

Abstract
Aplastic anemia (AA) is a marrow failure syndrome mediated by aberrant T-cell subsets. Mesenchymal stem cells (MSCs) play an important role in maintaining immune homeostasis through modulating a variety of immune cells. However, little is known about the immunomodulation potential of bone marrow MSCs (BM-MSCs) in AA. Here, we reported that BM-MSCs from AA patients were reduced in suppressing the proliferation and clonogenic potential of CD4+ T cells and the production of tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ), which was associated with decreased prostaglandin E2 (PGE2). Meanwhile, BM-MSCs from AA patients were defective to promote CD4+CD25+FOXP3+ regulatory T cells expansion through reduced transforming growth factor-β (TGF-β). No significant difference between AA and normal BM-MSCs was observed in affecting the production of interleukins (IL)-4, IL-10 and IL-17. Our data indicate that BM-MSCs were impaired in maintaining the immune homeostasis associated with CD4+ T cells, which might aggravate the marrow failure in AA.
Keywords: Aplastic anemia, Bone marrow mesenchymal stem cell, CD4+ T cells, Immunomodulation
1. Introduction
Aplastic anemia (AA) is mostly considered an immune-mediated bone marrow failure syndrome, characterized by hypoplasia and pancytopenia with fatty bone marrow and reduced angiogenesis. Previous investigations have demonstrated that acquired AA is manifested as abnormalities of hematopoietic stem/progenitors cells (HSCs/HPCs) and hematopoietic microenvironment, which are mediated by abnormal immunity [1,2]. T cell is the major factor mediating the pathogenesis of acquired AA, which is involved with imbalanced CD4+ and CD8+ T cells subpopulation. CD4+ T cells were commonly divided into helper T lymphocyte (Th1), Th2, Th17 and CD4+CD25+ FOXP3+ regulatory T cells (Tregs). Th1 cells and cytotoxic T lymphocytes (CTLs) are activated while Tregs are deficient, and Th2 cells are almost normal or expanded in AA [3–7]. Aberrant immune cells directly and indirectly destruct HSCs by secreting a variety of immune molecules including tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and interleukins (IL-2, 8, 12, 15, 17, 27) [1,8–10]. As a result, HSCs are severely impaired to be disabled cells leading to hypoplasia and pancytopenia. Lots of evidence has hinted that AA might be a syndrome characterized by stem/progenitor-cell disorders including HSCs/HPCs and bone marrow mesenchymal stem cells (BM-MSCs).
Previous studies have demonstrated that HSCs/HPCs from AA patients are defective in multiple biological properties and functions [11–13]. Besides the role of HSCs/HPCs in the process of hematopoisis, BM-MSCs as the key precursor cells of marrow microenvironment may also play an important role in the development of AA. MSCs differentiate into a variety of stromal cells to constitute HSC niche, which include endothelial cells, adipocytes, fibroblasts, osteoblasts and osteoclasts etc. MSCs and differentiated stromal cells support hematopoiesis and regulate almost overall immune cells function to maintain the hematopoietic and immune homeostasis [14,15]. MSCs can modulate the major immune cell functions including T, B, monocytes, dendritic cells (DCs), nature killer cells (NKs) and neutrophils [16,17]. MSCs possess remarkable immunosuppressive properties on Th1 and CTLs. MSCs inhibit the proliferation of T cells, IFN-γ and TNF-α secretion by Th1 cells while promoting IL-10 production by Th2 cells and the expansion of Tregs. However, it is controversial about the immunomodulation of MSCs on IL-4 and IL-17 production by Th2 and Th17 cells [18,19].
Recently, sporadic research showed that MSCs from AA patients had poor proliferation and deficient immune suppression of MLR, PHA-induced T cell activation and IFN-γ release [20,21]. T lymphocyte is known to be the major executor of the adaptive immune response and the arch-criminal of hematopoiesis destruction in AA. During the development of AA, Th1 and Th17 cells are expanded while Tregs are reduced [7]. However, it is still controversial about the levels and functions of Th2 cells [3,7]. It is necessary to determine whether BM-MSCs contribute to the aberrant immunomodulation process mediated by CD4+ T cells in AA.
Therefore, the present research was designed to elucidate the effect of BM-MSCs from AA patients on CD4+ T cells in order to obtain more evidence for the marrow microenvironment failure in the pathogenesis of AA. We found that BM-MSCs from AA patients were reduced in suppressing the proliferation and clonogenic potential of CD4+ T cells while promoting Tregs expansion. They were also defective to suppress the production of TNF-α and IFN-γ by CD4+ cells. However, there was no significant difference in regulating the production of IL-4, IL-10 and IL-17. Our data have demonstrated that BM-MSCs were abnormal in maintaining the CD4+ T cellular immune homeostasis in AA.
2. Materials and methods
2.1. Patients and controls
We analyzed bone marrow samples from 15 patients with AA (mean age 31 years, 8 men and 7 women), as well as from 11 healthy controls (mean age 33 years, 6 men and 5 women). The diagnosis of AA was established by morphological examination of bone marrow and blood after exclusion of any other marrow failure syndromes, such as paroxysmal nocturnal hemoglobinuria (PNH), myelodysplastic syndrome (MDS) and congenital bone marrow failure syndromes according to the international criteria [22]. All patients did not receive any specific therapy such as cyclosporine A and antithymocyte globulin (ATG) before enrollment. Controls were healthy donors who were also identified by morphology examinations of bone marrow and blood.
