

Abstract
Notwithstanding an extensive literature on assembly line polyketide synthases such as the 6-deoxyerythronolide B synthase (DEBS), a complete naturally occurring synthase has never been reconstituted in vitro from purified protein components. Here, we describe the fully reconstituted DEBS and quantitatively characterize some of the properties of the assembled system that have never been explored previously. The maximum turnover rate of the complete hexamodular system is 1.1 min−1, comparable to the turnover rate of a truncated trimodular derivative (2.5 min−1) but slower than a bimodular derivative (21 min−1). In the presence of similar concentrations of methylmalonyl- and ethylmalonyl-CoA substrates, DEBS synthesizes multiple regiospecifically modified analogs, one of which we have analyzed in detail. Our studies lay the foundation for biochemically interrogating and rationally engineering polyketide assembly lines in an unprecedented manner.
Multimodular polyketide synthases (PKSs) catalyze the biosynthesis of numerous structurally complex polyketide antibiotics1,2,3,4 via an assembly line mechanism.5 Although hundreds of these assembly lines have been sequenced and several have been extensively engineered,6,7,8,9 a naturally occurring multimodular PKS has never been fully reconstituted from purified protein components. Reconstitution of an entire metabolic pathway operating in an assembly line fashion opens up numerous opportunities, such as providing a platform for elucidating mechanistic details of polyketide biosynthesis and enabling engineering of polyketide synthases.
The 6-deoxyerythronolide B synthase (DEBS)10,11,12 is arguably the most-well studied member of this PKS family and produces the macrolactone 6-deoxyerythronolide B (6-dEB) 1 (Figure 1 and Figure S1). Its structure, mechanism, and promise for reprogrammed biosynthesis have been the focus of numerous reports.5,13,14,6,15,16 Both unimodular17 as well as bimodular18,19,20 derivatives have been functionally reconstituted from purified components. Two decades ago, we had observed cell-free synthesis of 1 in protein extracts harboring the three DEBS subunits.21 However, this system was not well defined since the DEBS proteins were not purified. Here we report the full, in vitro reconstitution and biochemical analysis of the full DEBS system along with two truncated derivatives.
Figure 1. Reconstituted DEBS and its truncated derivatives.

The domains corresponding to each polyketide synthase module are shown in a distinct color. LDD = loading didomain; KS = ketosynthase; AT = acyl transferase; ACP = acyl carrier protein; KR = ketoreductase; DH = dehydratase; ER = enoyl reductase; TE = thioesterase. Domains that are part of the same protein are shown contacting each other. Black tabs correspond to docking domains that contribute to non-covalent interactions between proteins harboring successive modules. For a more detailed description of the overall biosynthetic pathway, see Figure S1.
A major barrier to reconstituting fully active DEBS is the exceptionally large size of its constituent polypeptides, DEBS1, DEBS2, and DEBS3, each of which has a monomeric mass exceeding 300 kDa and is homodimeric in its catalytically active form.5 With considerable difficulty, we were able to develop expression and purification protocols for DEBS2 and DEBS3 from recombinant strains of E. coli. (See SI Methods for details). Since isolation of DEBS1 in its natural form proved intractable, we therefore expressed and reconstituted this multimodular protein as a dissociated complex of three proteins – a loading didomain (LDD), module 1 (M1), and module 2 (M2). Each dissociated protein was engineered to facilitate specific interaction with the next component of the assembly line, using complementary docking sites derived from the DEBS1-DEBS2 and DEBS2-DEBS3 interfaces17,19 (depicted as black tabs in Figure 1 and Figure S1). Specifically, LDD was fused to the C-terminal intermodular docking sequence of DEBS2, M1 was flanked by N- and C-terminal docking sequence from DEBS3 and DEBS1, respectively, and M2 was fused to the N-terminal docking sequence from DEBS2. The introduction of repetitive docking domains raised the potential for undesired crosstalk in the reconstituted system. Crosstalk would include translocation of the growing polyketide chain between M1 and DEBS2 or between two homodimers of M2. Based on recent evidence demonstrating the importance of specific protein-protein interactions between acyl carrier protein (ACP) and ketosynthase (KS) domains during chain translocation, we reasoned that matched docking domain interactions would be necessary but not sufficient for efficient intermodular association and chain translocation.22,23,24 Subsequent experiments confirmed the validity of our reasoning, as reported below.
