

Abstract
The t(12;21)(p13;q22) chromosomal translocation is the most frequent translocation in childhood B cell precursor-acute lymphoblastic leukemia and results in the expression of an ETV6/RUNX1 fusion protein. The frequency of ETV6/RUNX1 fusions in newborns clearly exceeds the leukemia rate revealing that additional events occur in ETV6/RUNX1-positive cells for leukemic transformation. Hitherto, the mechanisms triggering these second hits remain largely elusive. Thus, we generated a novel ETV6/RUNX1 transgenic mouse model where the expression of the fusion protein is restricted to CD19+ B cells. These animals harbor regular B cell development and lack gross abnormalities. We established stable pro-B cell lines carrying the ETV6/RUNX1 transgene that allowed us to investigate whether ETV6/RUNX1 itself favors the acquisition of second hits. Remarkably, these pro-B cell lines as well as primary bone marrow cells derived from ETV6/RUNX1 transgenic animals display elevated levels of reactive oxygen species (ROS) as tested with ETV6/RUNX1 transgenic dihydroethidium staining. In line, intracellular phospho-histone H2AX flow cytometry and comet assay revealed increased DNA damage indicating that ETV6/RUNX1 expression enhances ROS. On the basis of our data, we propose the following model: the expression of ETV6/RUNX1 creates a preleukemic clone and leads to increased ROS levels. These elevated ROS favor the accumulation of secondary hits by increasing genetic instability and double-strand breaks, thus allowing preleukemic clones to develop into fully transformed leukemic cells.
Introduction
The ETV6/RUNX1 (TEL/AML1) fusion gene is the most common chromosomal alteration in pediatric cancer and occurs in approximately 25% of children with B cell precursor-acute lymphoblastic leukemia (BCP-ALL) [1,2]. The t(12;21)(p13;q22) chromosomal translocation results in the fusion of two critical regulators of hematopoiesis, bringing together the 5′ portion of the ETV6 (TEL) gene on chromosome 12p13 and nearly the entire RUNX1 (AML1) gene on chromosome 21q22 [3,4]. Persuasive evidence that clearly shows the presence of the ETV6/RUNX1 fusion already prenatally and defines it as an early and initiating mutation is available. These insights were obtained from studies of identical twins with concordant ALL [5] and retrospective screening of archived neonatal blood spots of children diagnosed with ALL [6]. The early arise of the translocation indicates that it presents the first event (“hit”) in the process of leukemogenesis creating a preleukemic clone. This is further supported by the fact that the fusion gene may be present for up to 10 years before leukemia diagnosis [7,8]. Secondary genetic events are clearly required for the clinical manifestation of the leukemia that evolves from an ETV6/RUNX1+ preleukemic clone that is characterized by the surface markers CD34+CD38-/lowCD19+ [9,10].
At the time point of diagnosis, ETV6/RUNX1+ ALL is characterized by multiple copy number alterations [11,12]. So far, it is elusive how the presence of ETV6/RUNX1 induces the acquisition of these additional genetic events during pathogenesis of BCP-ALL.
Genomic instability is one of the characteristics of transformed cells [13] and may arise from increased DNA damage and/or impaired DNA repair [14]. DNA damage occurs due to spontaneous decay but also results from exposure to radiation or certain chemicals. Reactive oxygen species (ROS) are chemically reactive molecules that participate in self-propagating reactions, and, if allowed to accumulate, they can cause oxidative damage to intracellular macromolecules, including DNA, proteins, and lipids [15]. The increased mutation rate in a variety of cancers has been attributed to the accumulation of ROS [16]. The capability of ROS to induce DNA double-strand breaks (DSBs) stresses the potential of ROS for triggering chromosomal aberrations [17–20]. Additionally to ROS-mediated mutagenesis, the expression of leukemia-associated fusion genes may interfere with DNA repair as convincingly demonstrated for breakpoint cluster region/Abelson 1 (BCR/ABL1) leukemia [21]. DNA repair is also disturbed in leukemic cells characterized by translocation products such as RUNX1/ETO (AML1/ETO) [22,23] and in cases of ETV6/RUNX1+ leukemia [12]. ETV6/RUNX1+ cells also display a disrupted spindle checkpoint that may result in aneuploidy [24].
In this study, we describe a novel mouse model where ETV6/RUNX1 expression is restricted to CD19+ B lymphoid cells. In line with earlier reports [25–30], we did not observe leukemia formation in these animals. Here, we describe for the first time that the expression of ETV6/RUNX1 is associated with increased levels of ROS and the subsequent accumulation of DSBs, a prerequisite for mutagenesis. These findings suggest that the expression of ETV6/RUNX1 triggers mutagenesis through enhanced ROS production.
