic sections revealed that GnRH neurons formed from rostral to caudal in a ventromedial portion of the OP (∼150/200 μm) and were always juxtaposed to the mesenchymal source of Nog (Fig. 6A–D). In contrast, the lateral epithelium, positive for Fgf8 and Nog (Fig. 6E, white arrowhead, F) but not adjacent to Nog-expressing mesenchyme, did not generate GnRH or other neurons (Fig. 6F,H). Nog expression in the epithelium overlapped active BMP signaling (Fig. 6I) and was not sufficient alone to induce local neuronal formation. Thus, the ventromedial mesenchymal source of Nog (Fig. 6K) defined a “transitional” GnRH neurogenic niche between epidermal RE and developing VNO. A progressive increase of PAX6 and decrease of AP2α (Fig. 6F,G) in the niche proximal to the mesenchymal Nog source further supports the importance of this Nog source for neuronal formation. BMP/TGF-β signal transduction is known to repress SOX2 expression, a marker for stem cells/neural progenitors (Kawauchi et al., 2004; Domyan et al., 2011). MSX1 staining revealed that BMP signaling decreased in this transitional zone (Fig. 6I). Consistent with this, SOX2 expression abruptly increased in cells proximal to the ventromedial mesenchymal source of Nog, with little SOX2 expression found in the ventrolateral portion of the OP (Fig. 6H, black arrowhead and white arrowhead, respectively). Together, these data confirm that GnRH neurons form proximal to Fgf8-expressing epidermal cells in a specific neurogenic zone defined by mesenchymal Nog (Fig. 6K, scheme).
Figure 6.
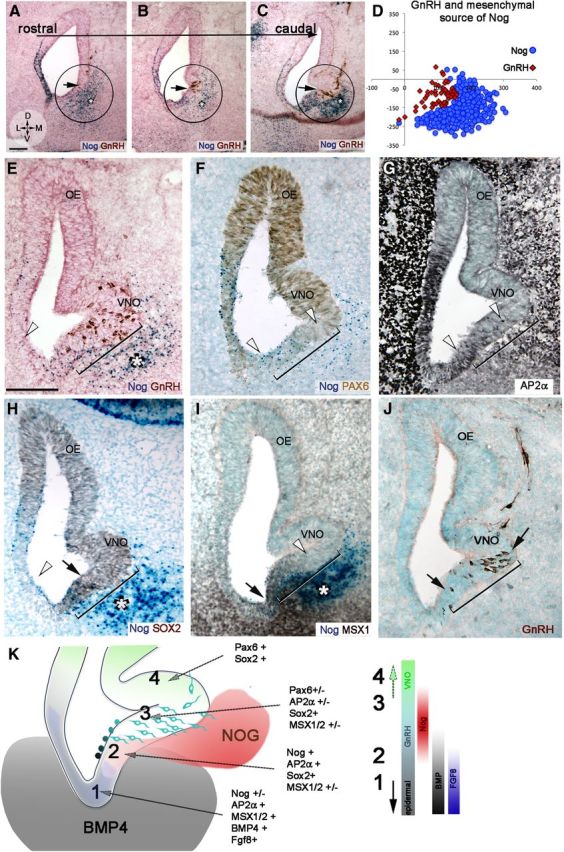
GnRH neurons form at the border between non-neuronal and neuronal epithelium in the developing OP. A–C, X-gal and GnRH immunostaining on serial coronal sections of E11.5 NognullLacz/WT embryo highlighting Nog (blue) and GnRH cells (brown). From rostral (A) to caudal (C), GnRH neurons originated only in ventral portions of the OP (circled), adjacent to the ventral mesenchymal source of Nog (asterisk). D, Scatter plot representing distribution of GnRH neurons (red) in four consecutive sections of NognullLacz/WT embryo in relation to mesenchymal source of Nog (blue). E–J, E11.5 coronal sections. E, GnRH neurons (brown) originate in a ∼250 mm area (bracket) that spans from the end of the Nog-expressing epithelium (blue dots) into the developing VNO and faces the mesenchymal source of Nog (asterisk). No GnRH neurons form in the lateral ventral OP (white arrowhead), a region that is not adjacent to Nog-expressing mesenchyme. F, PAX6 (brown) is not expressed in, or is expressed at very low levels along, the GnRH niche (bracket; compare with E). G, AP2α (black) is expressed in the GnRH neurogenic niche. AP2α expression decreased as PAX6 levels increased (compare with F). H, SOX2 (brown) levels increased along the GnRH niche (brackets), in correspondence to the ventral source of Nog (blue, asterisk). I, The GnRH niche (bracket, compare with J) was between the MSX1/Nog-positive epithelium and the MSX1-negative neurogenic VNO. MSX1 levels decreased coincident with mesenchymal Nog expression (asterisk). J, GnRH immunostaining (brown) as reference for H and I. K, Schematic summarizing molecular expression along the epidermal RE (1), transitional area (2), GnRH neurogenic area (3), and VNO (4). Molecules expressed in each area are listed. Color bars on right side indicate environmental factors associated with the transition from epidermal (1) to vomeronasal (4; +, high expression levels; +/−, low/nonuniform expression; −, nonexpressed/below detection). Scale bars, 100 μm.
Subthreshold Fgf8 expression leads to similar defects, independent of dose
To further delineate the relation between FGF8, BMP, and Nog in controlling neurogenesis, embryonic sections (E11.5) from mice carrying (1) two hypomorph Fgf8neo alleles (Fgf8neo/neo) or (2) one hypomorph and one null Fgf8null allele driving β-gal expression (Fgf8neo/nullLacZ) were analyzed (Figs. 7, 8). Surprisingly, regardless of the defective dosages (Storm et al., 2003), overlapping embryonic phenotypes were observed: (1) neurogenesis was only evident in the developing OE (Fig. 7A–F, area between white arrowheads) as reported previously for conditional KOs (Kawauchi et al., 2005), and (2) no GnRH immunoreactivity was detected as described previously for Fgf8neo/neo (Chung et al., 2008). In addition, it was obvious that neural formation was absent in the developing VNO and region proximal to the ventral RE (Fig. 7B,D,F). A complete lack of VNO formation in hypomorphs was confirmed by analyzing animals at later stages (E13.5–E15.5; data not shown). The overlapping phenotypes observed in the developing nasal area of the different hypomorph lines showed that subthreshold expression of Fgf8 led to similar defects, independent of dose.
