

Abstract
We previously demonstrated that, in ex vivo cultures, IFNα downregulates the expression of MHC class II (MHCII) genes in human non-professional APCs associated with pancreatic islets. IFNα has an opposing effect on MHCII expression in professional APCs. In this study, we found that the mechanism responsible for the IFNα-mediated MHCII's downregulation in human MHCII-positive non-professional antigen presenting human non-hematopoietic cell lines is the result of the negative feedback system that regulates cytokine signal transduction, which eventually inhibits promoters III and IV of CIITA gene. Because the CIITA-PIV isoform is mostly responsible for the constitutive expression of MHCII genes in non-professional APCs, we pursued and achieved the specific knockdown of CIITA-PIV mRNA in our in vitro system, obtaining a partial silencing of MHCII molecules similar to that obtained by IFNα. We believe that our results offer a new understanding of the potential significance of CIITA-PIV as a therapeutic target for interventional strategies that can manage autoimmune disease and allograft rejection with little interference on the function of professional APCs of the immune system.
Keywords: Non-professional antigen presenting cells, CIITA promoter IV, IFNα, MHCII
1. Introduction
The functioning of the immune response in infection, transplantation, cancer and autoimmunity is strictly dependent on the level of expression of MHC molecules on the surface of APCs [1]. Any degree of alterations in expression levels of MHC may influence various events downstream of TcR engagement [2,3]. On the basis of their potential for antigen presentation to T cells, APCs are frequently classified into two major categories: professional or non-professional. Professional APCs have been identified as cells of hematopoietic origin specialized in the priming of naive T cells. These cells, including dendritic cells (DCs), B lymphocytes, and cells of the monocyte/macrophage lineage, can induce both primary and memory immune responses because of their constitutive expression of MHC class II (MHCII) molecules and potent costimulatory molecules. Non-professional APCs have been identified as non-bone marrow-derived cells that do not express a complete range of costimulatory molecules. This definition applies to cell types that do not express basal levels of MHCII molecules but can be induced to express MHCII molecules in response to IFNγ [4], as well as to cell types that constitutively express MHCII molecules, such as thymic epithelial cells [5] and endothelial cells in various organs [6–8]. Spurious expression of MHCII molecules on non-bone marrow-derived cells has also been described in tumor cells from several neoplastic tissues, including glioma and melanoma [9–11]. Finally, the rejection of transplanted organs strictly depends on the MHCII expression in endothelial and epithelial cells in the transplant and in the host tissues [12].
MHCII expression is mainly regulated at the level of transcription by CIITA [4,13], a non-DNA-binding factor that exhibits a cell-type-specific, cytokine-inducible and differentiation-stage-specific expression profile [14]. In humans, four different CIITA transcription products have been identified, each of which is generated by one independent promoter (CIITA-PI, -PII, -PIII, and -PIV) and is active in an overlapping subset of cell types [15]. CIITA-PIV is generally regarded as being responsible for IFNγ-inducible expression of CIITA [16,17], but it has also been described as being constitutively active in many non-hematopoietic cells [1,6,8,10,18]. In several instances, the silencing of CIITA-PIV promoter as well as its transitory inhibition have been held responsible for failure of IFNγ to induce MHCII transcription and downregulation of basal MHCII expression [19–26]. Moreover, a study on the effects of CIITA-PIV knockout in transgenic mice demonstrated that the selective deletion of CIITA-PIV does not seem to dramatically affect MHCII expression in professional APCs while has a significant effect on MHCII expression in other APCs [27].
Interferon α (IFNα) is a type I IFN with an important role in the pathogenesis of several autoimmune diseases [28] and cancer immunotherapy [29]. In many cell types, type I IFNs block the induction of MHCII expression by IFNγ [30]. We recently demonstrated that the treatment with IFNα of human pancreatic islets ex vivo downregulates the CIITA-PIV-driven MHCII constitutive expression in non-professional APCs associated with islets [6]. In our system, the effect of IFNα-treatment on MHCII molecules was in contrast with the effect observed in professional APCs, where this cytokine upregulates the expression of MHCII genes. Other examples of discordance of IFNα-responsiveness in non-professional (melanoma cells) vs. professional APCs (immune cells) are described in human and mouse systems [31–33]. Apparently, similar to what happens with IFNγ, the biological effect of IFNα on MHCII expression is primarily mediated via the activation of the JAK/STAT pathway and the subsequent regulation of CIITA [30,34] by modulation of the promoter IV of this gene [6,35].
The aim of our study is to identify how the molecular system associated with the inhibitory function of IFNα on MHCII regulation in non-professional APCs is different from the system that mediates IFNα-induction of MHCII molecules in cells from the immune system (i.e., professional APCs). We believe that an understanding of these contrasting mechanisms can help in developing therapeutic strategies based on the tissue-specific regulation of MHCII gene expression in autoimmunity and transplantation.
The results presented in this paper provide experimental evidence supporting a simple mechanism that can account for the IFNα-mediated downregulation of MHCII in those non-professional APCs where the expression of these genes is mostly due to the constitutive activation of CIITA-PIV. We believe that this mechanism is due to the activation of the general negative feedback regulatory circuit of IFNα in the context of a constitutive weak expression of the target gene (CIITA-PIII and CIITA-PIV). On the basis of these results we formed the idea that it might be possible to mimic the IFNα-mediated downregulation of MHCII on these cells without the other (frequently unwanted) effects of this cytokine. To this purpose, we tested the effectiveness of using the RNA interference technology to selectively knock down the CIITA-PIV-driven expression of MHCII in non-professional APCs by specifically targeting CIITA-PIV mRNA.