Design and Methods
Generation of a CD19+ B Cell-Specific ETV6/RUNX1-Expressing Mouse
To generate a mouse that expresses ETV6/RUNX1 specifically in CD19+ cells, the CD19 bacterial artificial chromosome (BAC) RP23-407K24 was used. For BAC modification, the building vector pQS-CD19 containing an EcoRI and NheI restriction site was used [31]. The human ETV6/RUNX1 cDNA was amplified from an pMSCV-ETV6/RUNX1-internal ribosomal entry site (IRES)-green fluorescent protein (GFP) plasmid (a kind gift of O. Williams, University College London, Institute of Child Health, London, United Kingdom) using the primers 5′ GCGGAATTCATGTCTGAGACTCCTGCTCAG 3′ containing an EcoRI site and 5′ GCGGCTAGCTTAAGCGTAATCTGGAACATCGTATGGGTA 3′ containing an NheI site and a hemagglutinin (HA) tag. This fragment was inserted into the plasmid pQS-CD19 using EcoRI and NheI restriction sites. After NotI digestion, the linearized form of pQS-CD19-ETV6/RUNX1 was used to insert the ETV6/RUNX1-IRES-hCD2t expression cassette into the mouse CD19 BAC RP23-407K24 by standard BAC modification procedures as described earlier [32]. The expression cassette was thereby inserted into exon 1 of the CD19 BAC to express ETV6/RUNX1 and hCD2t under the control of the Cd19 promotor. Upon recombination, the start codon (ATG) of the Cd19 coding sequence in exon 1 is replaced by the ETV6/RUNX1-IRES-hCD2t-polyA expression cassette. Transcription from the Cd19 promoter is terminated by a polyadenylation signal just after the hCD2t coding sequence. Furthermore, the inserted bicistronic cDNA is not in frame with the Cd19 coding sequence. Transgenic mice were generated by injection of linearized BAC DNA at 1 ng/µl into pronuclei of C57BL/6xCBA F1 zygotes. The transgenic animals were backcrossed to C57BL/6 mice for seven generations. ETV6/RUNX1 transgenic (E/Rtg) mice were genotyped using the following oligos: forward—5′ GCCAGACATTGTGGCATATG 3′and reverse—5′ CGAGTCTTCCTCCATCCTGA 3′. Mice were kept at the animal facility at Research Institute of Molecular Pathology/Institute of Molecular Biotechnology (IMP/IMBA; Vienna, Austria). All animal experiments were carried out in accordance with protocols approved by the animal committee of the Medical University of Vienna and by the Federal Ministry of Science and Research.
Southern Blot Analysis
Twenty micrograms of tail genomic DNA of wt and E/Rtg litter-mates were digested with BamHI, separated on a 1% agarose gel, transferred to a nylon membrane (Macherey-Nagel, Düren, Germany), and hybridized with a probe downstream of the insertion site of the ETV6/RUNX1-IRES-hCD2t expression cassette (amplification primers, forward—5′ GGTATCGAGGTAACCAGTCAACACCC 3′ and reverse—5′ GTACCCCACAGGACAGCCAAAGTGTGG 3′).
mRNA Expression Analysis and Semiquantitative Reverse Transcription-Polymerase Chain Reaction Analysis
Total RNA from bone marrow (BM) and spleen was isolated using TRIzol (TRI Reagent; Sigma-Aldrich, Vienna, Austria) and subsequently treated with DNase I (DNase I recombinant; Roche Diagnostics, Vienna, Austria). cDNA was prepared with RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany) using random hexamer primers. Sequences of primer pairs used during the course of the study (5′-3′) are given as follows: ETV6/RUNX1, forward—CTCTGTCTCCCCGCCTGAA and reverse—CGGCTCGTGCTGGCAT; mouse Etv6, forward—GATAGTGGATCCCAACGGACT and reverse—ACGTTTGTTCATCCAGCACTT; and mouse Hprt, forward—GATACAGGCCAGACTTTGTTG and reverse—GGTAGGCTGGCCTATAGGCT.
Blood Smears and Blood Parameters
Blood smears were stained with Hemacolor Rapid staining of blood smear staining set (Merck Millipore, Billerica, MA). Confocal images (original magnification, x 100) were taken using a Carl Zeiss (Oberkochen, Germany) Axio Imager.Z1 microscope. Blood parameters were measured from EDTA blood by scil Vet ABC (Gurnee, IL) according to the manufacturers' instructions.
Flow Cytometric Analysis
Single-cell suspensions were preincubated with anti-CD16/CD32 (24G2) antibody to prevent nonspecific Fc receptor-mediated binding. Antibodies used for lineage determination included CD3e (145-2C11), Ter119 (Ter119), Gr-1 (RB6-8C5), CD11b (M1/70), and CD19 (1D3). An antibody against human CD2 (RPA 2.1; all BD Biosciences, San Jose, CA) as marker for ETV6/RUNX1 expression was used. Staining for free oxygen radicals was performed using dihydroethidium (DHE; Molecular Probes, Eugene, OR). For flow cytometric detection of phospho-histone H2AX (γH2AX; S139; Cell Signaling Technology; clone 20E3), B cells were fixed in paraformaldehyde (Sigma-Aldrich; 2%), permeabilized by exposure to ice-cold methanol (-20°C, 30 minutes), washed in phosphate-buffered saline containing 0.5% BSA (Sigma-Aldrich) and 0.2% Tween 20 (Sigma-Aldrich), and stained with an Alexa Fluor 647-conjugated γH2AX antibody (Cell Signaling Technology, Danvers, MA) for 60 minutes (room temperature). Subsequently, cells were washed and analyzed. Samples were acquired on a BD FACSCanto II FACS device (BD Biosciences), and data were analyzed with FlowJo software (TreeStar, Ashland, OR).
