

Abstract
Thiopurines are among the most successful chemotherapeutic agents used for treating various human diseases, including acute lymphoblastic leukemia and chronic inflammation. Although metabolic conversion and the subsequent incorporation of 6-thioguanine (SG) nucleotides into nucleic acids are considered important for allowing the thiopurine drugs to induce their cytotoxic effects, alternative mechanisms may also exist. We hypothesized that an unbiased analysis of SG-induced perturbation of the entire proteome might uncover novel mechanism(s) of action of the drug. We performed a quantitative assessment of global protein expression in control and SG-treated Jurkat T cells by employing stable isotope labeling by amino acids in cell culture and liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. LC-MS/MS quantification results uncovered substantially decreased expression of a large number of proteins in the mitochondrial respiratory chain complex, and Ingenuity Pathway Analysis of the significantly altered proteins showed that SG treatment induced mitochondrial dysfunction. This was accompanied by diminished uptake of MitoTracker Deep Red and the elevated formation of oxidatively induced DNA lesions, including 8,5′-cyclo-2′-deoxyadenosine and 8,5′-cyclo-2′-deoxyguanosine. Together, our results suggested that SG may exert its cytotoxic effect by inducing mitochondrial dysfunction and reactive oxygen species formation in acute lymphoblastic leukemia cells.
Thiopurines, including azathioprine, 6-mercaptopurine, and 6-thioguanine (SG),1 are widely used as chemotherapeutic and immunosuppressive agents (1–3), and 6-mercaptopurine and SG are commonly prescribed for the treatment of acute lymphoblastic leukemia (ALL) (3). A previous study showed that thiopurines may exert their cytotoxic effects via their metabolic activation and incorporation of SG into DNA, the spontaneous methylation of DNA SG by S-adenosyl-l-methionine to yield S6-methylthioguanine (S6mG), the misincorporation of thymine (T) opposite S6mG during DNA replication, and the triggering of post-replicative mismatch repair (MMR) by the ensuing S6mG:T mispair (4).
Recent studies suggest that the activation of MMR might not be the sole pathway contributing to the cytotoxic effects of the drug. In this vein, very low levels (<0.02%) of DNA SG were found to be methylated to S6mG in SG-treated leukemia cells (5), and SG itself can direct significant frequencies (about 10%) of the misincorporation of thymine during DNA replication in both E. coli and human cells (6, 7). These findings, in conjunction with the observation that the SG:T mispair can be recognized more readily by human MMR proteins than the S6mG:T mispair (8, 9), suggest that DNA SG itself may trigger the MMR pathway without being converted to S6mG. Additionally, leukemic cells deficient in MMR are also sensitive to thiopurine drugs (10), indicating that thiopurines may also induce their cytotoxic effects through other pathways.
Global transcriptome and proteome analyses are very useful for revealing novel mechanisms of action of anticancer drugs (11). Global gene expression analysis with microarray was used for assessing transcriptional responses of cancer cells to anticancer agents including thiopurines (12). However, results from mRNA-based gene expression analyses often exhibit poor correlation with protein expression data from proteomic measurements (13). This may be attributed to the fact that regulatory processes following mRNA production (i.e. posttranscriptional, translational, and protein degradation regulation) play substantial roles in controlling steady-state protein abundances (13). Mass spectrometry (MS)-based proteomics techniques, along with two-dimensional gel electrophoresis or stable-isotope labeling, have been widely used for the large-scale quantitative analysis of proteins in complex samples (14, 15). Among the many isotope-labeling methods, stable isotope labeling by amino acids in cell culture (SILAC), a metabolic-labeling technique, has the advantages of being simple, efficient, and capable of quantifying relatively small changes in protein expression (16).
Here we employed LC-MS/MS, in conjunction with SILAC, to assess, at the global proteome scale, the perturbation of protein expression in Jurkat T human ALL cells upon treatment with a clinically relevant concentration of SG. The results from the quantitative proteomic analysis enabled us to conclude that the exposure to SG resulted in mitochondrial dysfunction in ALL cells that was accompanied by a drug-induced loss of active mitochondria and the elevated generation of oxidatively induced DNA lesions.
MATERIALS AND METHODS
Cell Culture
Jurkat T, CEM, and HEK293T cells were purchased from ATCC (Manassas, VA). COS7 cells were kindly provided by Prof. F. M. Sladek (University of California Riverside). Jurkat T and CEM cells were cultured in RPMI 1640 medium (ATCC, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), 100 IU/ml penicillin. HEK293T and COS7 cells were cultured in Dulbecco's modified Eagle's medium (ATCC, Manassas, VA) under the same conditions without the addition of penicillin. Cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C, with medium renewal two to three times a week depending on the cell density. For SILAC experiments, RPMI 1640 medium without l-lysine or l-arginine was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). The complete light and heavy RPMI 1640 media were prepared by the addition of light or heavy lysine ([13C6,15N2]-l-lysine) and arginine ([13C6]-l-arginine), along with dialyzed FBS (Invitrogen, Carlsbad, CA) to the above lysine/arginine-depleted medium. We chose to label the proteome with lysine and arginine because all tryptic peptides except for the C-terminal peptides of some proteins carry a lysine or arginine, and this provides better proteome coverage than labeling the proteome using other amino acids such as leucine. The Jurkat T cells were cultured in heavy RPMI 1640 medium for at least 10 days to achieve complete stable isotope incorporation, and supplemental Fig. S1 displays representative mass spectra illustrating the complete incorporation of the heavy labeled lysine and arginine.