2.2. Isolation and identification of BM-MSCs
Bone marrow aspirates were taken from patients and healthy donors with informed consent in accordance with the Institutional Review Board of CAMS and PUMC. Bone marrow mononuclear cells (BMMNCs) were isolated from samples using the Ficoll-Hypaque (1.077 g/mL) (Tianjin Haoyang Biological Manufacture Co. Ltd., China) density gradient centrifugation. Isolated BMMNCs were cultured in Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (D-MEM/F-12) (Gibco, Carlsbad, CA, USA) supplemented with 40% MCDB-201 (Sigma, St. Louis, USA), 2% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), 1×insulin–transferrin–selenium (ITS) (Gibco, Carlsbad, CA, USA), 10−8 M dexamethasone (Sigma, St. Louis, USA), 100 U/mL penicillin/streptomycin, 2 mM l-glutamine (Sigma, St. Louis, USA), 2 ng/mL human basic-fibroblast growth factor (bFGF) and 10 ng/mL human EGF (PeproTech, Rocky Hill, NJ, USA). After 3 days, the culture medium was completely replaced and non-adherent cells were removed. At about 80–85% confluency, the adherent cells were detached by 0.125% trypsin and 0.1% EDTA (Sigma, St. Louis, USA) and replated at a 1:2 dilution under the same culture conditions. BM-MSCs were identified by the surface markers with monoclonal antibodies CD29 (MAR4), CD166 (3A6), CD44 (515), CD73 (AD2), CD49e (IIA1), CD34 (581), CD90 (5E10), CD45 (HI30), CD105 (266), HLA-DR (G46–6), isotype mAbs (BD Pharmingen, San Jose, CA, USA) using a FACScan flow cytometry (BD Biosciences, Mountain View, CA, USA). BM-MSCs (P3) were stained with β-tubulin for the morphology examination using a fluorescence confocal microscopy (Leica TCS SP2, Leica Microsystems,Wetzlar, Germany).
2.3. Adipogenic and osteogenic differentiation potential of BM-MSCs
BM-MSCs (P4) were induced to differentiate into adipocytes and osteoblasts. The induction medium for adipogenesis was Iscove's Modified Dulbecco's Medium (IMDM) (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS, 10−6 M dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 10 mg/mL insulin and 60 μM indomethacin (Sigma, St. Louis, USA). The induction medium for osteogenesis was IMDM supplemented with 10% FBS, 10−7 M dexamethasone, 0.2 mM ascorbic acid 2-phosphate and 10 mM glycerol 2-phosphate (Sigma, St. Louis, USA). Three days later, the culture medium was completely replaced. After the determined culture, the adipocytes were stained with Oil Red O, and the osteoblasts with von Kossa and alkaline phosphatase assays (Sigma, St. Louis, USA) according to the protocols.
2.4. Co-culture of BM-MSCs and peripheral blood (PB)-derived CD4+ T cells
Peripheral blood was obtained from healthy adult donors according to the Institutional Review Board of CAMS and PUMC. Peripheral blood mononuclear cells (PBMNCs) were isolated using Ficoll-Hypaque (1.077 g/mL) (Tianjin Haoyang Biological Manufacture Co. Ltd., China). CD4+ T cells were purified by positive selection with anti-CD4 mAb-conjugated microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. BM-MSCs (P4) and CD4+ T cells were co-cultured (MSC:CD4+ T cell ratio, 1:10) in the culture medium containing IMDM, 10% FBS, 100 U/mL penicillin/streptomycin and 2 mM l-glutamine in the presence of 5 mg/mL PHA (Roche, Penzberg, Germany) and 5 ng/mL of rIL-2 (PeproTech, Rocky Hill, NJ, USA) for 4 days.
2.5. Clonogenic and proliferation potential of CD4+ T cells
Clonogenic potential of CD4+ T cells was examined using an inverted microscope (OLYMPUS IX71S8F-2, Tokyo, Japan) after 4 days culture. CD4+ T cells proliferation was measured by incorporation of BrdU using cell proliferation ELISA assay after 4 days. CD4+ T cells were seeded in triplicate in 96-well plates. The optical density (OD) values were determined in triplicate against a reagent blank at a test wave length of 450 nm.
2.6. Cytokine enzyme-linked immunosorbent assay
Culture supernatants were harvested for cytokine determination by enzyme-linked- immunosorbent assay (ELISA). The concentrations of IFN-γ, TNF-α, IL-17A, IL-10, IL-4 and TGF-β (Neobioscience, Shanghai, China) and prostaglandin E2 (PGE2) (Cayman Chemicals, Ann Arbor, Michigan, USA) were measured according to the manufacturer's instructions. Samples were run in duplicate.