Protein Analysis
Protein expression has been determined from the fetal liver-derived CD19+ B cell lines of wt and E/Rtg mice. Cells were lysed in a buffer containing protease and phosphatase inhibitors [25 mM Hepes (pH 7.5), 150 mM NaCl, 10 mM EDTA, 0.1% Tween 20, 0.5% NP-40, 10 mM β-glycerophosphate, 40 µl/ml protease inhibitor cocktail (Roche, Basel, Switzerland), 0.1 mM PMSF, 0.1 mM NaVO4, and 1 mM DTT]. The protein concentration was determined by Bio-Rad protein assay kit as recommended by the manufacturer (Bio-Rad Laboratories, Hercules, CA). ETV6/RUNX1 was immunoprecipitated of 2 mg of cell lysate using an anti-RUNX1 antibody (N-20; Santa Cruz Biotechnology, Dallas, TX) and probed with a polyclonal antiserum directed against ETV6 [directed against helix-loop-helix (HLH) domain, a kind gift of Jan Cools, Center for Human Genetics, Faculty of Medicine, KU Leuven (University of Leuven; Leuven, Belgium)]. Proteins were electrophoretically resolved on an 8% polyacrylamide gel containing sodium dodecyl sulfate and transferred onto a polyvinylidene difluoride membrane (Whatman, Piscataway, NJ). Sites of antibody binding were detected using protein A-conjugated HRP (GE Healthcare, Little Chalfont, United Kingdom) with enhanced chemiluminiscence detection (GE Healthcare).
Infection of Fetal Livers and Cell Culture
Fetal liver cells of embryos (E14.5) were prepared, and single-cell suspensions were infected as previously described [33]. After outgrowth of cell lines, the cells were tested by flow cytometry for CD19 and hCD2t expression.
Transformed fetal liver and BM cells were maintained in RPMI (Sigma-Aldrich) containing 10% heat-inactivated fetal calf serum (PAA Laboratories, Pasching, Austria), 100 U/ml penicillin/streptomycin (Life Technologies, Carlsbad, CA), and 5 µM β-mercaptoethanol (Sigma-Aldrich). A010 cells that produce an ecotropic replication-deficient form of the Abelson virus were maintained in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal calf serum (PAA Laboratories), 100 U/ml L-glutamine (Life Technologies), and 100 U/ml penicillin/streptomycin (Life Technologies). Viral supernatant was prepared as previously described [33].
Comet Assay
DNA damage was detected using the CometAssay or single gel electrophoresis assay following the manufacturer's protocol (Trevigen, Gaithersburg, MD). Briefly, 106 cells/ml medium of freshly isolated BM cells [enriched for CD19+ B cells by magnetic activated cell sorting (MACS)] were mixed 1:10 (v.v-1) with low melting point agar at 38°C and immediately pipetted onto agar-covered slides. Slides were incubated at 4°C in the dark for 30 minutes, and the coverglass was removed, immersed in chilled lysis solution, and incubated again at 4°C for 60 minutes. Slides were placed in a horizontal electrophoresis chamber and electrophoresed with buffer (0.3 N NaOH and 1 mM EDTA) at 0.3 A for 20 minutes. Samples were dried and stained with CometAssay Silver Staining Kit (Trevigen). Comet tails were imaged using a model BA310 microscope (Motic, Hongkong) and quantified by TriTek CometScore (TriTek Corp, Sumerduck, VA). A minimum of 50 cells were scored per sample.
Statistics
All statistical analyses were done using GraphPad Prism 4 (San Diego, CA). Differences were assessed for statistical significance by unpaired Student's t test and one-way analysis of variance using the Tukey Multiple Comparison Test if not otherwise mentioned. Error bars represent means ± SD. P values are considered as follows: < .05: *, < .01: **, and < .001: ***.
Results
Generation of a CD19-Specific ETV6/RUNX1 Mouse
ETV6/RUNX1+ BCP-ALL is characterized by the expansion of B cells expressing the D-related antigens Cd19 and CD10 (equivalent to Hardy Fraction C) [34]. To express the ETV6/RUNX1 fusion protein in CD19+ B cells, a BAC containing the Cd19 locus was modified through homologous recombination in Escherichia coli [31,32]. A cassette encoding the human fusion gene ETV6/RUNX1 linked through an IRES to a truncated human CD2 (hCD2t) antigen reporter and followed by a polyadenylation signal (polyA) was inserted into the first exon of the Cd19 gene. As a result of the recombination strategy, expression of functional Cd19 from the BAC is prevented (Figure 1A). Several transgenic lines were obtained from oocyte injection of the CD19-ETV6/RUNX1 BAC. Southern blot analysis revealed that line 1, which was further analyzed in detail and used in the present study, harbors four copies of the CD19-ETV6/RUNX1 BAC (Figure 1B). To determine specific expression of ETV6/RUNX1 in B cells, we analyzed hCD2t reporter gene expression in various hematopoietic cell lineages isolated from BM by flow cytometry. hCD2t reporter expression was restricted to CD19+ cells and could not be detected in any other cell type analyzed (Figure 1C). CD19+ cell-specific expression of ETV6/RUNX1 mRNA was further confirmed by performing real-time PCR analysis (Figure W1). The uniform expression of the hCD2t reporter gene within the CD19+ cell population (Figure 1D) indicated that essentially all ETV6/RUNX1 transgene CD19+ cells express the ETV6/RUNX1 fusion protein. Consistently, using semiquantitative reverse transcription-polymerase chain reaction (PCR), we detected the ETV6/RUNX1 transgene expression both in hCD2thigh and hCD2tlow populations (Figure 1E).
Figure 1.