SG Treatment and Sample Preparation
Jurkat T cells, at a density of ∼7 × 105 cells/ml in light or heavy RPMI 1640 medium, were treated with 3 μm SG (Sigma, St. Louis, MO) for 24 h. The mean peak concentrations of SG in the plasma of ALL patients were (0.46 ± 0.68) and (2.7 ± 1.4) μm after oral SG administration at 60 mg/m2 and after 24 h of continuous intravenous infusion at 20 mg/m2/h, respectively (17). After treatment, the cells were harvested via centrifugation at 300g at 4 °C for 5 min and washed three times with ice-cold PBS to remove culture medium and FBS. To explore the role of reactive oxygen species in SG cytotoxicity, cells were also treated with 3 μm SG together with 500 μm allopurinol for 24 h. Cells were subsequently lysed with CelLyticTM M lysis buffer (Sigma) that was supplemented with 1 mm PMSF and a protease inhibitor mixture (Sigma). Cell lysates were centrifuged at 16,000g at 4 °C for 30 min, and the resulting supernatants were collected. The protein concentration in the cell lysate was measured using a Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA). SILAC labeling experiments were performed in three biological replicates: in the two forward SILAC experiments, the light-labeled drug-treated lysate and the heavy-labeled control lysate were combined at a 1:1 ratio (w/w) (Fig. 1A); in the reverse SILAC experiment, the heavy-labeled drug-treated cell lysate was mixed equally with the light-labeled control lysate.
Fig. 1.
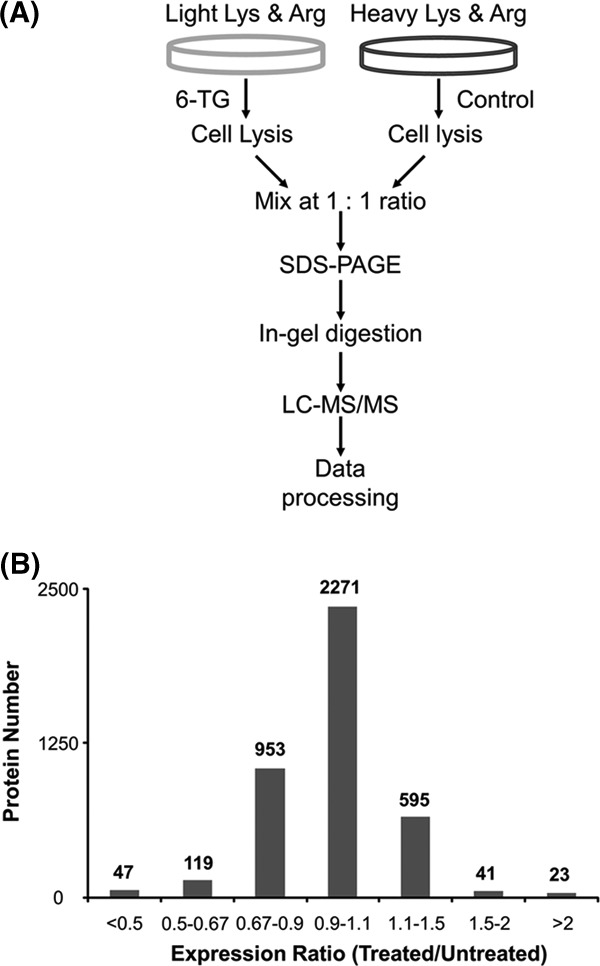
SILAC work flow and quantification results. A, a flowchart of forward SILAC combined with LC-MS/MS for the comparative analysis of protein expression in Jurkat T cells upon SG treatment. B, the distribution of expression ratios (treated/untreated) for the quantified proteins.
SDS-PAGE Separation and In-gel Digestion
The equi-mass mixture of light and heavy lysates was separated using 12% SDS-PAGE with a 4% stacking gel, and the gel was stained with Coomassie Blue. The gel was cut into 20 bands, and the proteins were reduced in-gel with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin (Promega, Madison, WI) at 37 °C overnight. Following the digestion, peptides were extracted into 5% acetic acid in H2O and CH3CN/H2O (1:1, v/v) and dried in a SpeedVac. The resulting peptide mixtures were stored at −80 °C until further analysis.
LC-MS/MS for Protein Identification and Quantification
On-line LC-MS/MS analysis was performed on an LTQ-Orbitrap Velos mass spectrometer coupled with an EASY n-LCII HPLC system and a nanoelectrospray ionization source (Thermo, San Jose, CA). The sample injection, enrichment, desalting, and HPLC separation were conducted automatically on a homemade trapping column (150 μm × 50 mm) and a separation column (75 μm × 120 mm, packed with ReproSil-Pur C18-AQ resin, 5-μm particle size, 300-Å pore size, Dr. Maisch HPLC GmbH, Ammerbuch, Germany). The peptide mixture was first loaded onto the trapping column with a solvent mixture of 0.1% formic acid in CH3CN/H2O (2:98, v/v) at a flow rate of 3.0 μl/min. The peptides were then separated using a 120-min linear gradient of 2%–40% acetonitrile in 0.1% formic acid at a flow rate of 300 nl/min.
The LTQ-Orbitrap Velos mass spectrometer was operated in the positive-ion mode, and the spray voltage was 1.8 kV. All MS/MS spectra were acquired in a data-dependent scan mode, where the 20 most abundant ions found in MS at a threshold above 500 counts were selected for fragmentation by collision-induced dissociation at a normalized collision energy of 35%. The full-scan mass spectra (from m/z 350 to 2000) were acquired with a resolution of 60,000 at m/z 400 after accumulation to a target value of 500,000.