2. Materials and methods
2.1. Reagents and cell lines
The Me10538 and M14 cell lines were both established from specimens obtained from primary tumors of melanoma patients [36,37]. The SK MEL-23 cell line was derived from a metastatic lesion of human melanoma [38]. The U87 cell line was derived from human malignant gliomas (ATCC HTB-14) [39]. All cell lines were cultured in RPMI Medium 1640 with 10% FCS (GIBCO) and 1% penicillin/streptomycin (Sigma). Recombinant human interferon gamma (IFNγ) was purchased from Peprotech, and recombinant human interferon alpha 2 b (IFNα) was purchased from PBL Biomedical Laboratories. Viability of cells after different treatments was measured through flow cytometry with 7-AAD and annexin V-FITC staining (BD Biosciences).
2.2. Flow cytometry analysis
Determination of cell surface expression of MHC class I (MHCI) and MHCII molecules was carried out by cytofluorimetric analysis using the FACS ARIA cell-sorting system and DIVA software (BD Biosciences). Direct immunofluorescence was executed using FITC mouse anti-human HLA-DR, -DQ and -ABC antibodies, along with the appropriate FITC mouse IgG isotype controls, all purchased from BD Biosciences. Staining, washing and analysis were performed as per the manufacturer's recommendations.
2.3. Measurement of specific transcripts by quantitative RT-PCR
Total RNA from cells was isolated using the RNeasy Mini Kit from QIAGEN. All Reverse Transcription reactions were performed using the QuantiTect RT Kit (QIAGEN). The accumulation of specific transcripts was measured by real-time PCR using the DNA Engine Opticon Real-Time PCR Detection System (BIORAD). The qPCR assays were performed using the quantity of cDNA obtained by reverse transcribing 10 ng of total RNA. The QuantiTect SYBR Green PCR Kit (QIAGEN) was used to perform all the reactions in the presence of 0.2 μM primers in a total volume of 25 μl. All primers used for qRT-PCR were synthesized by PRIMM, and their sequence and annealing temperature are presented in Table 1. Quantitative RT-PCR (qRT-PCR) reagent controls (reagents without any template or with 10 ng of not-reverse-transcribed RNA) were included in all the assays. Each assay was run in triplicate and the mean copy number from the three samples was used as the result of the single assay. Each assay was independently repeated at least three times and the mean copy number from the three assays was showed as the result of the experiment ± the standard error of the mean (SEM). The relative amount of specific transcripts was calculated by the comparative cycle threshold method presented by Livak and Schmittgen [40]. To correct for sample-to-sample variations in qRT-PCR efficiency and errors in sample quantitation, the level of GAPDH transcript was measured to normalize specific RNA levels. External standards were used to establish standard PCR curves for quantifying copies of transcripts that required an absolute, comparative quantitation. Fold-changes in expression were determined by dividing the normalized quantity of the gene of interest from IFNα-treated or IFNγ-treated cells by the normalized quantity of the gene of interest from untreated cells.
Table 1.
Primers for quantitative PCR used in the study
| Transcript ID | Primer 5′→ 3′ | Annealing temperature (°C) | Product size (bp) | |
|---|---|---|---|---|
| GAPDH | G-F | AACGGATTTGGTCGTATTGGGC | 60 | 216 |
| G-R | TCGCTCCTGGAAGATGGTGATG | |||
| HLA-DRA | DRA-F | GGACAAAGCCAACCTGGAAA | 60 | 120 |
| DRA-R | AGGACGTTGGGCTCTCTCAG | |||
| HLA-DQA1*01a | A1 | CGGTGGCCTGAGTTCAGCAA | 63 | 158 |
| A-R | GGAGACTTGGAAAACACTGTGACC | |||
| HLA-DQA1*03a | A3 | CTCTGTTCCGCAGATTTAGAAGA | 60 | 151 |
| A-R | GGAGACTTGGAAAACACTGTGACC | |||
| CIITA | C2TA-F | CCGACACAGACACCATCAAC | 58 | 222 |
| C2TA-R | CTTTTCTGCCCAACTTCTGC | |||
| CIITA-PIII | P3-F | CCTGGCTCCACGCCCTG | 55 | 230 |
| P-R | GAACTGGTCGCAGTTGATG | |||
| CIITA-PIV | P4-F | GAGCTGGCGGGAGGGAG | 55 | 244 |
| P-R | GAACTGGTCGCAGTTGATG | |||
| IRF1 | IRF1-F | CCTGATACCTTCTCTGATGGACTCA | 60 | 182 |
| IRF1-R | CTGTCCGGCACAACTTCCAC | |||
| IRF2 | IRF2-F | GTCTACCGAATGCTGCCCCT | 60 | 276 |
| IRF2-R | AATGTCTGGCGGATTGGTGA | |||
| SOCS1 | SOCS1-F | GCAGCTGCACGGCTCCT | 60 | 195 |
| SOCS1-R | GGAGACTGCATTGTCGGCTG | |||
| SOCS3 | SOCS3-F | GCGAAGGCTCCTTTGTGGAC | 60 | 250 |
| SOCS3-R | GGGAAACTTGCTGTGGGTGA | |||
| SMD3-R | GGCGAACTCACACAGCTCCA | |||
HLA-DQA1- allele specific primers and internal standards were used in our experiments to measure copy number of DQA1-specific cDNA. The quantity of DQA1 transcript accumulated in each sample is the results the sum of copy numbers obtained using allele-specific primers. HLA-DQA1*01-specific couple of primers has been used for samples derived from Me10538 (DQA1*01/*03), M14 (DQA1*01/*03), U87 (DQA1*01/*01). HLA-DQA1*03-specific couple of primers has been used for samples derived from Me10538 and M14.