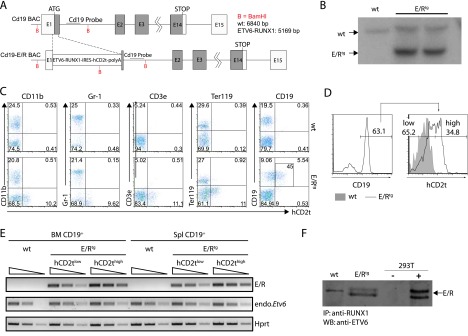
Design of BAC and expression analysis of the ETV6/RUNX1 transgene. (A) The ETV6/RUNX1-IRES-hCD2t expression cassette was inserted in the first exon of the Cd19 gene. (B) Southern blot analysis of the Cd19 locus in an ETV6/RUNX1 founder mouse. Genomic DNA was digested with BamHI, blotted on a nitrocellulose membrane, and hybridized with a radiolabeled DNA probe against the proximal region of the Cd19 intron 1. The expected insertion of the ETV6/RUNX1-IRES-hCD2t-polyA cassette gives rise to a labeled DNA fragment of 5169 bp in contrast to the wt Cd19 allele (6840 bp). (C) The reporter hCD2t is specifically expressed in CD19+ cells. BM cells of wt mice (upper panel) and E/R-expressing mice (lower panel) were isolated, stained for surface markers CD11b, Gr1, CD3e, Ter119 as well as CD19, and analyzed by flow cytometry. The reporter gene hCD2t is only expressed in CD19+ cells but not in other hematopoietic lineages. Black inlet shows percentage of hCD2t+ cells from the CD19-expressing population. (D) Distribution of the hCD2t expression levels on the entire CD19+ cell fraction. One representative histogram of hCD2thigh/low-expressing CD19+ cells is shown. (E) mRNA expression analysis for ETV6/RUNX1 and Etv6 in semiquantitative reverse transcription-PCR in FACS-sorted hCD2tlow/high cells of the BM and the spleen of wt and E/Rtg mice. The wedge shows five-fold dilution series. (F) ETV6/RUNX1 protein expression was analyzed by immunoprecipitation (anti-RUNX1) and Western blot analysis (anti-ETV6) from E/Rtg and wt fetal liver-derived B cell lines. As control, 293T cells transiently transfected with an E/R-expressing plasmid were used. An unspecific antibody cross-hybridization signal is visible in some of the samples (wt).
As the fusion gene is expressed under the control of the ETV6 promoter in patients with leukemia, we also tested whether the expression levels of the ETV6/RUNX1 transgene and the endogenous Etv6 gene are within the same range. As depicted in Figure 1E, ETV6/RUNX1 and Etv6 genes were expressed at comparable levels in CD19+hCD2tlow cells indicating that the transgene is expressed at a nearly physiological level. Finally, we confirmed ETV6/RUNX1 expression at protein level in CD19+ B cells (Figure 1F).
Unaltered Hematopoiesis in E/Rtg Mice
ETV6/RUNX1tg (E/Rtg) mice are born at normal mendelian ratio and do not display any gross abnormalities (Figure W2). We next tested whether the expression of ETV6/RUNX1 in the CD19+ B lymphoid compartment impacts on hematopoiesis in general and on B cell development in particular. Various hematopoietic lineages of wt and E/Rtg mice were assessed by flow cytometry. No consistent differences were detected between the transgenic and control mice (Figure W3, A and B). Similarly, we failed to detect any alterations in various blood parameters that may indicate an altered blood physiology as assessed by blood smears and a hematocytometer. Briefly, no differences in hematocrit levels, white blood cell counts, platelet numbers, or hemoglobin levels were detected (Figure 2, A and B). ETV6/RUNX1+ ALL cells are arrested at the pro-B cell stage [26,27]. To analyze B cell development in our CD19-specific E/Rtg mice, we stained BM and splenocytes for B cell surface markers corresponding to distinct developmental stages (Figure 2C). Gating of the early B cell fractions according to Hardy in the BM revealed no significant differences in pre-pro-B cells, early pro-B cells, and late pro-B cells, respectively, between wt and E/Rtg mice (Figure 2D, left panel). Interestingly, we observed slightly increased pre-B cell and immature B cell numbers in the spleen of E/Rtg animals although mature B cells were comparable; however, this did not meet the criteria of statistical significance (Figure 2D, right panel).
Figure 2.
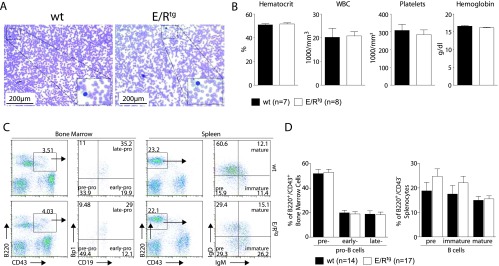
Effects of ETV6/RUNX1 expression on blood physiology and B cell development. (A) Representative blood smears of 8-week-old wt and E/Rtg mice. (B) Blood parameters of wt (n = 7) and E/Rtg (n = 8) mice as measured by scil Vet ABC. Statistical analysis using unpaired t test revealed no differences. (C) Flow cytometric analysis of B cell development. Shown are representative dot plots. Different developmental B cell stages are indicated in the quadrants of the dot plots as pre, early, and late and pre, immature, and mature. (D) Statistical analysis of B cell development of wt and E/Rtg mice using Student's unpaired t test. A summary of three experiments is shown.