Data Processing
The LC-MS/MS data were employed for the identification and quantification of the global proteome, which were conducted using MaxQuant, Version 1.2.0.18 (18), against the UniProt human database (with 538,585 sequence entries; release date: November 28, 2012), to which contaminants and reverse sequences were added. The maximum number of missed cleavages for trypsin was two per peptide. Cysteine carbamidomethylation and methionine oxidation were set as fixed and variable modifications, respectively. The tolerances in mass accuracy for MS and MS/MS were 25 ppm and 0.6 Da, respectively. Only those proteins with at least two distinct peptides discovered from LC-MS/MS analyses were considered reliably identified. The required false positive discovery rate was set to within 1% at both peptide and protein levels, with the minimal required peptide length being six amino acids. The quantification was based on three independent SILAC and LC-MS/MS experiments as noted above. To establish the threshold ratios for determining the significantly changed proteins, we calculated the standard scores (z-scores) for protein ratios as previously described (18, 19). Our analysis showed that at a 95% confidence level, those proteins with expression ratios greater than 1.22 or less than 0.73 could be considered significantly changed (20). To be stringent, however, we considered a protein as significantly altered only if its differential expression ratio (SG treated/control) was greater than 1.5 or less than 0.67.
Ingenuity Pathway Analysis
Ingenuity Pathway Analysis (IPA) (version 7.6, Ingenuity Systems Inc., Redwood City, CA) was employed to derive information about relationships, functions, and pathways of the differentially regulated proteins with the use of the Ingenuity Knowledge Base (Ingenuity Systems Inc.) (21, 22). The Ingenuity Knowledge Base consists of gene regulatory and signaling pathways that are integrated with other relevant databases such as NCBI Gene (www.ncbi.nlm.nih.gov/gene) and Gene Ontology. The proteins that were significantly changed upon SG treatment, including those quantified only in one cycle of the SILAC labeling experiment, were included for the pathway analysis. IPA determines the interaction by calculating a significance score using Fisher's exact test and exporting it as a p value. For canonical pathways, a p value of <0.05 is considered significant.
Plasmid Transfection
HEK293T cells were seeded in six-well plates at a 70% confluence level (∼3 × 105 cells/well), and SG was added to the culture medium at a final concentration of 3 μm. After a 24-h incubation, the cells were transfected with 1.5 μg Myc-Miro1 expression plasmid, which was kindly provided by Prof. E. Soriano (23), using Lipofectamine 2000 (Invitrogen). The cells were subsequently harvested for Western analysis 18 to 48 h after the transfection.
Western Blot and Flow Cytometry
Mouse monoclonal anti-Myc (9E10, 1:5000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) and mouse monoclonal anti-RHOT1 (1:10,000 dilution, Sigma) were used for Western analysis of ectopically expressed and endogenous Miro1, respectively. MitoProfile Total OXPHOS Human WB Antibody Mixture (1:10,000 dilution, ab110411, Abcam, Cambridge, United Kingdom) was employed to assess the expression level of five mitochondrial respiratory chain proteins.
Cellular uptake of MitoTracker Deep Red FM (Invitrogen) was quantified using flow cytometry on a BD FACS Aria I instrument (BD Biosciences, San Jose, CA). CEM, Jurkat T, and HEK293T cells, untreated or after a 24-h treatment with 3 μm SG and/or 500 μm allopurinol, were stained with MitoTracker Deep Red at a final concentration of 200 nm. After incubation for 15 to 20 min, the cells were washed thoroughly with prewarmed media and PBS. Flow cytometry analysis (BD FACS Aria I, NJ) was performed immediately after the cells were resuspended in a sorting buffer containing 1× PBS, 25 mm HEPES, 1 mm EDTA, and 1% FBS (pH 7.0). Counting was based on 20,000 cells per sample.
Trypan Blue Exclusion Assay
CEM, Jurkat T, and HEK293T cells were seeded in six-well plates at a density of ∼4 × 105 cells/ml and treated with SG and/or allopurinol at final concentrations of 3 μm and 500 μm, respectively. At 24 or 48 h after treatment, the cells were stained with trypan blue and counted on a hemocytometer to measure cell viability.
DNA Extraction and LC-MS/MS Measurement of Oxidatively Induced DNA Lesions
DNA was isolated from control and SG-treated cells using a salt extraction method and digested to nucleosides with a mixture of four enzymes (24). To the resulting mixture we subsequently added uniformly 15N-labeled 8,5′-cyclo-2′-deoxyadenosine (cdA) and 8,5′-cyclo-2′-deoxyguanosine (cdG), and the cdA and cdG were enriched from the nucleoside mixtures using HPLC, as described previously (24). The enriched fractions were subjected to LC-MS/MS analysis on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific) operated in multiple-reaction monitoring mode. A 3.0 × 100 mm Hypersil GOLD column (particle size = 5 μm; Thermo Scientific) was used for the on-line analyses of the fractions containing cdA and cdG, and the flow rate was 100 μl/min. A solution of 0.1% (v/v) formic acid in water (solution A) and a solution of 0.1% (v/v) formic acid in methanol (solution B) were employed as mobile phases, and a gradient of 0%–30% B over 40 min followed by 30%–80% B over 2 min was used for the separation. The spray voltage and S-lens radio frequency amplitude were 4.0 kV and 55 V, respectively, and the temperatures for the ion transport tube and vaporizer were set at 270 °C and 226 °C, respectively. The sheath and auxiliary gas flow rates were 15 and 10 arbitrary units, respectively. The respective multiple-reaction monitoring transitions for unlabeled and uniformly 15N-labeled cdG were m/z 266 → 180 and m/z 271 → 185. The corresponding multiple-reaction monitoring transitions for unlabeled and uniformly 15N-labeled cdA were m/z 250 → 164 and m/z 255 → 169. The collision energy was set at 17 V for cdG and 19 V for cdA, and the scan time was 300 ms.