2.4. Western blot analysis
Total levels of STAT1, STAT2, P-STAT1, and P-STAT2 molecules were measured by immunoblot in protein extracts from IFN-treated and untreated cells. Antibodies specific for STAT1 (C-terminus), P-STAT1 (pY701), STAT2, P-STAT2 (pY690), were purchased from BD Biosciences, while the anti-mouse IgG (Fc specific)-peroxidase secondary antibody and the monoclonal anti-alpha-tubulin were from Sigma-Aldrich. Lysates were prepared from cells plated at 5 × 105 cells /well in 6-well plates with 2 ml of medium. Adherent cells were removed by brief treatment with trypsin and EDTA (Sigma-Aldrich) and then combined with non-adherent cells from the same culture and washed in cold PBS prior to being resuspended in 100 μl of RIPA buffer (10 mM Tris–HCl, pH 7.6, 150 mM NaCl, 1% NP40). Protease inhibitor cocktail tablets from Roche were added at 1× concentration immediately prior to sample preparation. After 15 min of incubation at 4 °C with agitation, samples were centrifuged for 1 h at 4 °C and 12,500 rpm, and the recovered supernatant was divided into aliquots and stored at −80 °C until it was subjected to polyacrylamide gel electrophoresis. Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad Inc.) with bovine serum albumin standards, following the manufacturer's recommendations. Equal amounts of solubilized proteins (30 μg) were diluted in Laemmli sample buffer and subjected to electrophoresis on 12.5% acrylamide/bis gels. Proteins were then transferred onto PVDF membranes (Immobilon-P from Millipore) using an electroblotting system from Biometra. Membranes were prepared for immunoblotting by washing in TTBS (10 mM Tris–glycine, pH 8.0, 0.15 M NaCl, with 0.05% (w/v) Tween-20). Membranes were then blocked in TTBS plus 5% (w/v) non-fat dry milk for 1 h, followed by three 5 min washes in TTBS. Membranes were probed for specific proteins by 1 h incubations with the specific antibodies at the dilution suggested by the manufacturers. The membranes were then washed three times in TTBS and developed with the recommended dilution of the secondary antibody. After 1 h, the membranes were washed in TTBS, and the proteins on the nitrocellulose membrane were detected using the ECL Plus detection system (Healthcare/Amersham Biosciences), according to the manufacturer's protocol. Each membrane used for the immunoblot with the P-STAT-specific antibody was stripped once and reprobed with the antibody for the corresponding STAT protein. The stripping consisted of a single incubation of 20 min at 60 °C with agitation in the stripping solution (2% SDS, 62.5 mM Tris–HCl, pH 6.7, 100 mM β-mercaptoethanol), followed by six 2 min washes at RT in TTBS. Membranes were exposed to Fuji X-Ray Films (FUJIFILM Medical Systems). Enhanced chemiluminescent images of immunoblots were analyzed by scanning densitometry. Multiple exposures of each blot were used to obtain grayscale images of each chemiluminescent band and densitometric analysis was achieved by digital image analysis with NIH ImageJ 1.41 software ( rsb.info.nih.gov/ij/download) using the default settings for the background correction (rolling ball radius 50).
2.5. RNA interference-mediated gene silencing
The transfection of small interfering RNA (siRNA) for gene silencing was performed using HiPerFect Transfection Reagent (QIAGEN), as described in the Reagent Handbook. The siRNAs used in this study were all purchased from QIAGEN. Specifically, while those directed against the human CIITA and HLA-DRA (Hs_CIITA_2 HP, Hs_CIITA_3 HP, Hs_HLA-DRA_2 HP, and Hs_HLA-DRA_3 HP) were chosen from the list of predesigned siRNAs, the two siRNAs directed against CIITA-PIV (CtPIV-a and CtPIV-b) were custom-designed dXdY-overhang siRNAs, respectively, targeting the CCAGAGCTGGCGGGAGGGAGA and CAGCGGTAGGTGCAGCTCACA target DNA sequences. The AllStars Negative Control siRNA from QIAGEN was used as a negative control siRNA. The siRNA molecules selected for the experiments were those showing the highest and most specific interference potential against the intended targets (data not shown). The conditions for the experiments were chosen after we identified which siRNAs had the greatest efficiency of uptake with the lowest toxicity for the cell lines included in the study (data not shown). Transfection efficiency was monitored with Cy5-labeled negative control siRNA from QIAGEN. The toxicity of the different siRNAs at various concentrations was measured through flow cytometry with 7-AAD and annexin V-FITC staining (BD Biosciences). Based on the results of these tests, we selected one transfection protocol to be used with all cell lines. Briefly, cells were plated at 3×105 cells/well in 6-well plates and incubated overnight before transfection. A final concentration of 50 nM was used for the transfections of all siRNAs (including negative controls). Cells were collected by trypsinization at various times after the transfection for both flow cytometry analysis and RNA isolation.
2.6. Statistics
The statistical significance of differences among results between IFNα-treated or IFNγ-treated and untreated cells were evaluated by the Student's t test (Prism, GraphPad Software). p Values were determined using the one-tailed t test.