2.7. Induction the expansion of Tregs
BM-MSCs (P4) and CD4+ T cells were co-cultured (MSC:CD4+ T cell ratio, 1:10) in the culture medium containing IMDM, 10% FBS, 100 U/mL penicillin/streptomycin and 2 mM l-glutamine in the absence or presence of 300 U/mL rIL-2 (PeproTech, Rocky Hill, NJ, USA) for 5 days. After 5 days of co-culture, nonadherent T cells were harvested and evaluated for the proportion of Tregs with monoclonal antibodies FITC-CD4, APC-CD25 and PE-FOXP3 antibodies (BD Pharmingen, San Jose, CA, USA) using a FACScan flow cytometer (BD Biosciences, Mountain View, CA, USA).
2.8. Statistical analysis
Data were analyzed with the 15.0 SPSS software. Results are presented as mean ± SD. The statistical differences between groups were evaluated by One-Way ANOVA and Student's t test, defined as a value of P < 0.05, P < 0.01 and P < 0.001.
3. Results
3.1. Isolation and identification of BM-MSCs
BM-MSCs were isolated and cultured from 15 AA patients and 11 healthy controls. BM-MSCs were harvested at passage 3 to analyze the immunophenotype using flow cytometry. As shown in Fig. 1, BM-MSCs from both AA patients and healthy controls expressed CD105 (SH2), CD73 (SH3), CD90, CD29, CD44, CD49e, and CD166, but lack expression of CD34, CD45 and HLA-DR (HLA-II). There was no significant difference of the expression of BM-MSCs markers between AA patients and healthy controls (P > 0.05, data not shown). BM-MSCs from either AA patients or healthy controls could form a monolayer of bipolar spindle-like cells with a whirlpool-like array (Fig. 2A). After induction with different conditional media, BM-MSCs could differentiate into adipocytes and osteoblasts as detected by positive staining of Oil Red O for adipogenic differentiation (Fig. 2B and C), Allizarin Red, von Kossa and ALP for osteogenic differentiation (Fig. 2D, E and F), respectively. Interestingly, BM-MSCs from AA patients were easily induced to differentiate into adipocyte lineage, but difficultly induced to differentiate into osteoblast lineage.
Fig. 1.
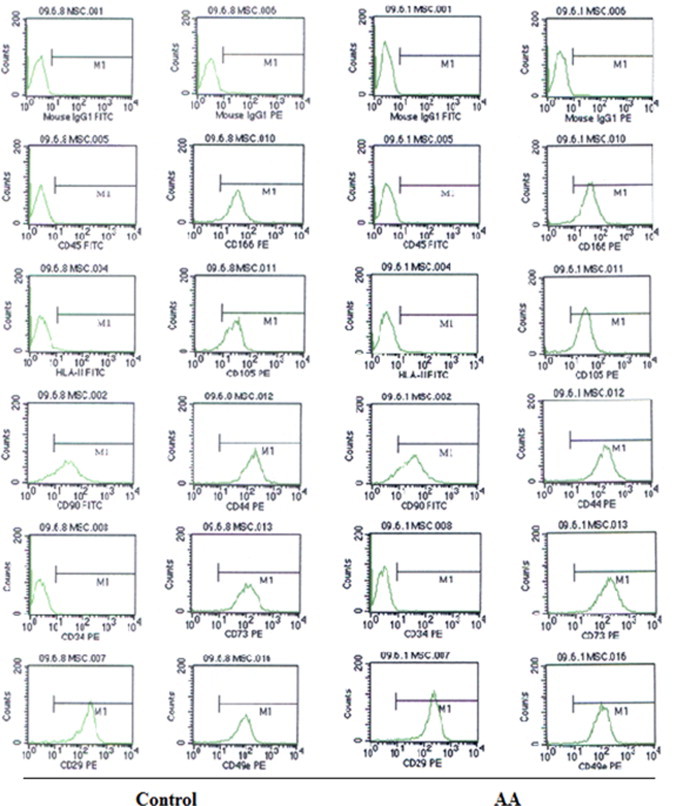
The representative for immnophenotype of BM-MSCs of healthy controls (n = 11) and AA patients (n = 15). The immnophenotype of BM-MSCs was assessed with monoclonal antibodies CD45, HLA-DR (HLA-II), CD90, CD34, CD29, CD166, CD105, CD44, CD73 and CD49e using a FACScan flow cytometry. All independent experiments were performed three times. Abbreviation: AA, aplastic anemia.
Fig. 2.
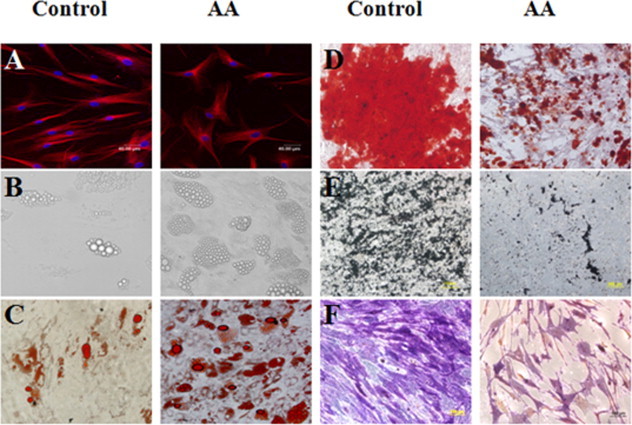
The representative morphology and differentiation capacity of BM-MSCs from AA patients and healthy controls. The morphology of BM-MSCs was shown after staining with β-tubulin (A). The adipogenic differentiation capacity of BM-MSCs detected by untaining (B) and positive staining of Oil Red O (C). The osteogenic differentiation capacity of BM-MSCs detected by positive staining of Allizarin Red (D), von Kossa (E) and ALP (F). Scale bar = 200μm. Abbreviation: AA, aplastic anemia.