RESULTS AND DISCUSSION
6-Mercaptopurine and SG are widely used for treating ALL and other human diseases (1–3). Although metabolic transformations of these prodrugs to SG nucleotides, their incorporation into nucleic acids, and triggering of the mismatch repair pathway are considered important (4), other mechanisms may also be at work. To exploit novel cellular pathways underlying the antineoplastic effects of the thiopurine drugs, we conducted a quantitative proteomic experiment, based on metabolic labeling using SILAC and LC-MS/MS, to exploit the SG-induced differential protein expression in human ALL cells.
SG Treatment, Protein Identification, and Quantification
We employed Jurkat T human ALL cells for the quantitative proteomic experiment. To minimize systematic errors and to assess the drug-induced alterations in protein expression reliably, we performed the SILAC experiments in three biological replicates, including two forward and one reverse SILAC labelings (Fig. 1A). The results from LC-MS/MS analyses of the three sets of SILAC samples allowed us to quantify a total of 4049 proteins (supplemental Table S1). Although the majority of the quantified proteins displayed similar levels of expression in the SG-treated and control cells, 230 exhibited significant changes (by >1.5-fold; see “Materials and Methods”) in expression levels after the drug treatment, with 166 and 64 being significantly down- and up-regulated, respectively (Fig. 1B). Among these 230 proteins, 100 were quantified in both forward and reverse SILAC labeling experiments (supplemental Table S2). Fig. 2 displays the representative LC-MS/MS results supporting the drug-induced down-regulation of Miro1, as reflected by the lower signal intensity for the tryptic peptide, LPLILVGNK, in the protein lysate from the SG-treated cells.
Fig. 2.

Example electrospray ionization MS data revealing the SG-induced down-regulation of Miro1. Shown are the MS results for the [M+2H]2+ ions of Miro1 peptides LPLILVGNK and LPLILVGNK* (K* denotes the heavy-labeled lysine) from the forward (left) and reverse (right) SILAC labeling experiments (A) and MS/MS results for the [M+2H]2+ ions of LPLILVGNK and LPLILVGNK* (B); heavy-labeled lysine-containing y-ions are labeled in bold.
SG Treatment Led to Mitochondrial Dysfunction
To explore the cellular pathways that are altered by SG treatment, we next subjected the significantly changed proteins to IPA. The IPA results revealed that the drug treatment led to the alteration of a number of canonical pathways (supplemental Table S3). Among them, mitochondrial dysfunction was the most significant and had the smallest p value (−log p = 7.41). In this context, most down-regulated proteins in this pathway are part of the mitochondrial respiratory chain complex, in which NDUFA2, NDUFA4, NDUFA10, and NDUFV1 are constituents of Complex I; UQCRC1, UQCRC2, and UQCRFS1 are components of Complex III; and MT-CO2, COX5B, and COX6C belong to Complex IV (supplemental Table S1). Additionally, we observed the diminished expression of cytochrome b5 (Table I), an outer mitochondrial membrane-bound hemoprotein functioning as an electron carrier for several membrane-bound oxygenases (25). Thus, the treatment of Jurkat T cells with SG led to the reduced expression of a large number of proteins involved in the electron transport chain in mitochondria. It is important to emphasize that diminished expression is not a general phenomenon for mitochondrial proteins. For instance, the expression levels of mitochondrial ATP synthase α subunit (ATP5A), the iron–sulfur subunit of complex II (SDH), and mitochondrial DNA-directed RNA polymerase did not change appreciably upon SG treatment (supplemental Table S1).
Table I. SG-induced alteration of expression of proteins indicating mitochondrial dysfunction.
| Gene symbol | Gene name | F1 | F2 | R1 | Fold change (SG-treated/control, mean ± S.D.) |
|---|---|---|---|---|---|
| MT-CO2 | Cytochrome c oxidase subunit II | 0.56 | 0.46 | 0.51 ± 0.07 | |
| COX5B | Cytochrome c oxidase subunit Vb | 0.39 | 0.51 | 0.42 | 0.45 ± 0.08 |
| COX6C | Cytochrome c oxidase subunit VIc | 0.57 | 0.51 | 0.54 ± 0.04 | |
| CPT1 | Carnitine palmitoyltransferase 1A | 2.01 | 2.10 | 1.63 | 1.91 ± 0.25 |
| CYB5A | Cytochrome b5 | 0.65 | 0.65 | ||
| NDUFA2 | NADH dehydrogenase α subcomplex 2 | 0.63 | 0.63 | ||
| NDUFA4 | NADH dehydrogenase α subcomplex 4 | 0.64 | 0.64 | ||
| NDUFA10 | NADH dehydrogenase α subcomplex 10 | 0.58 | 0.61 | 0.60 ± 0.02 | |
| NDUFV1 | NADH dehydrogenase flavoprotein 1 | 0.48 | 0.48 | ||
| UQCRC1 | Ubiquinol-cytochrome c reductase core protein I | 0.52 | 0.52 | 0.90 | 0.65 ± 0.22 |
| UQCRC2 | Ubiquinol-cytochrome c reductase core protein II | 0.47 | 0.47 | 0.96 | 0.63 ± 0.28 |
| UQCRFS1 | Ubiquinol-cytochrome c reductase rieske iron-sulfur polypeptide1 | 0.61 | 0.61 | ||
| RHOT1 | Mitochondrial RhoGTPase 1 (MIRO1) | 0.54 | 0.62 | 0.58 ± 0.06 |
Notes: Fold changes represent mean ± S.D. of quantification results from multiple SILAC labeling experiments. F1 and F2 refer to the results obtained from two forward cycles of SILAC labeling experiments, whereas R1 refers to the results from the reverse SILAC labeling experiment. Data without S.D. reflect that the proteins were quantified in only one set of SILAC labeling experiments, but at least two peptides were identified and quantified.