3. Results
3.1. Treatment with IFNα reduces the cell surface expression of MHCII molecules in human melanoma and glioma cell lines by downregulating CIITA-PIII and CIITA-PIV
Because treatment with IFNα downregulates constitutive MHCII expression in non-professional APCs associated with pancreatic islets [6] in an ex vivo system, we investigated whether IFNα regulation of the expression of MHCII molecules in human non-professional APCs also diverges from that of professional human APCs using an in vitro cell culture system. We selected cell lines representing bona fide non-professional APCs constitutively expressing MHCII molecules. Because constitutive expression (i.e., IFNγ-independent) of MHCII genes has been described in melanoma [41] and glioma cell lines [42] and because both non-bone marrow derived cell types possess the MHCII-mediated ability to present antigens to CD4+ T lymphocytes [43,44], we chose four already well-characterized cell lines that could be used for these experiments: three melanoma cell lines (SK MEL-23, Me10538 and M14) and one glioma cell line (U-87). All of them showed a strong IFNγ-dependent upregulation of MHCII expression (data not shown). To begin, we established the baseline level of MHCII expression in our cultures of SK MEL-23, Me10538, M14 and U-87 cells (see Fig. 1, no IFNα data). Flow cytometry analysis of SK MEL-23 confirmed the lack of expression of MHCII on the cell surface, matching the absence of HLA-DRA and HLA-DQA1 specific mRNA, as tested by quantitative RT-PCR qRT-PCR assay. Unlike SK MEL-23 cells, the other two melanoma cell lines Me10538 and M14, and the glioma cells U87 showed a significant level of HLA-DR and -DQ antigens, both at the protein and at the RNA level.
Fig. 1.
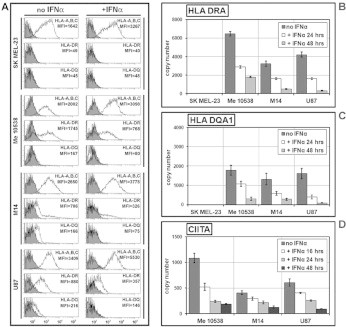
Quantitative analysis of the expression of MHCII molecules after IFNα-treatment of human tumor cell lines. (A) Cytofluorometric analysis of SK MEL-23, Me10538, M14, and U87 cells after 48 h with either no IFNα or 250U/ml of IFNα. Solid histograms represent isotype control mAb background fluorescence for each specific mAb investigated; open histograms represent specific fluorescence from cells stained with antibodies specific for MHCI (HLA-A,B,C), HLA-DRA, or HLA-DQ (as indicated next to the relative analysis). The x-axis of each histogram represents specific fluorescence on a five-decade logarithmic scale, and the y-axis represents the number of events. Mean fluorescence intensity (MFI) values are indicated next to the relative analysis. Data presented are from one representative experiment in a series of at least three. (B), (C), and (D) Quantification by RT-PCR in Me10538, M14, and U87 of HLA-DRA- (B) and HLADQA1-specific (C) mRNA after treatment for 24 and 48 h with either no IFNα or 250U/ml of IFNα, and of CIITA-specific mRNA (D) after treatment for 16, 24 and 48 h with either no IFNα or 250U/ml of IFNα. The results are expressed in copy number as the mean ± SEM of three independent experiments. In the absence of IFNα, the copy number of all transcripts did not vary significantly over the time tested.
For IFNα stimulation, cells were cultured with or without IFNα (250 U/ml) for 48 h. Because the ability of IFNα to upregulate the expression of MHCI molecules in melanoma cell lines is well established [45], the HLA-A,B,C immunophenotype was specifically measured as a positive control of the effects of this cytokine on gene expression in the cells used in the study. As shown in Fig. 1A, 48 h of incubation of each of the four cell lines with IFNα resulted in the expected significant increase in the density of MHCI molecules on the cell surface measured as MFI ( p < 0.01). On average, IFNα-treated MHCII-positive tumor cells showed a 1.5–2 fold increase of HLA-A,B,C molecules on the cell surface compared to untreated cells. In all MHCII-positive cells used as models of non-professional APCs, the flow cytometry analysis showed a consistent significant decrease of the level of MHCII molecules on IFNα-treated cells compared to untreated cells. The density of HLA-DR and HLA-DQ heterodimers on the surface of IFNα-treated cells was reduced to 40% and 50%, respectively, of the density of the corresponding molecules on untreated cells ( p < 0.05, for both), as seen in Fig. 1A. Incidentally, at no time did IFNα induce the expression of either HLA-DR or HLA-DQ on the cell surface of SK MEL-23.
To determine if the reduction in HLA-DR and HLA-DQ molecules on the surface of the IFNα-treated non-professional APC was due to diminished amount of the specific transcripts, we measured the accumulation of HLA-DRA and HLA-DQA1 in the total RNA from our set of samples after 24 and 48 h of culture either in the absence or presence of IFNα. Using a qRT-PCR assay with primers specific for the selected genes (Table 1), we observed a notable IFNα-induced decrease in the accumulation of HLA-DRA and HLA-DQA1 transcripts that was already significant ( p < 0.05) at 24 h and much more remarkable at 48 h (Fig. 1 B and C). Taken together, our data confirm that IFNα stimulation induces the downregulation of MHCII genes in different types of human non-professional APCs, in contrast with the observed IFNα-induced upregulation of these genes in professional APCs [6] and MHCI genes in both professional and non-professional APCs.