When all five proteins were mixed at concentrations of 2 μM each, along with the requisite propionyl-CoA, methylmalonyl-CoA, and NADPH substrates, LC-MS analysis revealed the presence of the expected product 1, as verified against an authentic standard (Figure S2).25 None of the plausible fall-off products, including the known products of modules 2, 3 or 526,27,28, were observed in detectable quantities. Thus, the reconstituted DEBS system produced the expected 14-membered lactone as its dominant product. Truncated trimodular (LDD-M1-M2-M3+TE) and bimodular (LDD-M1-M2+TE) derivatives of DEBS were also reconstituted similarly, yielding the predicted products 2 and 3, respectively, as verified against authentic standards27,26 (Figures S3 and Figure S4). In each case, the thioesterase (TE) domain of DEBS was fused to the terminal module (Figure 1) to facilitate assembly line turnover. No truncated polyketides were observed from the bimodular PKS, although the trimodular system also produced minor amounts of triketide 3 in quantities detectable by LC-MS, such that the integration of the extracted ions for 3 is approximately 10% of that for 2 (see Figure S5).
To facilitate kinetic analysis of reconstituted DEBS, we sought to develop a continuous assay in which NADPH consumption by its five catalytically active ketoreductase (KR) domains and one enoyl reductase (ER) domain could be measured by UV spectrophotometry. In order to validate this assay, stoichiometric equivalence between NADPH consumption and product formation had to be established. For this, a 6-dEB standard was prepared from an appropriate biosynthetic source, purified by silica gel chromatography, and used to establish an LC-MS calibration curve for 6-dEB quantification (Figure S6). For all five DEBS proteins at 2 μM, the amount of 6-dEB predicted by the UV assay and the amount measured directly by the LC-MS assay were in very close agreement (Figure S7.A). This equivalence allowed us to use UV spectrophotometry as the primary tool for continuous kinetic analysis of DEBS. The UV assay also verified that undesired crosstalk was insignificant. For the trimodular DEBS derivative, one might predict crosstalk between M1 and M3+TE or between two homodimers of M2 as a consequence of the repetitive docking domains. In control experiments, NADPH consumption above background was undetectable in either case (Figure S7.B).
By plotting the initial velocity of product formation against varying concentrations of DEBS2 or DEBS3, keeping all other proteins at a fixed concentration of 2 μM (Figure 2), the (V/[E]0)max of the complete assembly line was calculated to be 1.1 min−1 by direct fitting to the Michaelis-Menten equation (approximately the same maximum rate was achieved regardless of whether DEBS2 or DEBS3 was titrated), and the K50 values derived from these titrations were in the 2.5-4 μM range (Table 1). In the context of these experiments, K50 can be regarded as a measure of the interaction efficiency of the DEBS1-DEBS2 and/or DEBS2-DEBS3 interface. It was therefore reassuring to note that these experiments yielded very similar parametric values to those reported elsewhere using an alternative method.19
Figure 2. Effect of titrating DEBS2 or DEBS3 on the turnover rate of DEBS.

Except for the titrant, all other proteins were held at a constant concentration of 2 μM; the turnover rate is normalized to this concentration. Kinetic parameters are presented in Table 1.
Table 1.
Rate constants derived from kinetic analysis of the assembly lines shown in Figure 1.
| Assembly Line | Component Varied | (V/[E]0)max(min−1) | K50(μM) |
|---|---|---|---|
| LDD-M1-M2+TE | M1 | 21 ± 0.9 | 7.3 ± 0.7 |
| M2+TE | 15 ± 0.7 | 6.3 ± 0.9 | |
| LDD-M1-M2-M3+TE | M2 | 2.1 ± 0.1 | 2.6 ± 0.2 |
| M3+TE | 2.5 ± 0.1 | 3.2 ± 0.4 | |
| LDD-M1-M2-DEBS2-DEBS3 | DEBS2 | 1.1 ± 0.1 | 4.0 ± 1.1 |
| DEBS3 | 0.8 ± 0.1 | 2.9 ± 0.5 | |
Whereas the maximum turnover rate of 2.5 min−1 for the trimodular derivative of DEBS was comparable to that of the full hexamodular assembly line, the corresponding (V/[E]0)max of 21 min−1 for the bimodular derivative was significantly higher (Table 1 and Figure S8). While these results may superficially suggest that the primary rate limiting step in DEBS lies at the DEBS1-DEBS2 interface or in module 3, the titration data shown in Figure 2 suggest otherwise. Specifically, they indicate that flux control through DEBS is complex and distributed over reactions within DEBS2 and DEBS3 in a manner that cannot simply be alleviated by increasing the concentration of a single DEBS protein. Importantly, the maximum turnover rate of DEBS is consistent with its observed productivity in E. coli or S. erythraea,29,30 suggesting that the reconstituted system accurately reflects assembly line behavior under physiological conditions.