In agreement with the LC-MS/MS quantification results, Western blot analysis revealed that the SG-induced decrease in Miro1 expression was dose-dependent and occurred in multiple cell lines, including in two ALL lines (Jurkat T and CEM) and HEK293T human embryonic kidney epithelial cells (Figs. 3A and 3B). A similar observation was made for ectopically expressed Myc-tagged Miro1 in HEK293T cells (supplemental Fig. S2). Additionally, Western blot analysis with the use of a mixture of antibodies recognizing five mitochondrial respiratory chain proteins (see “Materials and Methods”) revealed that the drug treatment resulted in a significant reduction in the expression level of UQCRC2, whereas no change was found for ATP5A or SDH (Figs. 4A and 4B). These results are again consistent with the LC-MS/MS quantification data (supplemental Table S1). Western blot data did not reveal a significant alteration in the expression of NDUFB8 or MT-CO2 (Figs. 4A and 4B). The former protein was not quantified in LC-MS/MS experiments, whereas the latter displayed significant down-regulation based on both forward and reverse SILAC labeling experiments (Table I). The discrepancy between Western blot and LC-MS/MS results for the expression level of MT-CO2 likely can be attributed to inadequate specificity of the antibody toward this protein and/or co-migration of another protein or proteins with MT-CO2 under our SDS-PAGE conditions.
Fig. 3.
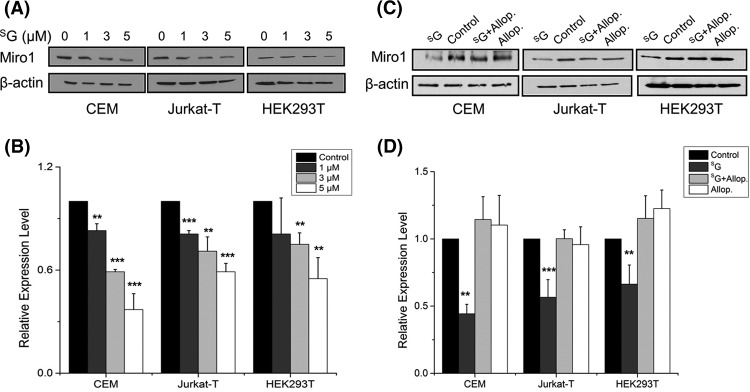
SG treatment resulted in diminished expression of Miro1. Western blot for monitoring the expression levels of endogenous Miro1 in CEM, Jurkat T, and HEK293T cells upon treatment with increasing concentrations of SG (A) or 24 h of treatment with SG and allopurinol, alone or in combination (C). β-actin served as the loading control. B, D, the quantification data representing the mean and S.D. of results from three independent drug-treatment and Western blot experiments. **, p < 0.01; ***, p < 0.001. The p values were calculated via two-tailed, unpaired Student's t test.
Fig. 4.
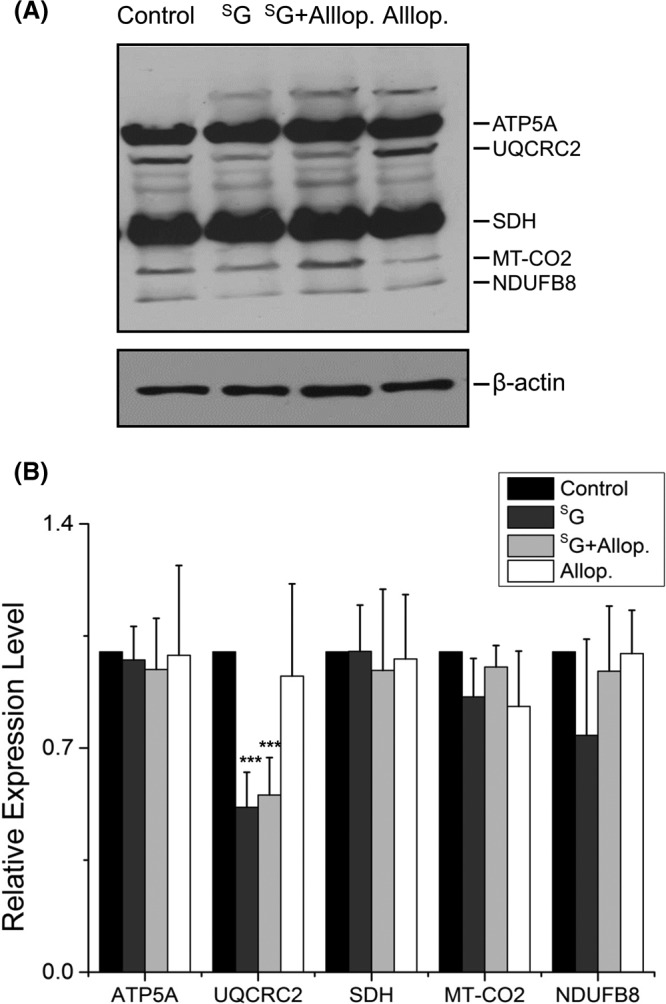
Western blot for monitoring the protein components of the mitochondria respiratory complex. A, Jurkat T cells were treated with 3 μM SG, 3 μM SG + 500 μM allopurinol, or 500 μm allopurinol for 24 h, analyzed via 12% SDS-PAGE, and probed for five representative proteins of the mitochondrial respiratory complex using an antibody mixture (see “Materials and Methods”). β-actin served as the loading control. B, bar graph displaying the quantification results, which represent the mean and S.D. of results from three independent drug treatment and Western blot experiments. ***, p < 0.001. The p values were calculated via two-tailed, unpaired Student's t test.