In light of our and other authors’ data on the IFNα-dependent transcriptional regulation of MHCII expression [6,35], we tested whether IFNα-induced downregulation of these genes observed in human melanoma and glioma cell lines was a direct consequence of CIITA downregulation. Using two primers annealing to sequences common to all the known transcripts generated by the four identified CIITA promoters (Table 1), we performed qRT-PCR assays on total RNA from non-treated cells and from MHCII-positive cells treated for 16, 24 or 48 h with IFNα. Our results revealed that treatment with 250 U/ml of IFNα reduced the CIITA-specific mRNA level in the cells used in our analysis (Fig. 1 D). This change was already evident after 16 h of treatment and became significant after 24 and 48 h ( p < 0.05). We next investigated whether IFNα leads to a selective decrease in transcription from specific CIITA promoters. To this purpose we used primer pairs that selectively amplify the cDNA derived from CIITA-PI, CIITA-PIII or CIITA-PIV mRNA isoforms (Table 1) to measure by qRT-PCR the number of copies of each CIITA isoforms in untreated cells and establish the pattern of usage of different CIITA promoters in Me10538, M14 and U-87 (Fig. 2 A). Our results showed that (i) CIITA-PI-specific cDNA was undetectable in all the cell lines tested, (ii) CIITA-PIV mRNA represented the greater part (70%) of the total CIITA transcripts in Me 10538 and was almost the exclusive isoform in M14 and U-87, and (iii) CIITA-PIII mRNA represented the 30% of the total CIITA transcripts in Me 10538 and was present at very low but reproducibly detectable levels (< 3% of the total amount of CIITA transcripts per cDNA sample) in both M14 and U-87.
Fig. 2.
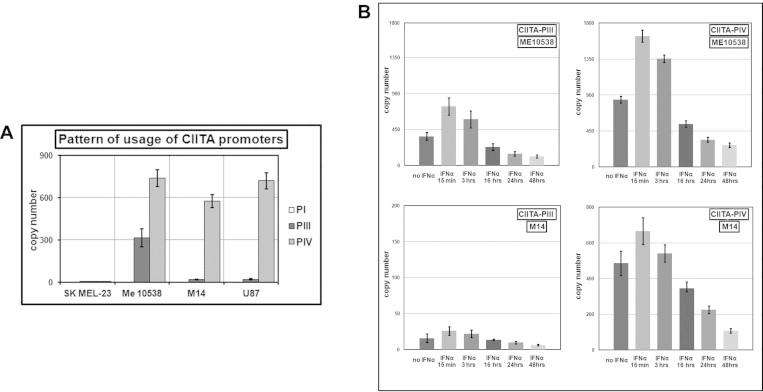
Quantitative analysis of the expression of the CIITA-PI, CIITA-PIII, and CIITA-PIV mRNA isoforms in human tumor cell lines. (A) Characterization of the pattern of usage of the different CIITA promoters in SK MEL-23, Me10538, M14, and U87 cells. (B) Measurement of CIITA-PIII and CIITA-PIV by qRT-PCR in Me10538 and M14 treated for 15 min, 3, 16, 24 and 48 h with either no IFNα or 250U/ml of IFNα. The results are expressed in copy number as the mean ± SEM of three independent experiments. In the absence of IFNα, the copy number of all transcripts did not vary significantly over the time tested.
Since it is known that CIITA-PIV as the major IFNγ responsive promoter for the induction of CIITA expression [15] and that CIITA-PIII is also regulated by IFNγ in a number of different cell types but with a level of inducibility weaker than that of CIITA-PIV [10,46], we thought that it was important to characterize the kinetics of IFNα-mediated change in level of both CIITA-PIII and CIITA-PIV transcripts. To this end, we measured the number of copies of each isoform in the cDNA samples obtained from Me10538 (as an example of a cell constitutively expressing significant levels of both molecules) and M14 (as an example of a cell expressing almost exclusively CIITA-PIV) after 15 min, 3 h, and 16 h of treatment with 250 U/ml of IFNα. The results of our qRT-PCR assays (Fig. 2 B) revealed that, shortly after stimulation (15 min), there was a rapid increase in the accumulation of CIITA-PIII and CIITA-PIV transcripts in both cell lines. This initial activation was quickly followed by a significant decline in the total copy number of both isoforms of CIITA, already evident at 3 h and more obvious at 16 h. After 48 h of culture in presence of IFNα, the expression of these molecules appeared drastically decreased in either Me10538 or M14, with the copy number of CIITA-PIII and CIITA-PIV molecules representing in treated cells respectively as little as the 27±5% and the 34±4% of the level in untreated control cells.
Together, these results demonstrate that the IFNα-mediated regulation of MHCII genes in human tumor cell lines clearly operate through the targeting of CIITA-PIII and CIITA-PIV by a mechanism inducing an early activation of both promoters prior to their eventual downregulation.
3.2. Kinetics of CIITA-PIII and CIITA-PIV expression in IFNα treated nonprofessional APCs suggest negative feedback regulation.
IFNα-induced changes in the expression of CIITA-PIII and CIITA-PIV showed similar kinetics characterized by a rapid but transient increase followed by a decrease in expression. This finding suggested a working hypothesis where IFNα-mediated MHCII's downregulation in nonprofessional APCs might be the result of the triggering of the negative feedback system that usually regulates cytokine signal transduction and that, in this instance, eventually reduce the accumulation of both CIITA isoforms to levels below those constitutive.