3.2. Inhibition of clonogenic and proliferation capacity of CD4+ T cells by BM-MSCs
Previous studies in our laboratory have showed that umbilical cord and fetal BM-derived MSCs modulate immune activities on different T subpopulations [18,23]. And the cellular immune mediated by CD4+ T cells has been considered as the major mechanism of HSCs destruction in acquired AA. Therefore, we examined the immune effect of BM-MSCs on healthy peripheral blood-derived CD4+ T cells to elucidate the immunomodulation capacity of BM-MSCs from AA patients. BM-MSCs from AA patients and healthy controls were paired to co-culture with PB CD4+ T cells sorted using microbeads from unrelated donors. As shown in Fig. 3, the presence of BM-MSCs from healthy controls (C) and AA patients (B) resulted in an obvious decrease in PHA-induced clonogenic capacity of CD4+ T cells. But the inhibition by BM-MSCs from AA patients was significantly attenuated in comparison with that of healthy controls. Meanwhile, the presence of BM-MSCs from healthy controls also resulted in a statistically significant decrease in PHA-induced proliferation capacity of CD4+ T cells (P = 0.002). The inhibition by BM-MSCs from AA patients (Fig. 3B and D) was significantly attenuated in comparison with that of healthy controls (P = 0.046) (Fig. 3C and D). There was no significant difference of proliferation rate between group CD4 and group AA-MSC+CD4 (P = 0.232) (Fig. 3D).
Fig. 3.
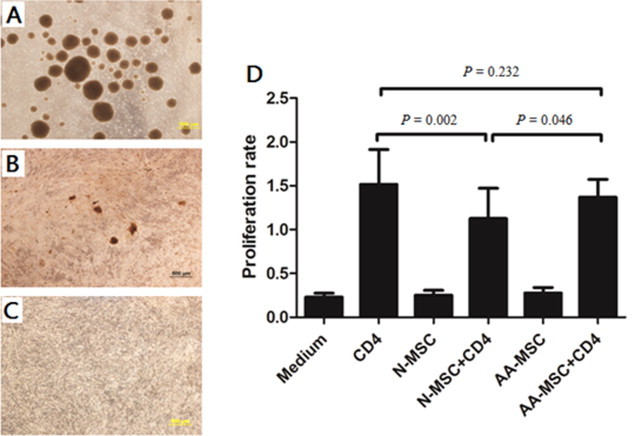
The inhibition of clonogenic and proliferation capacity of PB CD4+ T cells by BM-MSCs. The representative for clonogenic capacity of CD4+ T cells was examined without BM-MSCs (A), with BM-MSCs from AA patients (B) and healthy controls (C) using an inverted microscope (n = 10). Scale bar = 500μm. The proliferation capacity of CD4+ T cells was determined by incorporation of BrdU using cell proliferation ELISA assay (D). All data represented mean ± SD of triplicate of 10 independent experiments. The significance was shown as P < 0.05 and P < 0.01. N-MSC and BM-MSCs from healthy controls. AA-MSC and BM-MSCs from apalstic anemia patients.
3.3. Effects of BM-MSCs on the cytokines production by CD4+ T cells
The subpopulations of CD4+ T cells mostly exert their immune functions by secreting a variety of immune molecules. Recently, CD4+ T cells were divided into Th1, Th2, Th17 cells and Tregs according to their functions. Therefore, we compared the production of TNF-α, IFN-γ, IL-4, IL-10 and IL-17A by CD4+ T cells in the presence of BM-MSCs to elucidate the immune effect of BM-MSCs on different T cells subtypes such as Th1, Th2 and Th17 cells. We showed that both BM-MSCs inhibited the production of TNF-α and IFN-γ by CD4+ T cells (Fig. 4A and B) but promoted the production of IL-4 and IL-17A by CD4+ T cells (Fig. 4C and E) (P < 0.01). However, there was no significant effect on IL-10 production (Fig. 4D). Interesting, there was a significant decrease in inhibiting TNF-α and IFN-γ production mediated by BM-MSCs from AA patients compared with that of healthy controls (P < 0.05) (Fig. 4A and B). There was no significant difference in the production of IL-4, IL-10 and IL-17A by CD4+ T cells between AA patients and healthy controls (P > 0.05) (Fig. 4C–E). We further showed that PGE2 in the culture supernatant of BM-MSCs from AA patients was decreased compared with that of healthy controls (P < 0.05) (Fig. 4F).
Fig. 4.