The significant down-regulation of many proteins in the mitochondrial respiratory chain complex prompted us to ask whether SG treatment compromises mitochondrial integrity. Thus, we estimated the level of active mitochondria based on the cellular uptake of MitoTracker Deep Red. Our flow cytometry quantification results illustrated that treatment with SG led to the diminished uptake of MitoTracker Deep Red into Jurkat T, CEM, and HEK293T cells (Fig. 5A), suggesting decreased levels of active mitochondria after SG treatment. Moreover, the diminution in the level of active mitochondria in these cell lines paralleled their relative sensitivity toward SG (Fig. 5). In this context, we observed a more pronounced loss in active mitochondria for CEM than for Jurkat T and HEK293T cells (Fig. 5A), which mirrors the highest sensitivity of the CEM cells toward SG (Figs. 5B–5D).
Fig. 5.

Allopurinol could partially rescue SG-induced growth inhibition, but not the drug-elicited decline in active mitochondria. A, flow cytometry quantification data for the cellular uptake of MitoTracker Deep Red in CEM, Jurkat T, and HEK293T cells that were untreated (control) or treated with allopurinol (Allop.), SG, or both (SG+Allop.). Representative plots for flow cytometry analysis are shown in supplemental Fig. S2. ***, p < 0.001. The p values were calculated via two-tailed, unpaired Student's t test. B–D, survival rates of CEM, Jurkat T, and HEK293T cells that were untreated (control) or treated with Allop., SG, or SG+Allop. The data represent the mean and S.D. of results from three independent experiments.
SG Treatment Led to Elevated Formation of Oxidatively Induced DNA Lesions
We reasoned that the SG-induced down-regulation of many proteins in the mitochondrial respiratory chain complex may lead to decreased electron transport efficiency, a situation known to give rise to the elevated formation of reactive oxygen species (ROS) (26). Increased ROS production was found in a series of nuclear-encoded genetic defects of Complex I in the mitochondrial respiratory chain (27). Therefore, we next asked whether the SG-induced mitochondrial dysfunction could also lead to the increased formation of ROS and elevated generation of oxidatively induced DNA lesions in cells. Regarding this, we found recently that the oxidatively generated 8,5′-cyclo-2′-deoxyadenosine (cdA) and 8,5′-cyclo-2′-deoxyguanosine (cdG) can serve as robust biomarkers for oxidative stress (the mechanism for the formation of cdA is depicted in Fig. 6A) (24, 28, 29).
Fig. 6.

SG treatment led to elevated generation of oxidatively induced cdA and cdG lesions. Proposed mechanism for the formation of cdA (A) and SG-induced formation of the (5′R) and (5′S) diastereomers of cdA and cdG in Jurkat T (B), COS7 (C), and CEM (D) cells. The data represent the mean and S.D. of results from at least three independent drug treatments and LC-MS/MS measurements. *, p < 0.05; **, p < 0.01; ***, p < 0.001. The p values were calculated via two-tailed, unpaired Student's t test.
We measured, via LC-MS/MS, the levels of cdA and cdG in genomic DNA isolated from CEM, Jurkat T, and COS7 cells treated with SG (representative LC-MS/MS data and calibration curves are shown in supplemental Figs. S4–S9). The LC-MS/MS results showed that the treatment with SG indeed stimulated the formation of the (5′R) diastereomer of cdA and cdG (Figs. 6B–6D), though we failed to observe significant increases in the levels of the (5′S) diastereomers of the two lesions (with the exception of S-cdG in CEM cells). This could be attributed to the less efficient formation of the (5′S) diastereomers of cdA and cdG, as found previously for the ionizing-radiation-induced generation of these DNA lesions in isolated DNA (30).
In keeping with the elevated generation of oxidatively induced cdA and cdG lesions, we found that the co-treatment of cells with allopurinol, an ROS scavenger, could abrogate the SG-induced reduction in the expression level of Miro1 (Fig. 3B). Nevertheless, co-treatment with allopurinol was not able to rescue the SG-induced diminished expression of UQCRC2 (Fig. 4) or restore the SG-induced loss of active mitochondria, though allopurinol could rescue substantially the SG-induced growth inhibition of CEM, Jurkat T, and HEK293T cells (Fig. 5A). These results suggest that the decreased expression of proteins in the electron transport chain and mitochondrial dysfunction occur prior to ROS induction.
CONCLUSIONS
Thiopurines are among the most successful chemotherapeutic agents for treating acute lymphoblastic leukemia and other human diseases (1–3). Although triggering of the MMR pathway by S6mG in DNA was previously noted (4), it remains possible that other pathways may also contribute to the therapeutic efficacy of the drugs in ALL treatment (10). Thus, we set out to exploit novel mechanisms leading to the antileukemic effects of the thiopurine drugs by assessing the SG-induced alterations of global protein expression in Jurkat T cells. Our results revealed that the drug treatment led to the diminished expression of a large number of proteins that are components of the electron transport chain of the mitochondrial respiratory complex (Table I). In agreement with this observation, IPA results uncovered mitochondrial dysfunction as one of the major pathways altered by SG treatment (supplemental Table S3), and flow cytometry data showed the drug-induced loss of active mitochondria (Fig. 5A). In addition, we observed the elevated generation of oxidatively induced cdA and cdG lesions in SG-treated cells (Fig. 6). Consistent with these findings, a recent study showed that SG treatment can give rise to the enhanced formation of ROS, and the drug-induced ROS generation and cytotoxicity can be potentiated by glutathione depletion and rescued by allopurinol (31).
Mitochondrial dysfunction and the ensuing formation of oxidatively induced bulky cyclopurine DNA lesions may contribute to the cytotoxicity of the drug to ALL cells. cdA and cdG are known to strongly block DNA replication and transcription in cells (35–37). Additionally, cdA and cdG can lead to mutations during DNA replication and transcription (35–37). Therefore, the SG-induced formation of cdA and cdG, if left unrepaired, may also contribute to the development of thiopurine therapy-related cancers (38).