In humans, the signaling pathway for the transcriptional induction of IFN-stimulated genes (ISGs) involve, as matter of fact, the phosphorylation of STAT proteins by JAK kinases associated with the corresponding IFN receptor [28,47] and a negative feedback loop initiated by cytokine stimulation itself that eventually attenuates the cytokine signal transduction pathways by activating factors known as suppressors of cytokine signaling (SOCS) [48–50]. To test our hypothesis, we first examined the kinetics of IFNα-dependent STAT1 activation in our in vitro model of nonprofessional APCs. To this end, we evaluated by immunoblotting the level of the phosphorylated form of STAT1 in extracts from M14 cells cultured either in absence of IFNα or after 15 min, 3 hand 6 h of treatment with this cytokine. In every blot, each sample line was analyzed by densitometry and the signals specific to P-STAT1 were normalized to α-tubulin as a loading control and then to the corresponding signal of total STAT1. As shown in Fig. 3A, the quantity of P-STAT1 appeared almost undetectable in untreated cells, increased significantly after 15 min of IFNα stimulation and markedly diminished already after 3 h of treatment. Since functionally distinct biological responses are generated by IFNα and IFNγ through the activation of mostly overlapping signaling pathways, we believed it was important to compare the kinetics of IFNα-dependent STAT1 activation to that of IFNγ-dependent STAT1 activation in M14 cells. We measured the level of the P-STAT1 in extracts from M14 cells cultured either in absence of IFNγ or after 15 min and 6 h of treatment with this cytokine. As expected, also in this instance blots densitometry revealed a rapid and strong increase of PSTAT1 accumulation after 15 min of stimulation with IFNγ (Fig. 3 B). However, our analysis showed that IFNγ-dependent STAT1 activation in M14 was much more sustained than the one directed by IFNα, given that after 6 h of stimulation there was only the 50–60% reduction in quantity of PSTAT1 compared with the amount measured at 15 min.
Fig. 3.
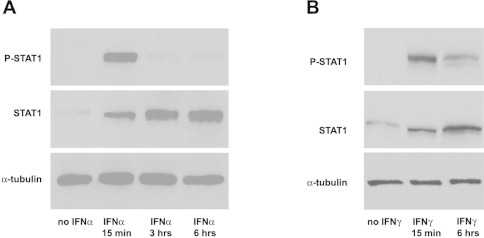
Kinetics of STAT1 activation after treatment of human tumor cell lines with IFNα and IFNγ. (A) A representative Western blot of protein extracts from M14 cultivated in the presence or absence of 250U/ml of IFNα for the indicated periods. Illustration is derived from high resolution scanning of films exposed for 30 min. Only control samples extracted from cells after 6 h of culture in the absence of IFNα are included in the picture because there was no significant difference (p > 0.5) in the intensity of chemiluminescent bands between samples after 15 min, 3 h, or 6 h of culture. (B) A representative Western blot of protein extracts from M14 cultivated in the presence or absence of 250U/ml of IFNγ for the indicated periods. Illustration is derived from high resolution scanning of films exposed for 30 min. Only control samples extracted from cells after 6 h of culture in absence of IFNγ are included in the picture because there was no significant difference (p > 0.5) in the intensity of chemiluminescent bands between samples after 15 min or 6 h of culture.
We wondered whether the kinetics of P-STAT1 activation by IFNα in MHCII-positive human tumor cells was consistent with our hypothesis that the IFNα-initiated negative feedback loop is responsible for the reduction of expression of MHCII molecules on these cells’ surface.
We examined the kinetics of expression of factors that drive IFN-induced P-STAT1 activation as well as its attenuation. We compared the effects of treatment with either IFNγ or IFNα on the expression of interferon regulatory factors 1 (IRF1) and 2 (IRF2) (because of their role in CIITA-PIV promoter activation [34,51]) and on suppressors of cytokine signaling 1 (SOCS1) and 3 (SOCS3) (because of their role in repressing IFNγ-dependent CIITA-PIV transcription [50,52]). We performed qRT-PCR assays using specific primer pairs (see Table 1) on total RNA isolated from Me10538, M14 and U-87 cells collected after 24 h of culture in absence of IFN and in presence of either IFNγ or IFNα. This interval of time was chosen to first detect any durable activation of these genes. In agreement with similar measurements performed in other systems [53], our results (showed in Fig. 4) indicated upregulation of IRF1 and IRF2 by both IFNs at the concentrations tested. Notably, IFNα relative to IFNγ induced significantly lower ( p < 0.05) accumulation of both IRF transcripts at 24 h in all cell lines tested. Measurements of the level of SOCS3 transcripts in IFNα-treated cells relative to IFNγ-treated cells revealed that IFNα induced a significantly lower ( p < 0.05) increase of SOCS3 expression in Me10538, M14 and U87. Finally, the measurement of SOCS1 transcript accumulation, which exhibited a very low basal constitutive expression in untreated cells, demonstrated a strong upregulation after 24 h of treatment with IFNγ in treated vs. untreated cells and a significantly stronger ( p < 0.05) upregulation after 24 h of treatment with IFNα.
Fig. 4.
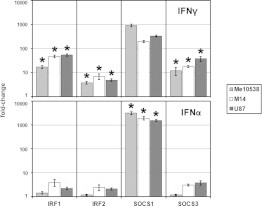
Comparison of the expression of the IRF1, IRF2, SOCS1 and SOCS3 genes following treatment of human tumor cell lines with either IFNα or IFNγ. Measurement of IRF1-, IRF2-, SOCS1-, and SOCS3-specific mRNA by qRT-PCR in Me10538, M14, and U87 after treatment for 24 h with either 250U/ml of IFNα or 250U/ml of IFNγ. The results are expressed as the mean ± SEM of fold change of the copy number of the IFN-treated sample relative to the untreated control in three independent experiments. The method of Student was used to evaluate the statistical significance of the differences between means of IFNγ- or IFNα-treated cell lines. The asterisks (*) indicate a p-value less than 0.05 using a two-tailed t test.