Figure 7.
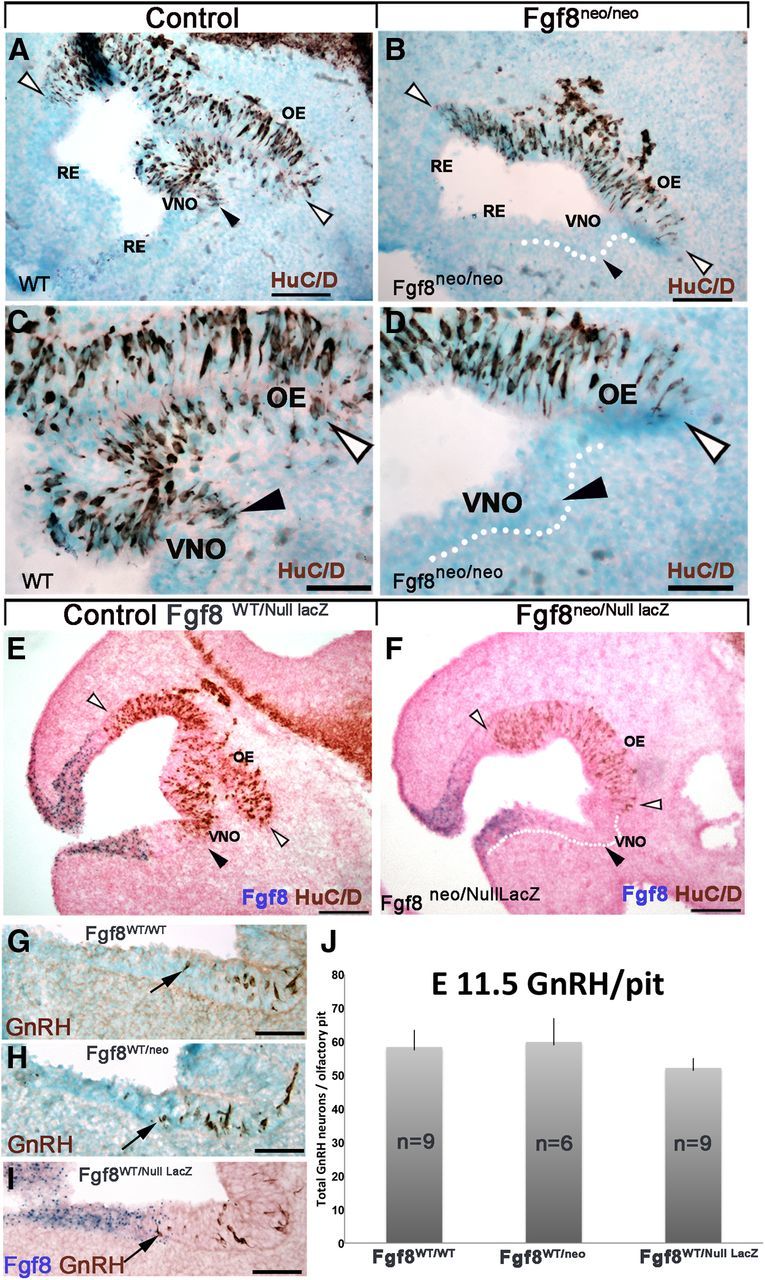
Fgf8 hypomorphs display complete loss of medioventral neurogenic milieus only in homozygosity. A–F, E11.5 parasagittal sections: HuC/D immunostaining on WT (A, C) and Fgf8neo/neo hypomorph (B, D). In control animals (A, C), positive neurons were detected in the OE (between white arrowheads) and VNO (black arrowhead). In Fgf8 hypomorphs (B, D) neurogenesis was limited to the OE (white arrowheads), no neurons were detected in the developing VNO (C vs D, black arrowheads). Similar results were observed comparing Fgf8WT/nullLacz(E) to Fgf8neo/nullLacz (F). No neurons were detected in the primordial VNO of either of the Fgf8 mutants (B, D, F). G–I, E11.5 immunostaining for GnRH showed comparable neurons (brown; black arrows) proximal to the VNO in wild-type (G), heterozygous Fgf8WT/neo (H), and Fgf8WT/null (I) embryos. J, Quantification of GnRH neurons in the nasal pit of all three genotypes showed no statistical differences (Fgf8WT/WT vs Fgf8WT/neo, p = 0.4; Fgf8WT/WT vs Fgf8WT/null LacZ, p = 0.9). Scale bars: A, B, E, F, 100 μm; C, D, G, H, I, 50 μm.
Figure 8.