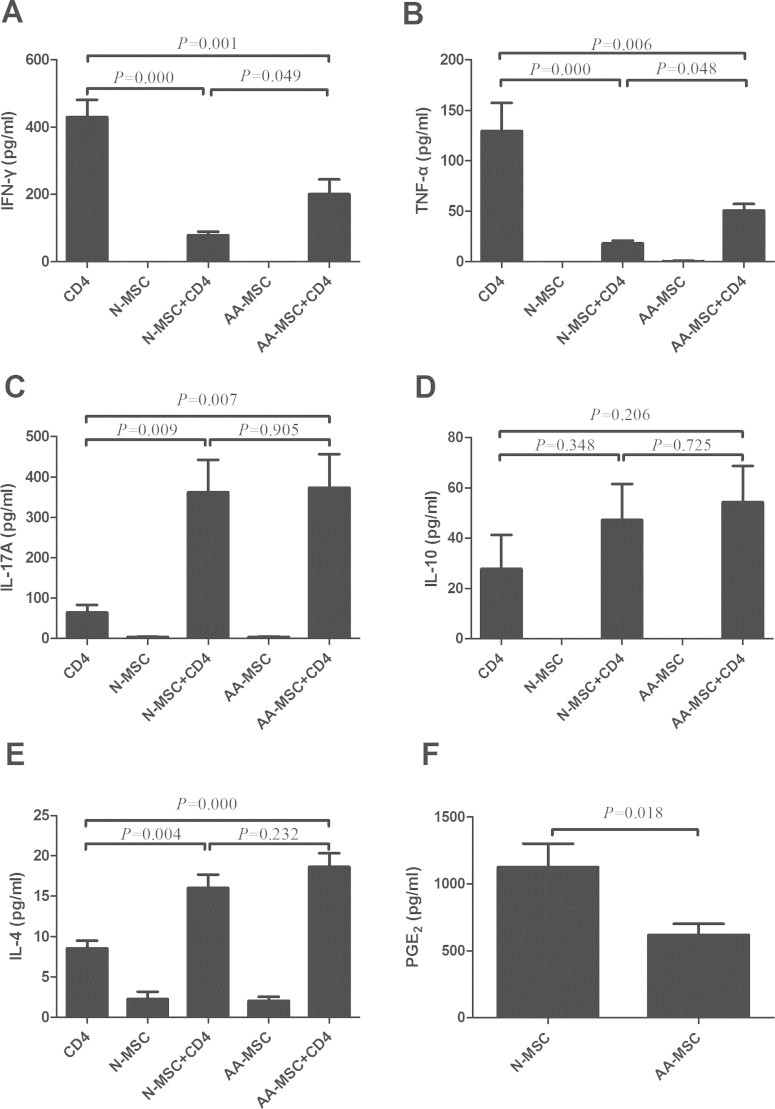
Effect of BM-MSCs on the secreted cytokines by CD4+ T cells. The production of IFN-γ (A), TNF-α (B), IL-17A (C), IL-10 (D) and IL-4 (E) by CD4+ T cells in the presence of BM-MSCs from AA patients and healthy controls was determined by ELISA. The concentration of PGE2 (F) in the culture supernatant of BM-MSCs was also determined by ELISA. All data represented mean ± SD of triplicate of 10 independent experiments. The significance was shown as P < 0.05, P < 0.01 and P < 0.001. N-MSC and BM-MSCs from healthy controls. AA-MSC and BM-MSCs from apalstic anemia patients.
3.4. BM-MSCs from AA patients were defective in promoting the expansion of Tregs.
Moreover, we compared the effect of BM-MSCs on the expansion of CD4+CD25+ FOXP3+ population (Tregs). We showed that BM-MSCs from healthy controls promoted the expansion of Tregs population by rhIL-2 (Fig. 5A and C) (P < 0.01). But, BM-MSCs from AA patients were defective in inducing Tregs expansion (5.82 ± 2.56%) compared with that of healthy controls (9.24 ± 1.61%) (P < 0.05). To demonstrate the possible mechanism, we examined TGF-β levels secreted by BM-MSCs and found that there was a relevant decrease in AA group (247.66 ± 117.23 pg/ml) than healthy controls (485.41 ± 99.27 pg/ml) (P < 0.05) (Fig. 5B).
Fig. 5.
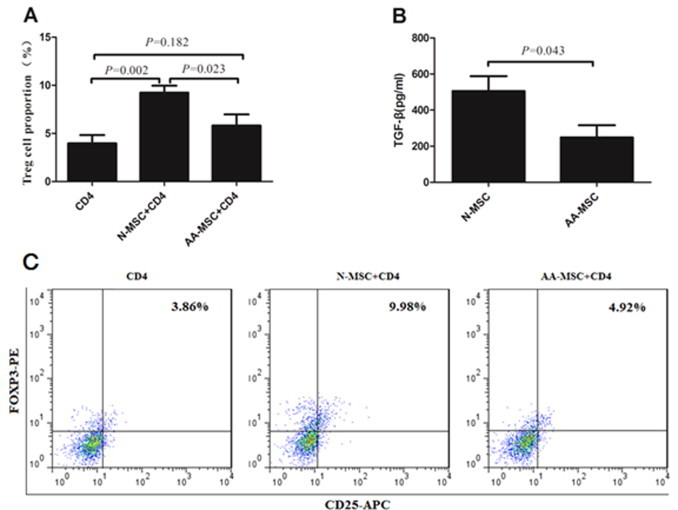
Effect of BM-MSCs on the expansion of Tregs. The proportion of Tregs in the presence of BM-MSCs from AA patients and healthy controls was determined using a FACScan flow cytometer (A) and (C). The production of TGF-β by BM-MSCs from AA patients and healthy controls was determined by ELISA (B). All data represented mean ± SD of triplicate of 10 independent experiments. The significance was shown as P < 0.05 and P < 0.01. N-MSC and BM-MSCs from healthy controls. AA-MSC and BM-MSCs from apalstic anemia patients.