To obtain further information on the kinetics of IRF1 and SOCS1 activation in our system, we analyzed by qRT-PCR the accumulation of IRF1 and SOCS1 transcripts in M14 cells treated with IFNα for various time periods (15 min, 3 h, 24 h, and 48 h) and from untreated cells. As illustrated in Fig. 5, our results showed no appreciable change in the accumulation of IRF1 transcript in M14 after either 15 min or 3 h of culture in presence of IFNα in comparison with untreated cells. However, a modest increase of IRF transcript is evident in IFNα-treated cells after 24 h, becoming a little more substantial after 48 h of stimulation. In contrast with the slow and weak IFNα-driven upregulation of IRF1 gene, IFNα-driven upregulation of SOCS1 in M14 was robust within 15 min, peaking at 24 h of treatment and still active after 48 h.
Fig. 5.
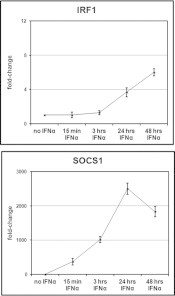
Kinetics of IRF1 and SOCS1 activation after IFNα-treatment of human tumor cell lines. Measurement of IRF1- and SOCS1-specific mRNA by qRT-PCR in M14 treated for 15 min, 3, 24 and 48 h with 250U/ml of IFNα. The results are expressed as the mean ± SEM of fold change of the copy number of the IFN-treated sample relative to the untreated control in three independent experiments.
In conclusion, we showed evidence that quantitative differences in the expression of SOCS1 and IRF1 are underlying the kinetics of a weak and transient activation of IFNα-dependent STAT1 activation as well as of CIITA-PII and CIITA–PIV activation.
3.3. MHCII downregulation can be achieved through the selective silencing of the CIITA-PIV isoform by RNA interference
The unique effects of IFNα on CIITA-PIV expression observed in these studies suggested that targeting the expression of this isoform in non-professional APCs might be an effective means of manipulation of MHCII expression without critically affecting professional APCs. We therefore tested the feasibility of utilizing CIITA-PIV specific RNA interference to downregulate MHCII expression. The effects of the gene silencing mediated by the specific interference with HLA-DRA, CIITA, and CIITA-PIV transcripts on the cell surface expression of HLA-DR and HLA-DQ molecules in Me10538, M14 and U87 cells are presented in Fig. 6. In summary, determination of cell surface expression of MHCII molecules was performed by direct immunofluorescence using anti-HLA-DR and -DQ Abs in all cells 72 h after transfection with 50 nM of the various siRNAs described in Section 2 (Material and methods). In all cell lines transfected with the control siRNA, the expression of either HLA-DR or HLA-DQ on the cell surface was not significantly modified. Transfection with siRNAs directed against the HLA-DRA sequence (indicated as HLA-DRA_2 and HLA-DRA_3 in Fig. 6) were used as positive controls of specificity. These siRNAs significantly reduced cell surface expression of HLA-DR in all cell lines tested, without significantly affecting the expression of HLA-DQ.
Fig. 6.

Quantitative analysis of the expression of MHCII molecules after siRNA-mediated specific knockdown of the CIITA-PIV isoform. Cytofluorometric analysis of Me10538, M14 and U87 cells transiently transfected with siRNAs specific for HLA-DRA, CIITA, CIITA-PIV, and a negative control siRNA. The density of HLA-DR and HLA-DQ molecules on the cell surface was measured as fluorescence from cells stained with specific antibodies and was expressed as MFI. The results are plotted as the mean ± SEM of fold change of MFI value relative to sham-transfected cells as a control in three independent experiments. The method of Student was used to evaluate the statistical significance of the differences between mean fold change of the MFI values. The asterisks (*) indicate a p-value less than 0.05 using a two-tailed t test.
Transfection with two different siRNAs (indicated as CIITA_2 and CIITA_3 in Fig. 6) each targeting all known human CIITA isoforms significantly reduced the surface expression of both HLA-DR and -DQ in all the MHCII-positive tumor cell lines. Finally, a similar efficiency of knockdown of both the MHCII molecules was achieved by the transfection of two different siRNAs specifically directed against CIITA-PIV (indicated as CtPIV-a and CtPIV-b in Fig. 6). To confirm our data on the MHCII cell-surface expression, we measured the amount of HLA-DRA, HLA-DQA1, CIITA, CIITA-PIV, and CIITA-PIII transcripts in the total RNA from our set of samples both 16 and 48 h after transfection with the different siRNAs used in our study (including the control siRNA) as well as in the sham-transfected samples (data not shown). Using an RT-PCR assay with primers specific for the selected genes, we confirmed that RNA interference with both HLA-DRA_2 and HLA-DRA_3 siRNAs exclusively affected the accumulation of DRA-specific RNA; silencing with the CIITA_2 and CIITA_3 siRNAs was effective on the accumulation of HLA-DRA-, HLA-DQA1- and all CIITA-specific RNA; and, finally, CtPIV-a and CtPIV-b siRNAs caused, in all instances, the reduction of the amount of transcripts from both HLA-DRA and HLA-DQA1 genes as well as the specific decline of only the CIITA-PIV mRNA isoform.
We conclude that by specifically knocking CIITA-PIV mRNA down in an in vitro model of non-professional APCs we achieved a level of MHCII gene downregulation reminiscent of that obtained by the IFNα treatment.