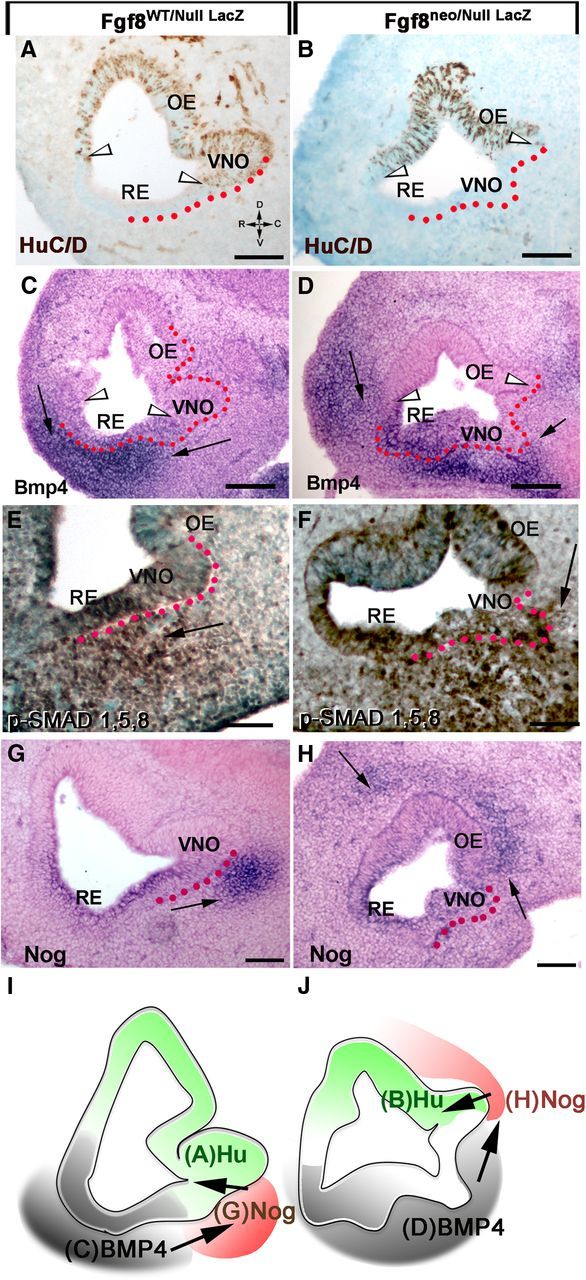
Changes in Fgf8 expression levels alter Bmp4, Nog expression in the mesenchyme and neurogenic pattern in the OP. Left, E11.5 control mice. Right, E11.5 Fgf8neo/nullLacz hypomorphs. A, B, HuC/D-positive cells (between arrowheads) identify the neurogenic areas in the nasal pit of control (A) and hypomorph (B). C, D, In situ hybridization for Bmp4. Bmp4 expression in the mesenchyme is indicated by arrows. In hypomorphs (D) a ventrocaudal expansion of Bmp4 expression correlated with lack of neuronal markers in the ventral portion of the OP (compare A, C to B, D). In Fgf8 mutants, Bmp4 expression along the RE appeared reduced compared with controls. E, F, p-SMAD 1,5,8 immunostaining on hypomorphs (F) showed a ventrocaudal expansion of mesenchymal Bmp4 signaling juxtaposed to the developing VNO and within the putative VNO (compare arrows). G, H, Nog in situ hybridization showed Nog expression in Fgf8 mutants was absent in the ventral mesenchyme juxtaposed to the VNO, but was detectable in a more dorsal portion of the developing pit (H, arrows). Expansion of Bmp4 expression (C vs D) in the hypomorphs correlated with changes in Nog expression in the nasal area (G vs H). Scale bars: A–D, 100 μm; E–H, 50 μm. I, J, Schematics summarizing results, with Bmp4 (gray) inducing mesenchymal Nog expression (red) that subsequently controls neurogenesis (green). The different pattern of Bmp and Nog expression translates in altered neurogenesis in Fgf8 mutants.
One mutant Fgf8 allele is not sufficient to disrupt GnRH neurons onset
Previous published data indicated that reduced functionality of one Fgf8 allele was sufficient to decrease FGF8 levels and affect GnRH immunoreactivity at postnatal stages (Chung et al., 2008). Quantification on E11.5 embryonic sections from Fgf8WT/WT, heterozygous null Fgf8null/WT and heterozygous hypomorphs Fgf8neo/WT revealed no differences in the location (Fig. 7G–I) or number of GnRH cells (Fig. 7J). Analysis at E15.5 confirmed similar numbers of GnRH cells in Fgf8neo hypomorphs and WT controls (742 ± 66 SD; 761 ± 111; p = 0.22). These data indicate that one fully functional Fgf8 allele is sufficient to allow normal olfactory/GnRH onset and GnRH peptide synthesis during embryonic development.
Altered mesenchymal signals lead to altered neurogenesis
The observations on Fgf8null/neo, Fgf8neo/neo, and heterozygous mice indicated that subthreshold Fgf8 expression leads to defects in VNO and GnRH specification. Since the neurogenic portion of the developing VNO is normally proximal to a Nog mesenchymal source that is induced by mesenchymal Bmp4 sources (Figs. 2, 4, 5), we hypothesized that the lack of ventral neurogenesis in the OP of Fgf8 mutants could result from variations in Bmp4 and Nog expression in this area. In situ hybridization for Bmp4 on control and Fgf8neo/nullLacZ hypomorph sections showed that the change in neuronal marker expression (HuC/D; Fig. 8A,B) was associated with a caudal expansion of mesenchymal BMP expression that included the area beneath the putative VNO (Fig. 8C,D). Confirmation of this expansion was obtained by p-SMAD 1,5,8 immunostaining (Fig. 8E,F). Consistent with the expansion of mesenchymal Bmp4 expression, Nog was ectopically expressed in the dorsal mesenchyme facing the putative OE (Fig. 8G,H) and nearly absent in the ventral area of the nasal mesenchyme proximal to the developing VNO. These experiments indicate that changes in Fgf8 levels altered expression of both Bmp4 and Nog, and, consequently, changed the neurogenic pattern (Fig. 8I,J, scheme).