Our study also provides new knowledge about the sequence of events underlying SG-induced toxicity. Based on the fact that genetic defects of Complex I in the mitochondrial respiratory chain are known to result in augmented ROS production (27), we deduce that the reduced expression of a large number of proteins in the mitochondrial respiratory complex may constitute an upstream event leading to mitochondrial dysfunction and the elevated generation of oxidatively induced DNA lesions. Additionally, we found that the reduced expression of mitochondrial electron transport chain protein UQCRC2 and the loss of active mitochondria, as reflected by the cellular uptake of MitoTracker Deep Red, could not be rescued by co-treatment of cells with an ROS scavenger, allopurinol (Figs. 4 and 5) (31). This result supported the conclusion that the loss of active mitochondria did not arise from ROS induction. We also found that the SG-induced down-regulation of Miro1 could be fully abolished by co-treatment of the cells with allopurinol (Figs. 3C and 3D), indicating that elevated numbers of ROS contribute to the decreased expression of Miro1. However, the molecular mechanisms underlying the SG-induced down-regulation of proteins in the mitochondrial respiratory chain complexes await further investigation.
Our findings also shed new light on the cellular effects of SG exposure observed in previous studies. In this vein, Kinsella and coworkers (32, 33) showed that SG treatment of cultured human cells can result in the activation of autophagy, a catabolic pathway for the lysosomal degradation of proteins and organelles. Considering the indispensable role of ROS, particularly those derived from mitochondria, in autophagy activation (34), our study also points to mitochondrial dysfunction as the potential origin for SG-induced autophagy.
Together, the results from the present study provide significant insights into the mechanisms of action of thiopurine drugs; namely, the drugs may exert their cytotoxic effect by triggering the diminished expression of proteins in the mitochondrial electron transport chain, inducing mitochondrial dysfunction and ROS generation and resulting in the augmented formation of oxidatively induced DNA lesions. Mitochondrial dysfunction may serve as a potential biomarker for monitoring the efficacy of thiopurine therapy.
Supplementary Material
Acknowledgments
We thank Ms. Holly Eckelhoefer for assistance with the flow cytometry experiments.
Footnotes
* This work was supported by the National Institutes of Health (R01 CA101864).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ALL
- acute lymphoblastic leukemia
- cdA
- 8,5′-cyclo-2′-deoxyadenosine
- cdG
- 8,5′-cyclo-2′-deoxyguanosine
- SG
- 6-thioguanine
- IPA
- Ingenuity Pathway Analysis
- MMR
- mismatch repair
- ROS
- reactive oxygen species
- SILAC
- stable isotope labeling by amino acids in cell culture
- S6mG
- S6-methylthioguanine.
REFERENCES
- 1. Elion G. B. (1989) The purine path to chemotherapy. Science 244, 41–47 [DOI] [PubMed] [Google Scholar]
- 2. Karran P. (2006) Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull. 79–80, 153–170 [DOI] [PubMed] [Google Scholar]
- 3. Pui C. H., Jeha S. (2007) New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat. Rev. Drug Discov. 6, 149–165 [DOI] [PubMed] [Google Scholar]
- 4. Swann P. F., Waters T. R., Moulton D. C., Xu Y. Z., Zheng Q. G., Edwards M., Mace R. (1996) Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 273, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 5. Wang H., Wang Y. (2010) LC-MS/MS coupled with stable isotope dilution method for the quantification of 6-thioguanine and S6-methylthioguanine in genomic DNA of human cancer cells treated with 6-thioguanine. Anal. Chem. 82, 5797–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yuan B., O'Connor T. R., Wang Y. (2010) 6-Thioguanine and S6-methylthioguanine are mutagenic in human cells. ACS Chem. Biol. 5, 1021–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yuan B., Wang Y. (2008) Mutagenic and cytotoxic properties of 6-thioguanine, S6-methylthioguanine, and guanine-S6-sulfonic acid. J. Biol. Chem. 283, 23665–23670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffin S., Branch P., Xu Y. Z., Karran P. (1994) DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA methylation damage. Biochemistry 33, 4787–4793 [DOI] [PubMed] [Google Scholar]
- 9. Waters T. R., Swann P. F. (1997) Cytotoxic mechanism of 6-thioguanine: hMutSa, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry 36, 2501–2506 [DOI] [PubMed] [Google Scholar]
- 10. Krynetski E. Y., Krynetskaia N. F., Gallo A. E., Murti K. G., Evans W. E. (2001) A novel protein complex distinct from mismatch repair binds thioguanylated DNA. Mol. Pharmacol. 59, 367–374 [DOI] [PubMed] [Google Scholar]
- 11. Shankavaram U. T., Reinhold W. C., Nishizuka S., Major S., Morita D., Chary K. K., Reimers M. A., Scherf U., Kahn A., Dolginow D., Cossman J., Kaldjian E. P., Scudiero D. A., Petricoin E., Liotta L., Lee J. K., Weinstein J. N. (2007) Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Mol. Cancer Ther. 6, 820–832 [DOI] [PubMed] [Google Scholar]
- 12. Cheok M. H., Yang W., Pui C. H., Downing J. R., Cheng C., Naeve C. W., Relling M. V., Evans W. E. (2003) Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat. Genet. 34, 231–231 [DOI] [PubMed] [Google Scholar]