ETV6/RUNX1 Elevates Cellular ROS Levels
Oncogenes such as BCR/ABL1 induce the production of ROS [17]. To investigate whether ETV6/RUNX1 affects the equilibrium of ROS production and scavenging, we generated stable pro-B cell lines from transgenic and control mice. This allowed us to address the impact of ETV6/RUNX1 on DNA damage in vitro in a stable and reproducible setting. Fetal liver cells from wt and E/Rtg littermate embryos were infected with the Abelson virus that immortalizes pro-B cells in vitro [35,36]. ETV6/RUNX1 transgene expression was confirmed by surface-marker staining of the reporter gene hCD2t (Figure 3A), followed by flow cytometric analysis. No changes in the expression of the pro-B cell surface markers CD19+B220+CD43+ and the proliferation rate of the cell lines were detected upon expression of ETV6/RUNX1 when analyzed by flow cytometry and when assessed by growth curves, respectively (Figures 3B and W4).
Figure 3.

E/R expression induces ROS production in vitro and in vivo. (A and B) Fetal liver cells of wt and E/Rtg mice were infected with a retrovirus encoding for p160v-ABL. Stably transformed pro-B cell lines were analyzed for the expression of the surface markers (A) CD19 and hCD2t as well as (B) CD19 and B220. (C) wt and E/Rtg cell lines were stained with DHE and subsequently analyzed by flow cytometry to measure differences in ROS level. One representative histogram is depicted. (D) A summary of all experiments using wt and E/Rtg cell lines stained with DHE is shown. Statistical analysis was carried out by one-tailed paired t test (P = .04). (E) wt and E/Rtg cell lines were treated for 24 hours with 5 mM ROS scavenger NAC and analyzed for ROS by flow cytometry. Depicted are histograms for wt (left panel) and E/Rtg (right panel) untreated cells and cells treated with NAC. (F) CD19+ BM cells of E/Rtg (n = 16) and control littermate mice (n =10) were analyzed for ROS levels. Bar graphs represent DHE-MFI ± SD; statistical differences were revealed using unpaired t test. (G) Indicated human and murine cell lines were transfected with a retrovirus encoding for E/R-IRES-GFP or GFP. The fold difference in DHE-MFI of GFP-positive versus GFP-negative cells is shown. (H) Bar graphs depict average fold change in DHE-MFI on E/R or GFP expression ± SD of all five cell lines analyzed. *P < .05 (unpaired t test).
Remarkably, the analysis of ROS levels in the E/Rtg pro-B cells consistently revealed about a two-fold increased mean fluorescence intensity (MFI) for the ROS-sensitive fluorescence probe DHE compared to control cells (wt DHE-MFI = 5700 vs E/Rtg DHE-MFI = 12,100; Figure 3, C and D). To verify that indeed an elevated ROS level accounts for the detected differences in DHE values, we treated our cells with the ROS scavenger N-acetyl-cystein (NAC) for 24 hours before measurement. A clear reduction in ROS levels was detected in E/Rtg as well as control cell lines (40% and 35% reduction in DHE-MFI, respectively; Figure 3E). To exclude the possibility of an Abelson oncogene-related impact on our experimental setup, we analyzed ex vivo-derived BM cells from our E/Rtg mice. CD19+ BM cells of E/Rtg mice (n = 16) displayed significantly elevated DHE fluorescence compared to littermate controls (n = 10; DHE-MFI = 1690 ± 138 and 1298 ± 31, respectively; P = .0365; Figure 3F), indicating that the observed differences are associated with ETV6/RUNX1 expression. Further evidence came from another experiment where we used five cell lines derived from different tissues of both murine and human origin (NIH3T3, HEPA1-6, RAW, MBA, and 32D). These cells were infected with a retrovirus encoding for ETV6/RUNX1-IRES-GFP or GFP alone. All cell lines tested except NIH3T3 reacted with an increase of ROS on ETV6/RUNX1 expression (P = .0285; Figure 3, G and H).
Accumulation of DNA DSBs on ETV6/RUNX1 Expression
It is a common feature of oncogenes to enhance DNA mutation rate [37]. The increased mutation rate may result from both an enhanced DNA damage and/or deregulated DNA repair mechanisms. Upon DNA DSBs, serine residue 139 of histone H2AX (γH2AX) becomes phosphorylated. This may be used as a sensitive marker to detect the presence of DSBs [38,39]. When we stained wt and E/Rtg cell lines with a γH2AX-specific antibody and analyzed them through intracellular flow cytometry, we found higher levels of DSBs in ETV6/RUNX1+ cells compared to control cells (γH2AX MFI = 3069 and 2012, respectively; Figure 4A). To assess DSBs in ex vivo-derived BM cells of wt and E/Rtg mice, we employed a comet assay. In such an assay, cells are embedded in agarose on microscope slides, lysed, and subjected to electrophoresis at high pH resulting in comet-like structures of the DNA. BM-derived and BM-sorted B cells were used directly ex vivo in this comet assay, which resulted in a higher percentage of comet tail DNA, an indicator of DNA damage, for E/Rtg-derived B cells compared to wt counterparts (43.3% vs 30.6%; Figure 4, B and C). These results suggested a link between E/Rtg expression and DSBs, a prerequisite for DNA mutations.
Figure 4.
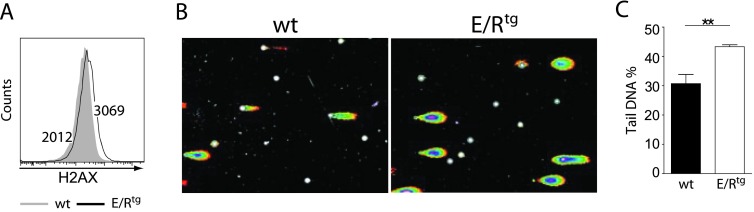
E/R expression induces DNA damage in wt and E/Rtg cells. (A) wt and E/Rtg cell lines were stained for γH2AX and analyzed by flow cytometry. One representative histogram is shown. (B and C) Comet assay of MACS-enriched CD19+ B cells derived from BM of wt and E/Rtg mice. Two mice of each genotype were pooled before MACS. (B) Representative pictures of comets of each genotype. (C) Statistical analysis (unpaired t test) of comet assay of wt and E/Rtg mice (n = 6 each) is depicted. Fifty pictures per sample were taken for statistical analysis of percentage of tail DNA. Average ± SD of each sample is shown.