Neurogenic permissive areas of the OP are defined by mesenchymal signals
Fgf8 hypomorphs exhibited a lack of neurogenesis in the ventromedial portion of the OP where GnRH and VNO neurons form, but no obvious defects in the early onset of cells in the lateral OE were observed (Fig. 9A,B). Reduction in local FGF8 has been described to lead to a direct decrease in SOX2 levels in the medial portion of the OP, causing loss of neuronal committed progenitors and transit amplifying cells (Tucker et al., 2010). However, BMP signaling can directly silence SOX2 expression (Domyan et al., 2011). To determine whether the variations in neurogenic pattern in Fgf8 hypomorphs correlated with altered mesenchymal signals, coronal sections of Fgf8neo/neo/NogLacZ/WT were analyzed. Changes in neurogenesis followed changes in mesenchymal expression of Nog (Fig. 9A,B). In control and mutant animals, comparable Nog expression and early neurogenic onset was observed in the dorsolateral nasal process and dorsolateral and dorsomedial OE; however, in medial nasal area, where neurogenesis was altered, a dramatic dorsomedial shift occurred in cells expressing Nog (Figs. 9, 10, schematic). Analysis of MSX1 on serial sections showed a dorsomedial shift in MSX1 expression in the medial mesenchyme (Fig. 9C,D). PAX6 was expressed in the neurogenic area of the developing OE, proximal to the mesenchymal sources of Nog in control animals. In the mutants, Nog was detectable, though at low levels, only in the area where the VNO and GnRH neurons normally form (Fig. 9E,F). In Fgf8 hypomorphs, an increase in SOX2 immunoreactivity occurred in tandem with the dorsalized mesenchymal source of Nog (Fig. 9G,H), moving farther from the ventral sources of Fgf8 (Fig. 1). In the lateral RE, AP2α expression patterns of Fgf8 hypomorphs (Fig. 9J) were similar to those of controls. In the medial RE epidermis, AP2α expression appeared reduced in intensity (Fig. 9I,J, arrowheads), in line with the previously described increased apoptosis in the mediorostral epidermis (Kawauchi et al., 2005). GnRH neurons normally form at the border between AP2α-expressing epidermis and the neurogenic area of the VNO (Fig. 9I,K). In the Fgf8 mutants, the reduced AP2α-expressing border was no longer proximal to sources of Nog (Fig. 9, compare A, B, I, J). As described by Chung and coworkers (Chung et al., 2008), GnRH neurons were not detected in the developing pit (Fig. 9K,L). However, the lack of GnRH neurons and the altered olfactory neurogenesis in Fgf8 mutants were the result of complex changes in placode/mesenchymal interactions rather than loss of a direct specification signal only. In fact, craniofacial defects in Fgf8 mutants resulted from aberrant Bmp4 and Nog expression, secondary to changes in Fgf8. These changes translated into the loss of neurogenic permissive conditions for the entire medioventral pit that included vomeronasal progenitors and GnRH progenitors, cells that were the most proximal to the FGF8 source (Fig. 9).
Figure 9.

Expression pattern of Bmp antagonists in nasal mesenchyme determines precursor identity and neurogenic potential of the OP. Coronal sections. Orientation indicated in A. D, Dorsal; V, ventral; L, lateral; M, medial. A–L, Sections from NognullLacz/WT control and Fgf8neo/neo/NognullLacz/WT mice showing neuron formation (HuC/D), Bmp4 signaling (MSX1), PAX6, SOX2, AP2α, and GnRH in relation to mesenchymal Nog (β-gal) expression (blue, arrows). In control animals (A) neuron formation (HuC/D brown, dorsal to dashed line) in the OE and VNO was defined by dorsolateral and medioventral sources of Nog (arrows). In Fgf8 hypomorphs (B) the medioventral source of Nog was found dorsalized with consequent loss of ventral neurogenesis in the VNO. C, D, MSX1/2 immunostaining (black) highlights the relation between mesenchymal Bmp signaling and Nog expression in control (C) and Fgf8 mutants (D). Reduction in MSX1 expression was noted in the RE (arrowheads) in mutant animals (D) compared with controls (C), while altered expression pattern was observed in the ventromedial mesenchyme with reduced expression proximal to the OP and broader expression in the medial mesenchyme. E, F, In control animals (E), PAX6 expression was high in the neurogenic areas of the pit (dorsal to dashed line; compare with A) in both OE and VNO proximal to the Nog sources (arrows) and distal to Bmp4 sources. In Fgf8 mutants (F), PAX6 expression was still detected in the medioventral OP where the VNO normally forms, though neurogenesis did not occur (arrow; compare with A). G, H, SOX2 expression was found to follow mesenchymal Nog expression in both controls (G) and Fgf8 hypomorphs (H). In the latter, high SOX2 levels were only detected in the areas proximal to Nog sources. Thus, Nog expression correlated with the neurogenic pattern. No SOX2 expression was found within, or proximal to, the developing VNO, where the GnRH normally form. I, J, Reduced AP2α expression was observed in the ventromedial RE of Fgf8 mutants (compare I, arrowhead, with J) while no obvious differences were observed in the lateral RE where transition from AP2α+ to HuC/D (compare with B) was similar to the controls (E, A). K, L, In control animals, the GnRH niche (K, arrowhead in circle) was found facing the mesenchymal source of Nog (arrow). In Fgf8 mutants GnRH niche (L, circled) was far from the mesenchymal source of Nog (arrow). Scale bars: (in A) A–J; (in K) K, L, 100 μm.
Figure 10.

Changes in FGF8, Bmp, Nog expression and neuronal formation in the OP of control and mutant animals. A, C, Summary schematics. Bmp4 expression defines (arrow) expression of Nog; in Fgf8 hypomorphs (C), changes in Bmp4 expression in the ventromedial developing mesenchyme, secondary to FGF8 changes, alter Nog expression (red) and therefore the neurogenic potential of progenitors in the OP (green). B, D, Scatter plots showing mesenchymal Nog expression (orange/red/brown) and HuC/D-positive neurons (shades of green) in serial coronal sections of controls (n = 3) and Fgf8 hypomorphs (n = 3) embryos at E11.5. Each data series represents cells plotted on three serial section/embryo. In the Fgf8 mutant, the ventromedial source of Nog was dorsalized and correlated with changes in neurogenesis.
Discussion
The “default model” of neural induction proposes that ectodermal stem cells, if protected from BMP/TGFβ signaling, are programmed to acquire a neural fate (Wilson and Hemmati-Brivanlou, 1995; Hemmati-Brivanlou and Melton, 1997). In contrast, other studies suggest that FGF8 signaling is necessary or sufficient for neural induction, independent of BMP (Kengaku and Okamoto, 1993; Lamb and Harland, 1995; Delaune et al., 2005; for review, see Stern, 2005). In the developing OP, we found that epidermal versus neural fate is in fact determined by the presence or absence of active BMP signals. The role played by FGF8 in controlling neurogenesis is largely indirect and is a consequence of its effect on craniofacial development and patterning of mesenchymal signals, such as Bmp4 and Nog.