The availability of fully active DEBS from purified protein constituents offers an opportunity to interrogate the cumulative specificity of the entire multimodular system for unnatural extender units. Previous studies have shown that unnatural extender units can be incorporated into polyketide products by engineering individual DEBS acyl transferase (AT) domains.31,32,33,34,35 While these findings imply a degree of tolerance of DEBS toward unnatural extender units, they do not address the more fundamental issue of the inherent extender unit specificity of the wild-type assembly line. To address this question, equimolar mixtures of malonate and methylmalonate, or alternatively methylmalonate and ethylmalonate were added to a reaction mixture containing reconstituted DEBS along with non-limiting concentrations of malonyl-CoA synthetase from S. coelicolor, which is capable of efficiently activating all of these dicarboxylates into their corresponding CoA thioesters.36In situ formation of extender units was necessary because, at the high substrate concentrations required to attain multiple turnovers, these CoA thioesters undergo rapid, AT-catalyzed hydrolysis.37 Only 6-dEB was observed in the assay mixture containing malonate and methylmalonate, a finding that is consistent with the strong ability of AT domains of DEBS to discriminate against malonyl-CoA.37,38 In contrast, the assay mixture containing methylmalonate and ethylmalonate produced at least three unnatural isomers derived from ethylmalonyl-CoA that were observed in quantities comparable to the natural product, 6-dEB, as measured by the MS ion count (Figure 3 and Figure S9). As described below, one such isomer was identified as having an ethyl substituent at C-8 of the macrolactone (compound 4, Scheme 1); structure elucidation of the other unnatural analogs is under way.
Figure 3. Product profile of DEBS incubated with equimolar concentrations of methylmalonyl and ethylmalonyl extender units.

(A) Extracted Ion Chromatogram (obtained by extraction of the [M+Na]+ species) for 6-dEB (top) and analogs with the molecular formula C22H40O6, corresponding to products derived from incorporation of a single ethylmalonyl extender unit (bottom). (B) Full mass spectrum of 6-dEB (13.5 min; top) and Peak II (14.0 min; bottom). In each case, ions corresponding to at least three of the following species were observed: [M+H-2H2O]+, [M+H-H2O]+, [M+H]+, and [M+Na]+.
Scheme 1.
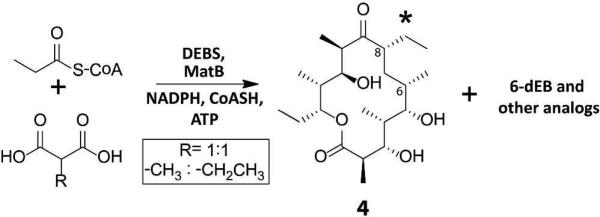
Incorporation of a non-natural ethyl extender unit into 6-dEB by DEBS under equimolar concentrations of methylmalonic acid and ethylmalonic acid.41
In comparison with a 6-dEB standard, ESI-MS/MS of 4 yielded a fragmentation pattern consistent with the results of a previous mass spectrometric analysis of the natural product 6-dEB (Figure S10 and Figure S11).39 This result established that the macrolactone ring was modified at either C-6 or C-8, resulting from incorporation of an ethylmalonyl extender unit by module 3 or by module 4. To distinguish between these possibilities and to assign the most likely site of modification, the truncated DEBS assembly lines shown in Figure 1 were assayed in the presence of equimolar concentrations of methylmalonate and ethylmalonate. The bimodular PKS corresponding to DEBS1 with a C-terminal-fused TE produced mainly triketide 3, accompanied by only a very small amount of a triketide analog, 5, in which one methylmalonyl extender was replaced with an ethylmalonyl building block (Figure S12). However, the analogous trimodular system produced the tetraketide analog, 6, corresponding to incorporation of a single ethylmalonyl unit that gave an ion count that was comparable to that for the natural tetraketide (Figure S13). As a final proof, module 3 of DEBS fused to the TE domain (i.e., M3+TE) was incubated with equimolar ratios of methylmalonate and ethylmalonate in the presence of the known diketide substrate, (2S,3R)-2-methyl-3-hydroxypentanoyl-SNAC. The resulting assay mixture produced the expected triketide lactone, 7, and its ethylmalonyl extender-derived analog, 8 (Figure S14). Taken together, these results conclusively demonstrate that module 3 is able to incorporate the alternative extender substrate, ethylmalonyl-CoA. ESI-MS/MS data and additional biochemical assays provide strong support that DEBS is capable of producing the putative analog 8-ethyl-8-desmethyl-6-deoxyerythronolide B without additional AT domain engineering.40