Discussion
The ETV6/RUNX1 fusion gene has been described to arise already in utero [5,9,40], producing a preleukemic clone that may progress to full-blown leukemia when acquiring additional genetic alterations [5,9,11]. Here, we report the generation of a mouse model for ETV6/RUNX1 where transgene expression is driven by a BAC containing the CD19-regulatory expression elements, restricting its expression to the B lymphoid lineage. We describe that ETV6/RUNX1-expressing B lymphoid cells display increased levels of ROS accompanied by increased DNA DSBs. Thus, we propose a model where ETV6/RUNX1 induces DNA instability through ROS deregulation, thereby facilitating mutagenesis and the appearance of additional genetic alterations.
Several reports described that enforced expression of the ETV6/RUNX1 fusion gene from retroviral promoters impairs B cell development and induces a block at the pro-B cell stage [26], leading to the expansion of the CD19+ B cell fraction [41]. Interestingly, we failed to detect any changes in B cell development at the pro-B cell stage. However, we observed a trend in elevated numbers of immature B cells as well as pre-B cells in the spleen of E/Rtg mice. Several reasons may account for these differences. Importantly, we succeeded in obtaining ETV6/RUNX1 expression at similar levels when compared to the endogenous Etv6 gene. Thus, it is attractive to speculate that higher ETV6/RUNX1 levels are required to inflict a B cell differentiation block. In addition, one may reason that the onset of transgenic expression accounts for the different impact on B cell development; it may be of relevance at which stage of B cell development the transgene is expressed. In our case, transgene expression is under the control of the Cd19 promoter and thereby restricted to already committed B cells. In contrast, other studies employed promoters that allowed for ETV6/RUNX1 expression already at earlier developmental stages such as in hematopoietic stem and/or progenitor cells. This may support the concept that the expression of ETV6/RUNX1 before B lymphoid development is required.
ETV6/RUNX1 is commonly assumed to be the first hit during leukemogenesis, and it is assumed that secondary alterations have to occur for the clinical manifestation of the disease. In line, animal models for ETV6/RUNX1 in mice and zebra fish revealed that the expression of the fusion gene does not suffice to allow for leukemogenesis [25–30]. Van der Weyden et al. used random mutagenesis using a Sleeping Beauty transposon to induce BCP-ALL in ETV6/RUNX1 knock-in mice [42]. Several cooperating mutations were identified in these mice, some of which resembled alterations in BCP-ALL cases (including regulators of B cell development). Although a few mutations prevail, genome-wide profiling of genetic alterations in ETV6/RUNX1+ BCP-ALL revealed that the mutational spectrum is highly heterogeneous, with numerous recurrent lesions generating mutations or copy number alterations in only a few patients [11,43].
Although the animal model for ETV6/RUNX1+ leukemia that Van der Weyden et al. used in their study relied on the usage of knock-in mice [42], a few reports also suggest that the human ETV6/RUNX1 gene fusion—as used for the generation of our transgenic animals—could lead to leukemia development if combined with a secondary hit in mice. It has been reported either for BM transplantation experiments where the human gene fusion was combined with loss of p16/p19 [25] or also in a recent study on inducible E/R transgenic mice in combination with loss of p16 [44] that the human fusion gene was able to induce leukemogenesis in mice.
Intracellular redox homeostasis is maintained by balancing ROS production with ROS scavenging through cellular antioxidant defense systems [45]. Several pathophysiological conditions including leukemia [46] have been associated with unbalanced ROS metabolism. Moreover, ROS is known to oxidize DNA that further provokes mutation of genes that may promote carcinogenesis [47]. In this report, we show for the first time that ETV6/RUNX1 expression in B cells is associated with elevated endogenous ROS levels and an increase in DSBs (Figures 3 and 4). The finding that a chromosomal translocation product induces DNA damage through ROS production is not entirely surprising as it has already been shown to be true for other chromosomal alterations and oncogenes; ample evidence supports this concept {summarized in [48]}. The expression of RUNX1/ETO or RUNX1 itself in human fibroblasts suffices to elevated intracellular ROS levels when compared to empty vector control [49]. These data have been supported in a Drosophila model where the expression of RUNX1/ETO resulted in high levels of ROS [50]. In our hands, the expression of the ETV6/RUNX1 fusion product in B cells, but also various other cell types, increased ROS levels in the respective cells. This observation also suggests that increased ROS production induced by RUNX1 containing translocation products could be a common mechanism driving leukemogenesis.
ROS is viewed as an important signaling mediator [51] and may be implicated in gene-regulatory pathways such as the one involving hypoxia-inducible factor 1 (HIF-1) [52,53]. Interestingly, ETV6/RUNX1 was shown to downregulate expression of DNA damage-inducible transcript 4 (DDIT4) (also known as REDD1), an important HIF-1 effector [54], whereas the loss of DDIT4 was reported to induce HIF-1 stabilization and tumorigenesis through a ROS-dependent mechanism [55], suggesting a possible link between ETV6/RUNX1 and increased ROS accumulation in patients.