The epidermis and RE controls craniofacial mesenchymal development
The outgrowth and patterning of facial mesenchyme, as for the limbs, is dependent on signals from the overlying ectoderm (Wedden, 1987; Richman and Tickle, 1992; LaMantia et al., 2000), with FGF8 being one of the key signals in these processes (Crossley and Martin, 1995; Kawauchi et al., 2005). Studies examining facial development in mouse models with reduced Fgf8 expression reported increased apoptosis limited to the Fgf8-expressing rostral portions of the OP together with olfactory defects (Kawauchi et al., 2005; Chung et al., 2008). Based on these observations, the FGF8-expressing region of the OP was proposed to be the stem cell reservoir, giving rise to neurons in the OE and VNO. Using Fgf8 knock-in models and Fgf8Cre lineage tracing, we demonstrated that neither the OE nor VNO originate from progenitors belonging to Fgf8-expressing domains. These data indicate that neurogenic defects in Fgf8 mutants are not derived from loss of stem cells in the rostral OP region. Fgf8 was found to be coexpressed with Bmp4 in ectodermal progenitors that gave rise to parts of the RE and rostral facial epidermis, lips, olfactory mucosa, and teeth enamel, but did not give rise to neurogenic OP stem cells. Based on these observations, we propose that reduced FGF8 signal directly affects growth of specific portions of the non-neurogenic ectoderm, including the skin of the lip, palate, and RE. The Fgf8-expressing ectoderm that is affected by reduced Fgf8 is also the morphogenic center controlling craniofacial development. Based on these data, we propose that neurogenic defects in the OP of Fgf8 mutants are largely indirect.
Mesenchymal signals regulate neurogenesis
By contextualizing the expression of Fgf8, Bmp4, and Nog, we uncovered spatial expression patterns that indicate the neurogenic changes in the OP of Fgf8 mutants are mainly a reflection of epithelial/mesenchymal cross-talk defects. In fact, the expression of neuronal stem/progenitor markers (SOX2, PAX6, and ASCL-1) did not directly correlate with Fgf8 levels or proximity to the Fgf8-expressing ectoderm but rather with an absence of BMP/TGFβ signaling, similar to that proposed by Gokoffski et al. (2011) for the mature OE. Thus, delineation of epidermal and neurogenic permissive areas in the developing OP (Fig. 4, schematic) appear to be established by an equilibrium between sources of BMP and the induction of BMP/Tgf-β inhibitors, such as Nog and/or Follistatin in the nasal mesenchyme (Gazzerro et al., 1998; Kearns and Demay, 2000; Stottmann et al., 2001; Kawauchi et al., 2009). Consistent with this idea, Nog transfections in the OP of chick had been shown to induce neurogenesis in areas that would normally acquire epidermal fate (Maier et al., 2010, 2011). Analyzing neurogenesis in relation to Nog and Fgf8 expression using NogLacZ and Fgf8LacZ embryos, we observed that in physiological conditions neuronal formation did not occur within the Fgf8-expressing and/or Nog-expressing domains. In the OP, Nog and Fgf8 were for the most part expressed by epidermal/respiratory cells that were also positive for Bmp4 expression and downstream signals. These data indicate that if FGF8 exerts a proneurogenic effect in the developing OP, this happens indirectly only in neurogenic permissive areas delineated by mesenchymal sources of BMP/TGF-β inhibitors.
In the OP, a direct role for FGF8 in controlling neuronal cell fate through restriction of BMP4 expression/signaling has been proposed (Wilson et al., 2000; Pera et al., 2003; Maier et al., 2010). Our data indicate that autocrine as well as paracrine FGF8 (Kawauchi et al., 2005) is not sufficient to antagonize BMP signaling and protect ectodermal progenitors of the OP from acquiring “epidermal” cell fate. A primary role played by mesenchymal signals in controlling neurogenesis in the OP and GnRH neurogenesis (Fig. 9, model) found further support from examination of Fgf8 hypomorphs. Partial loss of FGF8 severely affected the development of medial nasal process (Kawauchi et al., 2005) and mesenchymal tissue that later gives rise to the palate roof and the nasal septum. Analysis of the medial process of Fgf8 mutants revealed changes in Bmp4 expression together with Nog expression going from ventral to dorsal areas. These changes correlated with shifts in the neurogenic pattern (Figs. 7–9). Notably in the lateral nasal process, where no major changes in either BMP4 or Nog occurred, the neurogenic pattern remained unaltered. These data indicate that Fgf8 is not the main neurogenic inducer for the OE and suggest that factors other than Fgf8 are crucial in controlling morphogenesis of the lateral nasal process. In Fgf8 conditional KOs and in Fgf8 hypomorphs, changes in neuronal markers and cell proliferation in the ventromedial OP had been previously noticed but whether or not this effect was direct or indirect had not been addressed (Chung et al., 2008; Tucker et al., 2010). We observed that changes in SOX2, PAX6, and HuC/D expression in Fgf8 mutants reflected mesenchymal expression of Nog and were not in proximity to FGF8 sources (Tucker et al., 2010). These data are consistent with mesenchymal Bmp expression and silencing of BMP/TGFβ signaling being a key element in defining the proliferative characteristics of OP progenitors.
Reduced FGF8 and Kallmann syndrome
A decrease in Fgf8/FgfR1 signaling has been associated with various forms of HH, including cases of Kallmann (Falardeau et al., 2008; Trarbach et al., 2010). Moreover, variable penetrance of craniofacial defects, including cleft palate, which can derive from interrupted fusion of the maxillary and medial nasal process, dental agenesis, and cleft lips, have been described in pedigrees of families carrying Fgf8/FgfR1 mutations and HH (Tompach and Zeitler, 1995; Mølsted et al., 1997; Dode et al., 2007; Riley et al., 2007; Falardeau et al., 2008; Bailleul-Forestier et al., 2010). Notably all the affected structures listed above were found to be positive for Fgf8 lineage tracing in this report.