4. Discussion
The present study was aimed to elucidate the effect of BM-MSCs from AA patients on CD4+ T cells and obtain more evidence for the marrow microenvironment failure in AA. We found that BM-MSCs from AA patients were reduced in suppressing the proliferation and clonogenic potential of CD4+ T cells and the production of TNF-α and IFN-γ by CD4+ T cells, which might be associated with decreased PGE2. Meanwhile, BM-MSCs from AA patients were defective in promoting Tregs expansion through reduced TGF-β. However, there was no significant difference between normal and AA BM-MSCs in their ability to affect the production of interleukins IL-4, IL-10 and IL-17 by CD4+ T cells.
Both HSCs and MSCs are key stem cells responsible for normal hematopoiesis. HSCs maintain hematopoiesis through self-renewal and differentiation. MSCs, as non-hematopoietic stem cells, support hematopoiesis and maintain the immune homeostasis in the bone marrow. Previous investigations have demonstrated that HSCs were damaged by T cells-mediated immune during the development of AA. Various evidence showed that BM-MSCs in AA were also abnormal when compared with healthy controls [20,21]. Transplantation of MSCs can enhance the reconstruction of hematopoiesis and immune systems of AA patients [24–26]. However, it remains unclear about the comprehensive abnormality of BM-MSCs in AA.
Immune regulation is one of the most important functions of MSCs. In recent years, MSCs have attracted significant attention from basic and clinical investigators because of their effective immunomodulation potential. MSCs affect a variety of immune cells maturation and functions. Especially, they exhibit remarkable regulation on CD4+ T cells including Th1, Th2, Th17 and Tregs to maintain the immune balance. MSCs inhibit the proliferation of T cells, IFN-γ and TNF-α secretion by Th1 cells while promote IL-10 production by Th2 cells and the expansion of Tregs. Acquired AA is considered as T cell-mediated marrow failure. Abnormal CD4+ T cells population and secreted cytokines play very important roles in the destruction of HSCs/HPCs. During the development of AA, Th1 and Th17 cells are expanded and activated [7]. A variety of immune molecules including IFN-γ, TNF-α and ILs (IL-2, 8, 12, 15, 17, and 27) compose a cytokine network to destruct stem/progenitor cells as well as HSCs/HPCs and MSCs [1,10]. Inversely, deficient Tregs diminish the capacity of immune regulation and the support of hematopoiesis [4,7]. It is obvious that aberrant CD4+ T cells immunity in AA is in accordance with the regulation of MSCs on CD4+ T cells. Therefore, we co-cultured BM-MSCs together with PB-derived CD4+ cells to demonstrate whether deficient MSCs aggravate the immune imbalance in AA.
In the present study, we found that BM-MSCs from AA patients were reduced in suppressing the proliferation and clonogenic potential of CD4+ T cells. BM-MSCs from AA patients were also defective in inhibiting the production of TNF-α and IFN-γ by CD4+ T cells while there was no significant difference in modulating the production of IL-4, IL-10 and IL-17A compared with healthy controls. Therefore, BM-MSCs could not inhibit the over-production by activated CD4+ T cells. Finally, excessive TNF-α and IFN-γ accumulated in the bone marrow to further destruct the normal hematopoiesis. Previous studies have demonstrated that BM derived MSCs and CD4+ T cells might interact with each other through direct contact and a variety of molecules such as PGE2, galectin-1, indoleamine 2,3 dioxygenase (IDO), nitric oxide (NO), HLA-5G and TGF-β [23,27–30]. PGE2 and galectin-1 are of great importance in the immunosuppresive process of MSCs. Researches in our laboratory showed that PGE2 played important roles in the immunosuppression of various MSCs on Th1 and the production of TNF-α and IFN-γ. In order to demonstrate the possible mechanism, we compared and found that PGE2 in the culture supernatant of BM-MSCs from AA patients was significantly decreased. We inferred that these defects might be associated with decreased PGE2. In addition, BM-MSCs from AA patients were also defective in promoting Tregs expansion. In AA, reduced total number of Tregs could not maintain the immune homeostasis and might aggravate the immune destruction of hematopoiesis. Interestingly, TGF-β as a key cytokine in inducing Tregs expansion was decreased in the culture supernatant of BM-MSCs from AA patients. Reduced TGF-β might be one of the possible factors for reduced contribution of BM-MSCs to Tregs expansion. In short, BM-MSCs from AA patients were limited either in the suppression of Th1 (TNF-α and IFN-γ) or in the contribution on Tregs expansion, but have no obvious defect on the regulation of Th2 (IL-4 and IL-10) and Th17 (IL-17A) cells. As a result, the polarization of Th1 and increased IFN-γ and TNF-α might destruct the hematopoiesis in the process of AA. When BM-MSCs were destructed to be defective, bone marrow suffered more severe loss in maintaining the immune homeostasis which would further aggravate the aberrant immunity in AA. Maybe our data also provides one of the key persuasive annotations for the effective MSC transplantation in AA.