Irradiation and topoisomerase II inhibitors are therapeutic modalities that induce DSBs [56]. This predicts that cells harboring ETV6/RUNX1 ought to be more sensitive to topoisomerase inhibitors. In fact, this prediction has been verified with patient-derived cells; BCP-ALL cells are highly sensitive to doxorubicine and etoposide, if they are ETV6/RUNX1+. However, these cells do not differ from ETV6/RUNX1- BCP-ALL cells with respect to their susceptibility to other therapeutically relevant agents (vincristine, glucocorticoids, cytarabine, alkylating agents, and thioguanin) [57]. Taken together, our data propose a novel mechanism of how additional mutations could be acquired in an ETV6/RUNX1+ preleukemic clone that then could cause leukemia development.
Supplementary Material
Acknowledgments
We thank Shinya Sakaguchi, Michael Freissmuth, and Richard Moriggl for continuous discussions and for critically reading the manuscript. We also thank Renate Panzer-Grümayer for providing scientific advice. We are also grateful to Hans-Christian Theussl for technical assistance in the generation of the CD19-specific E/Rtg mice. We also thank Reimar David, Peter Martinek as well as Jaqueline Horvath for technical assistance.
Abbreviations
- BCP-ALL
B cell precursor-acute lymphoblastic leukemia
- ROS
reactive oxygen species
- DSB
double-strand break
- DHE
dihydroethidium
- E/Rtg
ETV6/RUNX1 transgene
Footnotes
Financial support for this project was provided by the St Anna Kinderkrebsforschung, Children's Cancer Research Institute, financing the position of H.-P.K. and E.B. Further support came from the Austrian Science Fund Fonds zur Förderung der wissenschaftlichen Forschung (FWF) SFB-F28 to V.S. The authors declare no conflict of interests.
This article refers to supplementary materials, which are designated by Figures W1 to W4 and are available online at www.neoplasia.com.
References
- 1.Fears S, Gavin M, Zhang DE, Hetherington C, Ben-David Y, Rowley JD, Nucifora G. Functional characterization of ETV6 and ETV6/CBFA2 in the regulation of the MCSFR proximal promoter. Proc Natl Acad Sci USA. 1997;94:1949–1954. doi: 10.1073/pnas.94.5.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fears S, Vignon C, Bohlander SK, Smith S, Rowley JD, Nucifora G. Correlation between the ETV6/CBFA2 (TEL/AML1) fusion gene and karyotypic abnormalities in children with B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 1996;17:127–135. doi: 10.1002/(SICI)1098-2264(199610)17:2<127::AID-GCC8>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 3.Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley JD, Gilliland DG. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romana SP, Mauchauffé M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard OA. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 5.Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2003;102:2321–2333. doi: 10.1182/blood-2002-12-3817. [DOI] [PubMed] [Google Scholar]
- 6.Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3:639–649. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 7.Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood. 1999;94:1057–1062. [PubMed] [Google Scholar]
- 8.Maia AT, Koechling J, Corbett R, Metzler M, Wiemels JL, Greaves M. Protracted postnatal natural histories in childhood leukemia. Genes Chromosomes Cancer. 2004;39:335–340. doi: 10.1002/gcc.20003. [DOI] [PubMed] [Google Scholar]
- 9.Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319:336–339. doi: 10.1126/science.1150648. [DOI] [PubMed] [Google Scholar]
- 10.Castor A, Nilsson L, Astrand-Grundström I, Buitenhuis M, Ramirez C, Anderson K, Strömbeck B, Garwicz S, Békássy AN, Schmiegelow K, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005;11:630–637. doi: 10.1038/nm1253. [DOI] [PubMed] [Google Scholar]
- 11.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 12.Kuster L, Grausenburger R, Fuka G, Kaindl U, Krapf G, Inthal A, Mann G, Kauer M, Rainer J, Kofler R, et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood. 2011;117:2658–2667. doi: 10.1182/blood-2010-03-275347. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531:231–251. doi: 10.1016/j.mrfmmm.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive oxygen species (ROS)-induced genetic and epigenetic alterations in human carcinogenesis. Mutat Res. 2011;711:167–173. doi: 10.1016/j.mrfmmm.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, Blasiak J, Skorski T. BCR/ABL kinase induces selfmutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–327. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, Nieborowska-Skorska M, Blasiak J, Skorski T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 19.Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, Small D, Rassool F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111:3173–3182. doi: 10.1182/blood-2007-05-092510. [DOI] [PubMed] [Google Scholar]
- 20.Walz C, Crowley BJ, Hudon HE, Gramlich JL, Neuberg DS, Podar K, Griffin JD, Sattler M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J Biol Chem. 2006;281:18177–18183. doi: 10.1074/jbc.M600064200. [DOI] [PubMed] [Google Scholar]
- 21.Burke BA, Carroll M. BCR-ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia. 2010;24:1105–1112. doi: 10.1038/leu.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E, Casciari C, et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Invest. 2003;112:1751–1761. doi: 10.1172/JCI17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krejci O, Wunderlich M, Geiger H, Chou FS, Schleimer D, Jansen M, Andreassen PR, Mulloy JC. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood. 2008;111:2190–2199. doi: 10.1182/blood-2007-06-093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krapf G, Kaindl U, Kilbey A, Fuka G, Inthal A, Joas R, Mann G, Neil JC, Haas OA, Panzer-Grümayer ER. ETV6/RUNX1 abrogates mitotic checkpoint function and targets its key player MAD2L1. Oncogene. 2010;29:3307–3312. doi: 10.1038/onc.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernardin F, Yang Y, Cleaves R, Zahurak M, Cheng L, Civin CI, Friedman AD. TEL-AML1, expressed from t(12;21) in human acute lymphocytic leukemia, induces acute leukemia in mice. Cancer Res. 2002;62:3904–3908. [PubMed] [Google Scholar]