GnRH neurons originate from heterogeneous precursors in a well defined portion of the ventral OP (Tucker et al., 2010; Forni et al., 2011a) proximal to FGF8, BMP, and Nog sources. Loss of function of one Fgf8 allele has previously been proposed to compromise embryonic GnRH neurons onset (Chung et al., 2008; Sabado et al., 2012). However, our analysis of heterozygous null Fgf8null/WT and heterozygous hypomorph Fgf8neo/WT mice, where no craniofacial defects occurred, found no differences in the onset or number of GnRH neurons during embryonic development. This analysis indicates that one functional Fgf8 allele is sufficient for normal GnRH neuronal development and, therefore, loss of function of one Fgf8 allele is not per se autosomal dominant. Our data also suggest that Fgf8 point mutations in heterozygous states, which have been found to correlate with variable penetrance of craniofacial defects and HH in humans, are likely to act in a competitive/dominant-negative fashion if not in association with other genetic mutations (Trarbach et al., 2010). In fact, in mice, we observed that defects in GnRH development only occurred at subthreshold Fgf8 levels that also affected craniofacial development and neuronal formation in the ventromedial OP. Though we cannot rule out a priming effect for Fgf8 in GnRH neuronal formation, expansion, or survival, (Sabado et al., 2012) or in modulating GnRH peptide synthesis levels (Falardeau et al., 2008), our data indicate that the absence of GnRH neurons in Fgf8 hypomorph mutants is one aspect of a much larger loss of a neurogenic permissive milieu that also includes the VNO rather than the lack of a specification signal.
Altered FGF8 can affect Bmp, Msx1 (Storm et al., 2006), and consequently BMP antagonist expression pattern. Genetic mutations that directly or indirectly affect Bmp expression have been shown to affect normal plate fusion and to increase apoptosis in the fusion lip ectoderm (Heikinheimo et al., 1994; Liu et al., 2005; He et al., 2010), an area where Fgf8 is expressed and increased apoptosis occurs in Fgf8 mutants (Kawauchi et al., 2005). These data suggest an important diagnostic value of craniofacial defects (e.g. cleft palate, cleft lip, dental agenesis, and mandibular and maxillary abnormalities) seen in human Kallmann cases with genetic mutations affecting FGF8 signal transduction (Mølsted et al., 1997; Kawauchi et al., 2005; Riley et al., 2007; Trarbach et al., 2010). Our data indicate that specific pathological conditions affecting the FGF8 axis, including autosomal-dominant negative mutations in the receptors (Kim et al., 2005) or truncated forms of the ligand, lead to syndromic defects, e.g., Kallmann syndrome, as a result of dysmorphic mesenchymal growth and loss of neurogenic milieus. A systematic analysis of early vomeronasal and GnRH development in animal models with defective development of medial nasal process could lead to the identification of important FGF8 target genes in this area and candidate genes responsible for the etiology of forms of HH and Kallmann syndrome.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS002824-13). Several monoclonal antibodies used in these studies were obtained from the Developmental Studies Hybridoma Bank developed under National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology, Iowa City, Iowa.
References
- Bailleul-Forestier I, Gros C, Zenaty D, Bennaceur S, Leger J, de Roux N. Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. Int J Paediatr Dent. 2010;20:305–312. doi: 10.1111/j.1365-263X.2010.01056.x. [DOI] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti K, Liu W, Csermely T, Bertuzzi S, Arnheiter H. Alternative promoter use in eye development: the complex role and regulation of the transcription factor MITF. Development. 2008;135:1169–1178. doi: 10.1242/dev.014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U, Zou Z, Buck LB. Feedback loops link odor and pheromone signaling with reproduction. Cell. 2005;123:683–695. doi: 10.1016/j.cell.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- Chiba S, Lee YM, Zhou W, Freed CR. Noggin enhances dopamine neuron production from human embryonic stem cells and improves behavioral outcome after transplantation into parkinsonian rats. Stem Cells. 2008;26:2810–2820. doi: 10.1634/stemcells.2008-0085. [DOI] [PubMed] [Google Scholar]
- Chmielnicki E, Benraiss A, Economides AN, Goldman SA. Adenovirally expressed noggin and brain-derived neurotrophic factor cooperate to induce new medium spiny neurons from resident progenitor cells in the adult striatal ventricular zone. J Neurosci. 2004;24:2133–2142. doi: 10.1523/JNEUROSCI.1554-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WC, Tsai PS. Role of fibroblast growth factor signaling in gonadotropin-releasing hormone neuronal system development. Front Horm Res. 2010;39:37–50. doi: 10.1159/000312692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WC, Moyle SS, Tsai PS. Fibroblast growth factor 8 signaling through fibroblast growth factor receptor 1 is required for the emergence of gonadotropin-releasing hormone neurons. Endocrinology. 2008;149:4997–5003. doi: 10.1210/en.2007-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Delaune E, Lemaire P, Kodjabachian L. Neural induction in Xenopus requires early FGF signalling in addition to BMP inhibition. Development. 2005;132:299–310. doi: 10.1242/dev.01582. [DOI] [PubMed] [Google Scholar]
- Dode C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, Kottler ML, Chabrolle C, Gancel A, Francois I, et al. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat. 2007;28:97–98. doi: 10.1002/humu.9470. [DOI] [PubMed] [Google Scholar]