In summary, the present study demonstrated that BM-MSCs from AA patients were impaired in immunomodulation ability on CD4+ T cells, which might cause more severe imbalance of immune regulation and aggravate the bone marrow failure. Meanwhile, out data suggest that transplantation of MSCs alone or in combination with HSCs could be an effective therapeutic strategy for AA in the future.
Acknowledgments
This study was supported by the National Basic Research Program of China (nos. 2011CB964800 and 2011CB964802), the 863 project (Grant no. 2011AA020118) from the Ministry Science & Technology of China, the National Natural Science Foundation of China (nos. 30872330 and 81160071), Tianjin Research Program of Application Foundation and Advanced Technology (no. 12JCZDJC25000) and West Light Foundation of The Chinese Academy of Sciences (2010).
References
- 1.Li JP, Zheng CL, Han ZC. Abnormal immunity and stem/progenitor cells in acquired aplastic anemia. Critical Reviews in Oncology/Hematology. 2010;75:79–93. doi: 10.1016/j.critrevonc.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giannakoulas NC, Karakantza M, Theodorou GL, Pagoni M, Galanopoulos A, Kakagianni T, Kouraklis-Symeonidis A, Matsouka P, Maniatis A, Zoumbos NC. Clinical relevance of balance between type 1 and type 2 immune responses of lymphocyte subpopulations in aplastic anaemia patients. British Journal of Haematology. 2004;124:97–105. doi: 10.1046/j.1365-2141.2003.04729.x. [DOI] [PubMed] [Google Scholar]
- 4.Solomou EE, Rezvani K, Mielke S, Malide D, Keyvanfar K, Visconte V, Kajigaya S, Barrett AJ, Young NS. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood. 2007;110:1603–1606. doi: 10.1182/blood-2007-01-066258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zoumbos NC, Gascon P, Djeu JY, Trost SR, Young NS. Circulating activated suppressor T lymphocytes in aplastic anemia. New England Journal of Medicine. 1985;312:257–265. doi: 10.1056/NEJM198501313120501. [DOI] [PubMed] [Google Scholar]
- 6.de Latour RP, Visconte V, Takaku T, Wu C, Erie AJ, Sarcon AK, Desierto MJ, Scheinberg P, Keyvanfar K, Nunez O, Chen J, Young NS. Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood. 2010;116:4175–4184. doi: 10.1182/blood-2010-01-266098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kordasti S, Marsh J, Al-Khan S, Jiang J, Smith A, Mohamedali A, Abellan PP, Veen C, Costantini B, Kulasekararaj AG, Benson-Quarm N, Seidl T, Mian SA, Farzaneh F, Mufti GJ. Functional characterization of CD4+ T cells in aplastic anemia. Blood. 2012;119:2033–2043. doi: 10.1182/blood-2011-08-368308. [DOI] [PubMed] [Google Scholar]
- 8.Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood. 2002;100:1185–1191. doi: 10.1182/blood-2002-01-0035. [DOI] [PubMed] [Google Scholar]
- 9.Verma A, Deb DK, Sassano A, Kambhampati S, Wickrema A, Uddin S, Mohindru M, Van Besien K, Platanias LC. Cutting edge: activation of the p38 mitogen-activated protein kinase signaling pathway mediates cytokine-induced hemopoietic suppression in aplastic anemia. Journal of Immunology. 2002;168:5984–5988. doi: 10.4049/jimmunol.168.12.5984. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Zhao Q, Xing W, Feng J, Wu H, Li H, Ge M, Tian K, Li X, Zhou J, Liu B, Zhang L, Zheng Y, Han ZC. Interleukin-27 enhances the production of tumour necrosis factor-alpha and interferon-gamma by bone marrow T lymphocytes in aplastic anaemia. British Journal of Haematology. 2011;153:764–772. doi: 10.1111/j.1365-2141.2010.08431.x. [DOI] [PubMed] [Google Scholar]
- 11.Manz CY, Nissen C, Wodnar-Filipowicz A. Deficiency of CD34+ c-kit+ and CD34 + 38- hematopoietic precursors in aplastic anemia after immunosuppressive treatment. American Journal of Hematology. 1996;52:264–274. doi: 10.1002/(SICI)1096-8652(199608)52:4<264::AID-AJH5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 12.Matsui WH, Brodsky RA, Smith BD, Borowitz MJ, Jones RJ. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458–462. doi: 10.1038/sj.leu.2404119. [DOI] [PubMed] [Google Scholar]
- 13.Rizzo S, Scopes J, Elebute MO, Papadaki HA, Gordon-Smith EC, Gibson FM. Stem cell defect in aplastic anemia: reduced long term culture-initiating cells (LTC-IC) in CD34+ cells isolated from aplastic anemia patient bone marrow. Hematology Journal. 2002;3:230–236. doi: 10.1038/sj.thj.6200187. [DOI] [PubMed] [Google Scholar]