- 26.Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc Natl Acad Sci USA. 2004;101:8443–8448. doi: 10.1073/pnas.0402063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, Bergholz U, Greaves M, Löhler J, Stocking C. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene. 2005;24:7579–7591. doi: 10.1038/sj.onc.1208931. [DOI] [PubMed] [Google Scholar]
- 28.Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD. TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2006;103:15166–15171. doi: 10.1073/pnas.0603349103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, Hock H. TEL-AML1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell Stem Cell. 2009;5:43–53. doi: 10.1016/j.stem.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 30.Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet. 2001;130:93–104. doi: 10.1016/s0165-4608(01)00518-0. [DOI] [PubMed] [Google Scholar]
- 31.Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. 2006;24:269–281. doi: 10.1016/j.immuni.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27:1555–1557. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, Rothammer K, Roussel MF, Ihle JN. Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of Stat5. Blood. 2000;96:2277–2283. [PubMed] [Google Scholar]
- 34.Romana SP, Poirel H, Leconiat M, Flexor MA, Mauchauffé M, Jonveaux P, Macintyre EA, Berger R, Bernard OA. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood. 1995;86:4263–4269. [PubMed] [Google Scholar]
- 35.Witte ON. Functions of the abl oncogene. Cancer Surv. 1986;5:183–197. [PubMed] [Google Scholar]
- 36.Rosenberg N, Witte ON. The viral and cellular forms of the Abelson (abl) oncogene. Adv Virus Res. 1988;35:39–81. doi: 10.1016/s0065-3527(08)60708-3. [DOI] [PubMed] [Google Scholar]
- 37.Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21:8591–8604. doi: 10.1038/sj.onc.1206087. [DOI] [PubMed] [Google Scholar]
- 38.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 39.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 40.Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci USA. 2002;99:8242–8247. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with pre-leukemic activity. Blood. 2004;103:3890–3896. doi: 10.1182/blood-2003-10-3695. [DOI] [PubMed] [Google Scholar]
- 42.van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, Corcoran AE, Greaves MF, Mullighan CG, Huntly BJ, et al. Modeling the evolution of ETV6-RUNX1-induced B-cell precursor acute lymphoblastic leukemia in mice. Blood. 2011;118:1041–1051. doi: 10.1182/blood-2011-02-338848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lilljebjörn H, Soneson C, Andersson A, Heldrup J, Behrendtz M, Kawamata N, Ogawa S, Koeffler HP, Mitelman F, Johansson B, et al. The correlation pattern of acquired copy number changes in 164 ETV6/RUNX1-positive childhood acute lymphoblastic leukemias. Hum Mol Genet. 2010;19:3150–3158. doi: 10.1093/hmg/ddq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Jones L, Gaillard C, Binnewies M, Ochoa R, Garcia E, Lam V, Wei G, Yang W, Lobe C, et al. Initially disadvantaged, TEL-AML1 cells expand and initiate leukemia in response to irradiation and cooperating mutations. Leukemia. 2013;27:1570–1573. doi: 10.1038/leu.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 47.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 48.Skorski T. Chronic myeloid leukemia cells refractory/resistant to tyrosine kinase inhibitors are genetically unstable and may cause relapse and malignant progression to the terminal disease state. Leuk Lymphoma. 2011;52(suppl 1):23–29. doi: 10.3109/10428194.2010.546912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolyniec K, Wotton S, Kilbey A, Jenkins A, Terry A, Peters G, Stocking C, Cameron E, Neil JC. RUNX1 and its fusion oncoprotein derivative, RUNX1-ETO, induce senescence-like growth arrest independently of replicative stress. Oncogene. 2009;28:2502–2512. doi: 10.1038/onc.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinenko SA, Hung T, Moroz T, Tran QM, Sidhu S, Cheney MD, Speck NA, Banerjee U. Genetic manipulation of AML1-ETO-induced expansion of hematopoietic precursors in a Drosophila model. Blood. 2010;116:4612–4620. doi: 10.1182/blood-2010-03-276998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 52.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 53.Goyal P, Weissmann N, Grimminger F, Hegel C, Bader L, Rose F, Fink L, Ghofrani HA, Schermuly RT, Schmidt HH, et al. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free Radic Biol Med. 2004;36:1279–1288. doi: 10.1016/j.freeradbiomed.2004.02.071. [DOI] [PubMed] [Google Scholar]
- 54.Fuka G, Kauer M, Kofler R, Haas OA, Panzer-Grümayer R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PloS One. 2011;6:e26348. doi: 10.1371/journal.pone.0026348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, Ellisen LW. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci USA. 2010;107:4675–4680. doi: 10.1073/pnas.0907705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 57.Frost BM, Forestier E, Gustafsson G, Nygren P, Hellebostad M, Jonsson OG, Kanerva J, Schmiegelow K, Larsson R, Lönnerholm G. Translocation t(12;21) is related to in vitro cellular drug sensitivity to doxorubicin and etoposide in childhood acute lymphoblastic leukemia. Blood. 2004;104:2452–2457. doi: 10.1182/blood-2003-12-4426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.