In summary, we have reconstituted in vitro the complete DEBS system, and have established a kinetic assay for the megasynthase that stoichiometrically relates NADPH consumption to polyketide production. Using this assay, the turnover rate of the assembly line and selected truncated derivatives was analyzed, leading to preliminary but unexpected insights into metabolic flux control on PKS assembly lines. In addition, the overall substrate specificity of DEBS for smaller as well as bulkier extender units was studied, leading to the surprising discovery that a putative analog, 8-ethyl-8-desmethyl-6-deoxyerythronolide B, is generated by the assembly line in the presence of non-limiting concentrations of ethylmalonyl-CoA and is produced in comparable amounts to the natural 6-dEB product. This result indicates a previously unrecognized promiscuity of the wild-type DEBS assembly line for an unnatural extender unit. The utility of this reconstituted system to probe the mechanism and substrate specificity of DEBS has thus been clearly demonstrated, and additional experiments are currently underway to provide further mechanistic insight into the biochemical function of the complete DEBS system.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 GM087934, C.K. and GM022172, D.E.C.). B.L. is a recipient of the National Science Foundation Graduate Research Fellowship. R.V.O. is a recipient of a National Institute of General Medical Sciences Postdoctoral Fellowship (GM103165-01A1) and is a fellow of the Center for Molecular Analysis and Design (CMAD) at Stanford University. We thank Dr. Elizabeth Sattely for the use of her lab's LC/MS instrument and Dr. Gulbenk Anarat Capillino for assistance with LC/MS-MS analysis. We also thank Dr. Katherine Watts for assistance in LC/MS sample preparation and Briana Dunn for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
All materials, methods, and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- 1.Walsh C. Antibiotics : actions, origins, resistance. ASM Press; Washington, D.C.: 2003. [Google Scholar]
- 2.Khosla CJ. Org. Chem. 2009;74:6416–6420. doi: 10.1021/jo9012089. [DOI] [PubMed] [Google Scholar]
- 3.Fischbach MA, Walsh CT. Chem. Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 4.Wenzel SC, Muller R. Curr. Opin. Chem. Biol. 2005;9:447–458. doi: 10.1016/j.cbpa.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu. Rev. Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 6.McDaniel R, Thamchaipenet a., Gustafsson C, Fu H, Betlach M, Ashley G. Proc. Natl. Acad. Sci. U. S. A. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon YJ, Beck BJ, Kim BS, Kang HY, Reynolds KA, Sherman DH. Chem. Biol. 2002;9:203–214. doi: 10.1016/s1074-5521(02)00095-9. [DOI] [PubMed] [Google Scholar]
- 8.Tang L, Chung L, Carney JR, Starks CM, Licari P, Katz LJ. Antibiot. 2005;58:178–184. doi: 10.1038/ja.2005.20. [DOI] [PubMed] [Google Scholar]
- 9.Kim W, Lee D, Hong SS, Na Z, Shin JC, Roh SH, Wu CZ, Choi O, Lee K, Shen YM, Paik SG, Lee JJ, Hong YS. Chembiochem. 2009;10:1243–1251. doi: 10.1002/cbic.200800763. [DOI] [PubMed] [Google Scholar]
- 10.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. Nature. 1990;348:176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 11.Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- 12.Caffrey P, Bevitt DJ, Staunton J, Leadlay PF. FEBS Lett. 1992;304:225–228. doi: 10.1016/0014-5793(92)80624-p. [DOI] [PubMed] [Google Scholar]
- 13.Staunton J, Wilkinson B. Expert. Opin. Investig. Drugs. 1998;7:1369–1387. doi: 10.1517/13543784.7.9.1369. [DOI] [PubMed] [Google Scholar]
- 14.Weissman KJ, Leadlay PF. Nat. Rev. Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 15.Katz L. Methods. Enzymol. 2009;459:113–142. doi: 10.1016/S0076-6879(09)04606-0. [DOI] [PubMed] [Google Scholar]