- 14.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nature Reviews Immunology. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 15.Le Blanc K, Samuelsson H, Gustafsson B, Remberger M, Sundberg B, Arvidson J, Ljungman P, Lonnies H, Nava S, Ringden O. Transplantation of mesenchymal stem cells to enhance engraftment of hematopoietic stem cells. Leukemia. 2007;21:1733–1738. doi: 10.1038/sj.leu.2404777. [DOI] [PubMed] [Google Scholar]
- 16.Rasmusson I. Immune modulation by mesenchymal stem cells. Experimental Cell Research. 2006;312:2169–2179. doi: 10.1016/j.yexcr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Armstrong MA, Li G. Mesenchymal stem cells in immunoregulation. Immunology and Cell Biology. 2006;84:413–421. doi: 10.1111/j.1440-1711.2006.01458.x. [DOI] [PubMed] [Google Scholar]
- 18.Guo Z, Zheng C, Chen Z, Gu D, Du W, Ge J, Han Z, Yang R. Fetal BM-derived mesenchymal stem cells promote the expansion of human Th17 cells, but inhibit the production of Th1 cells. European Journal of Immunology. 2009;39:2840–2849. doi: 10.1002/eji.200839070. [DOI] [PubMed] [Google Scholar]
- 19.Tatara R, Ozaki K, Kikuchi Y, Hatanaka K, Oh I, Meguro A, Matsu H, Sato K, Ozawa K. Mesenchymal stromal cells inhibit Th17 but not regulatory T-cell differentiation. Cytotherapy. 2011;13:686–694. doi: 10.3109/14653249.2010.542456. [DOI] [PubMed] [Google Scholar]
- 20.Bacigalupo A, Valle M, Podesta M, Pitto A, Zocchi E, De Flora A, Pozzi S, Luchetti S, Frassoni F, Van Lint MT, Piaggio G. T-cell suppression mediated by mesenchymal stem cells is deficient in patients with severe aplastic anemia. Experimental Hematology. 2005;33:819–827. doi: 10.1016/j.exphem.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Chao YH, Peng CT, Harn HJ, Chan CK, Wu KH. Poor potential of proliferation and differentiation in bone marrow mesenchymal stem cells derived from children with severe aplastic anemia. Annals of Hematology. 2010;89:715–723. doi: 10.1007/s00277-009-0892-6. [DOI] [PubMed] [Google Scholar]
- 22.Marsh JC, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, Keidan J, Laurie A, Martin A, Mercieca J, Killick SB, Stewart R, Yin JA. Guidelines for the diagnosis and management of aplastic anaemia. British Journal of Haematology. 2009;147:43–70. doi: 10.1111/j.1365-2141.2009.07842.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen K, Wang D, Du WT, Han ZB, Ren H, Chi Y, Yang SG, Zhu D, Bayard F, Han ZC. Human umbilical cord mesenchymal stem cells hUC-MSCs exert immunosuppressive activities through a PGE2-dependent mechanism. Clinical Immunology. 2010;135:448–458. doi: 10.1016/j.clim.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 24.Jaganathan BG, Tisato V, Vulliamy T, Dokal I, Marsh J, Dazzi F, Bonnet D. Effects of MSC co-injection on the reconstitution of aplastic anemia patient following hematopoietic stem cell transplantation. Leukemia. 2010;24:1791–1795. doi: 10.1038/leu.2010.164. [DOI] [PubMed] [Google Scholar]
- 25.Fang B, Li N, Song Y, Li J, Zhao RC, Ma Y. Cotransplantation of haploidentical mesenchymal stem cells to enhance engraftment of hematopoietic stem cells and to reduce the risk of graft failure in two children with severe aplastic anemia. Pediatric Transplantation. 2009;13:499–502. doi: 10.1111/j.1399-3046.2008.01002.x. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Wang Z, Xue M, Liu J, Yan H, Guo Z. Co-transfusion of haplo-identical hematopoietic and mesenchymal stromal cells to treat a patient with severe aplastic. Cytotherapy. 2010;12:563–565. doi: 10.3109/14653241003695059. [DOI] [PubMed] [Google Scholar]
- 27.Gieseke F, Bohringer J, Bussolari R, Dominici M, Handgretinger R, Muller I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood. 2010;116:3770–3779. doi: 10.1182/blood-2010-02-270777. [DOI] [PubMed] [Google Scholar]
- 28.Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 29.Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, Muroi K, Ozawa K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood. 2007;109:228–234. doi: 10.1182/blood-2006-02-002246. [DOI] [PubMed] [Google Scholar]
- 30.Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, Borg C, Saas P, Tiberghien P, Rouas-Freiss N, Carosella ED, Deschaseaux F. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+ CD25 high FOXP3+ regulatory T cells. Stem Cells. 2008;26:212–222. doi: 10.1634/stemcells.2007-0554. [DOI] [PubMed] [Google Scholar]