- 13. Vogel C., Marcotte E. M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 15. Cravatt B. F., Simon G. M., Yates J. R. (2007) The biological impact of mass-spectrometry-based proteomics. Nature 450, 991–1000 [DOI] [PubMed] [Google Scholar]
- 16. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 17. Lowe E. S., Kitchen B. J., Erdmann G., Stork L. C., Bostrom B. C., Hutchinson R., Holcenberg J., Reaman G. H., Woods W., Franklin J., Widemann B. C., Balis F. M., Murphy R. F., Adamson P. C. (2001) Plasma pharmacokinetics and cerebrospinal fluid penetration of thioguanine in children with acute lymphoblastic leukemia: a collaborative Pediatric Oncology Branch, NCI, and Children's Cancer Group study. Cancer Chemother. Pharmacol. 47, 199–205 [DOI] [PubMed] [Google Scholar]
- 18. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 19. Graumann J., Hubner N. C., Kim J. B., Ko K., Moser M., Kumar C., Cox J., Scholer H., Mann M. (2008) Stable isotope labeling by amino acids in cell culture (SILAC) and proteome quantitation of mouse embryonic stem cells to a depth of 5,111 proteins. Mol. Cell. Proteomics 7, 672–683 [DOI] [PubMed] [Google Scholar]
- 20. Waanders L. F., Hanke S., Mann M. (2007) Top-down quantitation and characterization of SILAC-labeled proteins. J. Am. Soc. Mass Spectrom. 18, 2058–2064 [DOI] [PubMed] [Google Scholar]
- 21. Muller T., Schrotter A., Loosse C., Helling S., Stephan C., Ahrens M., Uszkoreit J., Eisenacher M., Meyer H. E., Marcus K. (2011) Sense and nonsense of pathway analysis software in proteomics. J. Proteome Res. 10, 5398–5408 [DOI] [PubMed] [Google Scholar]
- 22. Thomas S., Bonchev D. (2010) A survey of current software for network analysis in molecular biology. Hum. Genomics 4, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lopez-Domenech G., Serrat R., Mirra S., D'Aniello S., Somorjai I., Abad A., Vitureira N., Garcia-Arumi E., Alonso M. T., Rodriguez-Prados M., Burgaya F., Andreu A. L., Garcia-Sancho J., Trullas R., Garcia-Fernandez J., Soriano E. (2012) The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat. Commun. 3, 814. [DOI] [PubMed] [Google Scholar]
- 24. Wang J., Yuan B., Guerrero C., Bahde R., Gupta S., Wang Y. (2011) Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography-tandem mass spectrometry coupled with stable isotope-dilution method. Anal. Chem. 83, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schenkman J. B., Jansson I. (2003) The many roles of cytochrome b5. Pharmacol. Ther. 97, 139–152 [DOI] [PubMed] [Google Scholar]
- 26. Lenaz G. (2001) The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52, 159–164 [DOI] [PubMed] [Google Scholar]
- 27. Pitkanen S., Robinson B. H. (1996) Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Invest. 98, 345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mitra D., Luo X., Morgan A., Wang J., Hoang M. P., Lo J., Guerrero C. R., Lennerz J. K., Mihm M. C., Wargo J. A., Robinson K. C., Devi S. P., Vanover J. C., D'Orazio J. A., McMahon M., Bosenberg M. W., Haigis K. M., Haber D. A., Wang Y., Fisher D. E. (2012) An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 491, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tilstra J. S., Robinson A. R., Wang J., Gregg S. Q., Clauson C. L., Reay D. P., Nasto L. A., St Croix C. M., Usas A., Vo N., Huard J., Clemens P. R., Stolz D. B., Guttridge D. C., Watkins S. C., Garinis G. A., Wang Y., Niedernhofer L. J., Robbins P. D. (2012) Inhibition of IKK/NF-κB delays the onset of senescence and aging-related degenerative diseases caused by DNA damage. J. Clin. Invest. 122, 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Belmadoui N., Boussicault F., Guerra M., Ravanat J. L., Chatgilialoglu C., Cadet J. (2010) Radiation-induced formation of purine 5′,8-cyclonucleosides in isolated and cellular DNA: high stereospecificity and modulating effect of oxygen. Org. Biomol. Chem. 8, 3211–3219 [DOI] [PubMed] [Google Scholar]
- 31. Brem R., Karran P. (2012) Oxidation-mediated DNA cross-linking contributes to the toxicity of 6-thioguanine in human cells. Cancer Res. 72, 4787–4795 [DOI] [PubMed] [Google Scholar]
- 32. Zeng X., Kinsella T. J. (2008) Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 68, 2384–2390 [DOI] [PubMed] [Google Scholar]
- 33. Zeng X., Yan T., Schupp J. E., Seo Y., Kinsella T. J. (2007) DNA mismatch repair initiates 6-thioguanine-induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 13, 1315–1321 [DOI] [PubMed] [Google Scholar]
- 34. Scherz-Shouval R., Elazar Z. (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 36, 30–38 [DOI] [PubMed] [Google Scholar]
- 35. Yuan B., Wang J., Cao H., Sun R., Wang Y. (2011) High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 39, 5945–5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jasti V. P., Das R. S., Hilton B. A., Weerasooriya S., Zou Y., Basu A. K. (2011) (5′S)-8,5′-cyclo-2′-deoxyguanosine is a strong block to replication, a potent pol V-dependent mutagenic lesion, and is inefficiently repaired in Escherichia coli. Biochemistry 50, 3862–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. You C., Dai X., Yuan B., Wang J., Wang J., Brooks P. J., Niedernhofer L. J., Wang Y. (2012) A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol. 8, 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Relling M. V., Rubnitz J. E., Rivera G. K., Boyett J. M., Hancock M. L., Felix C. A., Kun L. E., Walter A. W., Evans W. E., Pui C. H. (1999) High incidence of secondary brain tumours after radiotherapy and antimetabolites. Lancet 354, 34–39 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.