- 16.Cane DE. J. Biol. Chem. 2010;285:27517–27523. doi: 10.1074/jbc.R110.144618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gokhale RS, Tsuji SY, Cane DE, Khosla C. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 18.Pieper R, Gokhale R, Luo G, Cane D, Khosla C. Biochemistry. 1997;2960:1846–1851. doi: 10.1021/bi962324z. [DOI] [PubMed] [Google Scholar]
- 19.Tsuji SY, Cane DE, Khosla C. Biochemistry. 2001;40:2326–2331. doi: 10.1021/bi002463n. [DOI] [PubMed] [Google Scholar]
- 20.Roberts G. a., Staunton J, Leadlay PF. Eur. J. Biochem. 1993;214:305–311. doi: 10.1111/j.1432-1033.1993.tb17925.x. [DOI] [PubMed] [Google Scholar]
- 21.Pieper R, Pieper G, Luo D, Cane C. Khosla Nature. 1995;378:263–266. doi: 10.1038/378263a0. [DOI] [PubMed] [Google Scholar]
- 22.Wu N, Cane DE, Khosla C. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]
- 23.Kapur S, Chen A, Cane D, Khosla C. Proc. Natl. Acad. Sci. U. S. A. 2010;107:22066. doi: 10.1073/pnas.1014081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapur S, Lowry B, Yuzawa S, Kenthirapalan S, Chen AY, Cane DE, Khosla C. Proc. Natl. Acad. Sci. U. S. A. 2012;109:4110–4115. doi: 10.1073/pnas.1118734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao CM, Katz L, Khosla C. Science. 1994;265:509–512. doi: 10.1126/science.8036492. [DOI] [PubMed] [Google Scholar]
- 26.Kao CM, Luo GL, Katz L, Cane DE, Khosla CJ. Am. Chem. Soc. 1994;116:11612–11613. [Google Scholar]
- 27.Kao CM, Luo G, Katz L, Cane DE, Khosla CJ. Am. Chem. Soc. 1996;118:9184–9185. [Google Scholar]
- 28.Kao CM, Luo GL, Katz L, Cane DE, Khosla CJ. Am. Chem. Soc. 1995;117:9105–9106. [Google Scholar]
- 29.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 30.Minas W, Brunker P, Kallio PT, Bailey JE. Biotechnol. Prog. 1998;14:561–566. doi: 10.1021/bp980055t. [DOI] [PubMed] [Google Scholar]
- 31.Oliynyk M, Brown MJ, Cortés J, Staunton J, Leadlay PF. Chem. Biol. 1996;3:833–839. doi: 10.1016/s1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]
- 32.Stassi DL, Kakavas SJ, Reynolds KA, Gunawardana G, Swanson S, Zeidner D, Jackson M, Liu H, Buko A, Katz L. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7305–7309. doi: 10.1073/pnas.95.13.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau J, Fu H, Cane DE, Khosla C. Biochemistry. 1999;38:1643–1651. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]
- 34.Reeves CD. Crit. Rev. Biotechnol. 2003;23:95–147. doi: 10.1080/713609311. [DOI] [PubMed] [Google Scholar]
- 35.Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. ACS Chem. Biol. 2013;8:443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- 36.Hughes AJ, Keatinge-Clay A. Chem. Biol. 2011;18:165–176. doi: 10.1016/j.chembiol.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 37.Dunn BJ, Cane DE, Khosla C. Biochemistry. 2013;52:1839–1841. doi: 10.1021/bi400185v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. Science. 1994;263:378–380. doi: 10.1126/science.8278811. [DOI] [PubMed] [Google Scholar]
- 39.Ashley GW, Carney JR. J Antibiot. 2004;57:579–589. doi: 10.7164/antibiotics.57.579. [DOI] [PubMed] [Google Scholar]
- 40.The tolerance of DEBS module 3 for incorporation of an ethylmalonyl-CoA extender unit in competition with the natural methylmalonyl-CoA stands in intriguing contrast to the apparent intrinsic substrate specificity of the heptamodular tylactone synthase, TYLS, for which the product tylactone, in common with many other 16-membered macrolide lactones, carries a 6-ethyl group and an 8-methyl group. Module 5 of TYLS (corresponding to module 4 of DEBS) must therefore be strictly specific for ethylmalonyl-CoA while module 4 of TYLS (corresponding to module 3 of DEBS) must be specific for only methylmalonyl-CoA. (cf. O'Hagan D, Robinson JA, Turner DL. J. Chem. Soc. Chem. Commun. 1983:1337–1340. TylG has been heterologously expressed in S. venzuelae: Jung WS, Lee SK, Hong JS, Park SR, Jeong SJ, Han AR, Sohng JK, Kim BG, Choi CY, Sherman DH, Yoon Y. J. Appl. Microbiol. Biotechnol. 2006;72:763–769. doi: 10.1007/s00253-006-0318-5.
- 41.The C-8 stereochemistry has been provisionally assigned based on biosynthetic analogy to 6-dEB.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.