- Domyan ET, Ferretti E, Throckmorton K, Mishina Y, Nicolis SK, Sun X. Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development. 2011;138:971–981. doi: 10.1242/dev.053694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornaro M, Geuna S, Fasolo A, Giacobini-Robecchi MG. HuC/D confocal imaging points to olfactory migratory cells as the first cell population that expresses a post-mitotic neuronal phenotype in the chick embryo. Neuroscience. 2003;122:123–128. doi: 10.1016/j.neuroscience.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Forni PE, Wray S. Neural crest and olfactory system: new prospective. Mol Neurobiol. 2012;46:349–360. doi: 10.1007/s12035-012-8286-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni PE, Fornaro M, Guénette S, Wray S. A role for FE65 in controlling GnRH-1 neurogenesis. J Neurosci. 2011a;31:480–491. doi: 10.1523/JNEUROSCI.4698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni PE, Taylor-Burds C, Melvin VS, Williams T, Wray S. Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. J Neurosci. 2011b;31:6915–6927. doi: 10.1523/JNEUROSCI.6087-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzerro E, Gangji V, Canalis E. Bone morphogenetic proteins induce the expression of Noggin, which limits their activity in cultured rat osteoblasts. J Clin Invest. 1998;102:2106–2114. doi: 10.1172/JCI3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokoffski KK, Wu HH, Beites CL, Kim J, Kim EJ, Matzuk MM, Johnson JE, Lander AD, Calof AL. Activin and GDF11 collaborate in feedback control of neuroepithelial stem cell proliferation and fate. Development. 2011;138:4131–4142. doi: 10.1242/dev.065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshammer U, Cebrián C, Ilagan R, Meyers E, Herzlinger D, Martin GR. FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development. 2005;132:3847–3857. doi: 10.1242/dev.01944. [DOI] [PubMed] [Google Scholar]
- He F, Xiong W, Wang Y, Matsui M, Yu X, Chai Y, Klingensmith J, Chen Y. Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev Biol. 2010;347:109–121. doi: 10.1016/j.ydbio.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikinheimo M, Lawshé A, Shackleford GM, Wilson DB, MacArthur CA. Fgf-8 expression in the post-gastrulation mouse suggests roles in the development of the face, limbs and central nervous system. Mech Dev. 1994;48:129–138. doi: 10.1016/0925-4773(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Hemmati-Brivanlou A, Melton D. Vertebrate embryonic cells will become nerve cells unless told otherwise. Cell. 1997;88:13–17. doi: 10.1016/S0092-8674(00)81853-X. [DOI] [PubMed] [Google Scholar]
- Hu JS, Doan LT, Currle DS, Paff M, Rheem JY, Schreyer R, Robert B, Monuki ES. Border formation in a Bmp gradient reduced to single dissociated cells. Proc Natl Acad Sci U S A. 2008;105:3398–3403. doi: 10.1073/pnas.0709100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, Klingensmith J, Meyers EN. Fgf8 is required for anterior heart field development. Development. 2006;133:2435–2445. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Mental Deficiency. 1944;48:203–236. [Google Scholar]
- Kawauchi S, Beites CL, Crocker CE, Wu HH, Bonnin A, Murray R, Calof AL. Molecular signals regulating proliferation of stem and progenitor cells in mouse olfactory epithelium. Dev Neurosci. 2004;26:166–180. doi: 10.1159/000082135. [DOI] [PubMed] [Google Scholar]
- Kawauchi S, Shou J, Santos R, Hébert JM, McConnell SK, Mason I, Calof AL. Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development. 2005;132:5211–5223. doi: 10.1242/dev.02143. [DOI] [PubMed] [Google Scholar]
- Kawauchi S, Kim J, Santos R, Wu HH, Lander AD, Calof AL. Foxg1 promotes olfactory neurogenesis by antagonizing Gdf11. Development. 2009;136:1453–1464. doi: 10.1242/dev.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns AE, Demay MB. BMP-2 induces the expression of activin betaA and follistatin in vitro. J Cell Biochem. 2000;79:80–88. [PubMed] [Google Scholar]
- Kengaku M, Okamoto H. Basic fibroblast growth factor induces differentiation of neural tube and neural crest lineages of cultured ectoderm cells from Xenopus gastrula. Development. 1993;119:1067–1078. doi: 10.1242/dev.119.4.1067. [DOI] [PubMed] [Google Scholar]
- Kim HG, Herrick SR, Lemyre E, Kishikawa S, Salisz JA, Seminara S, MacDonald ME, Bruns GA, Morton CC, Quade BJ, Gusella JF. Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J Med Genet. 2005;42:666–672. doi: 10.1136/jmg.2004.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenke U, Taylor-Burds C. Culturing embryonic nasal explants for developmental and physiological study. Curr Protoc Neurosci. 2012:1–16. doi: 10.1002/0471142301.ns0325s59. Chapter 3: Unit 3.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PR, Guerrero G, Krishnamurthy R, Mitchell PJ, Wray S. Ectopic expression of luteinizing hormone-releasing hormone and peripherin in the respiratory epithelium of mice lacking transcription factor AP-2alpha. Mech Dev. 2000;94:79–94. doi: 10.1016/S0925-4773(00)00316-6. [DOI] [PubMed] [Google Scholar]
- LaMantia AS, Bhasin N, Rhodes K, Heemskerk J. Mesenchymal/epithelial induction mediates olfactory pathway formation. Neuron. 2000;28:411–425. doi: 10.1016/S0896-6273(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Lamb TM, Harland RM. Fibroblast growth factor is a direct neural inducer, which combined with Noggin generates anterior–posterior neural pattern. Development. 1995;121:3627–3636. doi: 10.1242/dev.121.11.3627. [DOI] [PubMed] [Google Scholar]
- Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132:1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- Maier E, von Hofsten J, Nord H, Fernandes M, Paek H, Hébert JM, Gunhaga L. Opposing Fgf and Bmp activities regulate the specification of olfactory sensory and respiratory epithelial cell fates. Development. 2010;137:1601–1611. doi: 10.1242/dev.051219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier E, Nord H, von Hofsten J, Gunhaga L. A balance of BMP and notch activity regulates neurogenesis and olfactory nerve formation. PloS One. 2011;6:e17379. doi: 10.1371/journal.pone.0017379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal L, Luxardi G, Thomé V, Kodjabachian L. BMP inhibition initiates neural induction via FGF signaling and Zic genes. Proc Natl Acad Sci U S A. 2009;106:17437–17442. doi: 10.1073/pnas.0906352106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino R, Gañan Y, Macias D, Economides AN, Sampath KT, Hurle JM. Morphogenesis of digits in the avian limb is controlled by FGFs, TGFbetas, and Noggin through BMP signaling. Dev Biol. 1998;200:35–45. doi: 10.1006/dbio.1998.8946. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Mølsted K, Kjaer I, Giwercman A, Vesterhauge S, Skakkebaek NE. Craniofacial morphology in patients with Kallmann's syndrome with and without cleft lip and palate. Cleft Palate Craniofac J. 1997;34:417–424. doi: 10.1597/1545-1569_1997_034_0417_cmipwk_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Ogata T, Fujiwara I, Ogawa E, Sato N, Udaka T, Kosaki K. Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr J. 2006;53:741–743. doi: 10.1507/endocrj.k06-099. [DOI] [PubMed] [Google Scholar]
- Pera EM, Ikeda A, Eivers E, De Robertis EM. Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 2003;17:3023–3028. doi: 10.1101/gad.1153603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman JM, Tickle C. Epithelial-mesenchymal interactions in the outgrowth of limb buds and facial primordia in chick embryos. Dev Biol. 1992;154:299–308. doi: 10.1016/0012-1606(92)90069-s. [DOI] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dodé C, Mohammadi M, Marazita ML, Murray JC. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 2007;104:4512–4517. doi: 10.1073/pnas.0607956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabado V, Barraud P, Baker CV, Streit A. Specification of GnRH-1 neurons by antagonistic FGF and retinoic acid signaling. Dev Biol. 2012;362:254–262. doi: 10.1016/j.ydbio.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern CD. Neural induction: old problem, new findings, yet more questions. Development. 2005;132:2007–2021. doi: 10.1242/dev.01794. [DOI] [PubMed] [Google Scholar]
- Storm EE, Rubenstein JL, Martin GR. Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci U S A. 2003;100:1757–1762. doi: 10.1073/pnas.0337736100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK, Martin GR, Rubenstein JL. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development. 2006;133:1831–1844. doi: 10.1242/dev.02324. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Anderson RM, Klingensmith J. The BMP antagonists Chordin and Noggin have essential but redundant roles in mouse mandibular outgrowth. Dev Biol. 2001;240:457–473. doi: 10.1006/dbio.2001.0479. [DOI] [PubMed] [Google Scholar]
- Streit A, Berliner AJ, Papanayotou C, Sirulnik A, Stern CD. Initiation of neural induction by FGF signalling before gastrulation. Nature. 2000;406:74–78. doi: 10.1038/35017617. [DOI] [PubMed] [Google Scholar]
- Tang J, Song M, Wang Y, Fan X, Xu H, Bai Y. Noggin and BMP4 co-modulate adult hippocampal neurogenesis in the APP(swe)/PS1(DeltaE9) transgenic mouse model of Alzheimer's disease. Biochem Biophys Res Commun. 2009;385:341–345. doi: 10.1016/j.bbrc.2009.05.067. [DOI] [PubMed] [Google Scholar]
- Tompach PC, Zeitler DL. Kallmann syndrome with associated cleft lip and palate: case report and review of the literature. J Oral Maxillofac Surg. 1995;53:85–87. doi: 10.1016/0278-2391(95)90511-1. [DOI] [PubMed] [Google Scholar]
- Toyoda R, Assimacopoulos S, Wilcoxon J, Taylor A, Feldman P, Suzuki-Hirano A, Shimogori T, Grove EA. FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development. 2010;137:3439–3448. doi: 10.1242/dev.055392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trarbach EB, Abreu AP, Silveira LF, Garmes HM, Baptista MT, Teles MG, Costa EM, Mohammadi M, Pitteloud N, Mendonca BB, Latronico AC. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J Clin Endocrinol Metab. 2010;95:3491–3496. doi: 10.1210/jc.2010-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AS, Yamada G, Grigoriou M, Pachnis V, Sharpe PT. Fgf-8 determines rostral-caudal polarity in the first branchial arch. Development. 1999;126:51–61. doi: 10.1242/dev.126.1.51. [DOI] [PubMed] [Google Scholar]
- Tucker ES, Lehtinen MK, Maynard T, Zirlinger M, Dulac C, Rawson N, Pevny L, Lamantia AS. Proliferative and transcriptional identity of distinct classes of neural precursors in the mammalian olfactory epithelium. Development. 2010;137:2471–2481. doi: 10.1242/dev.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bolotin D, Chu DH, Polak L, Williams T, Fuchs E. AP-2alpha: a regulator of EGF receptor signaling and proliferation in skin epidermis. J Cell Biol. 2006;172:409–421. doi: 10.1083/jcb.200510002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedden SE. Epithelial-mesenchymal interactions in the development of chick facial primordia and the target of retinoid action. Development. 1987;99:341–351. doi: 10.1242/dev.99.3.341. [DOI] [PubMed] [Google Scholar]
- Wilson PA, Hemmati-Brivanlou A. Induction of epidermis and inhibition of neural fate by Bmp-4. Nature. 1995;376:331–333. doi: 10.1038/376331a0. [DOI] [PubMed] [Google Scholar]
- Wilson SI, Graziano E, Harland R, Jessell TM, Edlund T. An early requirement for FGF signalling in the acquisition of neural cell fate in the chick embryo. Curr Biol. 2000;10:421–429. doi: 10.1016/s0960-9822(00)00431-0. [DOI] [PubMed] [Google Scholar]
- Wray S. Gonadotropin-releasing hormone: GnRH-1 system. In: Squire LR, editor. Encyclopedia of neuroscience. Vol 4. Oxford: Academic; 2009. [Google Scholar]
- Wray S. From nose to brain: development of gonadotrophin-releasing hormone-1 neurones. J Neuroendocrinol. 2010;22:743–753. doi: 10.1111/j.1365-2826.2010.02034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S, Gähwiler BH, Gainer H. Slice cultures of LHRH neurons in the presence and absence of brainstem and pituitary. Peptides. 1988;9:1151–1175. doi: 10.1016/0196-9781(88)90103-9. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- Zimmerman LB, De Jesús-Escobar JM, Harland RM. The Spemann organizer signal Noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/S0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]