4. Discussion
Several studies show that treating melanoma patients with IFNα results in prolonged disease free survival, although the mechanism of this cytokine remains speculative [54]. Besides its effects on the host immune cells and its antiangiogenic properties, the antitumor action of IFNα treatment depends on the direct antiproliferative and proapoptotic characteristics of IFNα on the cancer cells [33]. Interestingly, MHCII-positive melanoma cells that behave as non-professional APCs exhibit a different response to IFNα-induced Jak-STAT signaling compared to immune cells (i.e., professional APCs) [33]. This fact, coupled with our data indicating the opposing effect of IFNα on MHCII expression in non-professional vs. professional APCs [6], suggested that a further definition of the mechanism responsible for the IFNα-mediated downregulation of MHCII expression in non-professional APCs was needed.
The role IFNs as modulators of MHCII gene expression has been studied in a variety of systems. It is well established that IFNγ induction of MHCII gene expression operates at the transcriptional level by upregulating the expression of the CIITA gene. Induction is accomplished mostly through the activation of CIITA-IV promoter [4,55], but also by way of less well characterized mechanisms of activation of CIITA-PI and CIITA-PIII promoters [46,56–58]. Studies with STAT2 knockout mice have demonstrated that IFNα modulates MHCII expression differently in different cell types through the CIITA-PIV promoter [35,59]. We have previously described that IFNα downregulates the PIV-driven expression of CIITA in human non-professional APCs associated with pancreatic islets cultured ex vivo [6]. The opposite, upregulatory effect on MHCII expression that we observed in professional APCs was mostly due to IFNα-mediated persistent activation of CIITA-PIII and CIITA-PI isoforms, respectively, in B lymphoblastoma cell lines and DCs, along with the moderate activation of CIITA-PIV in both cell types. Indeed, MHCII-mediated antigen presentation by professional APCs is not affected in CIITA-PIV knockout mice [27].
CIITA-PIII and PIV promoters are constitutively active in some melanoma and glioma cells [9,10,42]. The findings of the study we describe here reveal that in MHCII-positive non-hematopoietic cells, which act as non-professional APCs, IFNα inhibits promoters PIII and PIV of CIITA by a mechanism inducing an early activation of both promoters prior to their eventual downregulation. The decrease in the expression of CIITA-PIII and CIITA-PIV, in turn induces downregulation of MHCII genes, and results in a reduction of the density HLA-DR and HLA-DQ molecules to 40–50% of the density of the corresponding molecules on untreated cells. The significance of this finding, as per the conclusion of Christinck et al. and DiMolfetto et al. [2,3], is based on the notion that even subtle differences in the level of peptide/MHC density on APCs can significantly influence the nature of the immune response.
In all the systems so far characterized, IFNα induces the expression of ISGs through the activation of STAT1 and STAT2 and the consequent assembly of two different DNA-binding complexes: IFN-stimulated gene factor-3 (ISGF-3) and AAF (alpha-activated factors). ISGF-3 complexes interact with response elements in the promoters of ISGs called ISREs and are composed by P-STAT1, P-STAT2 (responsible for the unique properties of type I IFNs-dependent STAT1 activation [60]) and, interferon regulatory factor (IRF9) [61]. AAF are P-STAT1 homodimers, indicated as GAF (gamma-activated factors) when they are produced as signaling molecules for IFNγ, that interact with the GAS (gamma-interferon-activated) sites in the promoters of ISGs [62,63].
Our central interest in this study was to investigate possible qualitative or quantitative differences in the IFNα-induced mechanisms bringing about CIITA-PIII and CIITA-PIV activation in professional vs. non-professional APCs. Lymphoblastoid cell lines (LBCL) are often used as in vitro models of professional APCs. In preliminary studies, we found that LBCLs were unsuitable as a model because of their constitutive level of IFNα production [64] and resulting activation of STAT1 and STAT2 (data not shown). Of note, we did not detect any IFNα-induced P-STAT2 accumulation at different times of stimulation in all three the MHCII-positive extrahematopoietic cell lines selected for our study. Because of the absence of STAT2 activation we concluded that the GAS box present in both CIITA-PIII and CIITA-PIV promoters [65] must be the DNA cis-element targeted by the IFNα-mediated MHCII downregulation. Because the GAS box is also the DNA cis-element targeted by the IFNγ-mediated MHCII upregulation we proceeded to directly comparing the signaling pathways and the expression of genes targeting CIITA-PIII and CIITA-PIV after treatment of these cells with either IFNα or IFNγ. Based on the general pattern for the course of gene regulation by cytokine activation, we concentrated our attention on the duration of signaling and activation of IFN-triggered signal transduction pathways and their effect on the expression of CIITA-PIII and CIITA-PIV.
It is well documented that the effect of stimulation with either type of IFN on the transcription of ISGs relies on the expression and the extent of the activation of STAT proteins (reviewed in [66]). For example, IFNγ-mediated induction of CIITA gene expression does not occur at the expected levels when there is only a transient stimulation with IFNγ, possibly because of the falloff of P-STAT1 levels resulting in a short-lasting occupancy of CIITA promoter and/or a premature ending of the IRF1 synthesis [51]. When we measured the accumulation of P-STAT1 in non-professional APCs after treatment with either IFNγ or IFNα we observed that, while IFNγ directed both the rapid (i.e., within 15 min) and sustained (i.e. after 6 h) activation of STAT1, IFNα-treatment induced a rapid but transient activation of STAT1, becoming unappreciable after 3 h of stimulation.
To investigate the effect of the treatment with either IFNγ or IFNα on the transcription of known factors that have been shown to be inducible by both cytokines and are important in the regulation of CIITA-PIV [34,50-52], we measured the accumulation in Me10538, M14 and U87 of mRNA specific for IRF1, IRF2, SOCS1, and SOCS3 after treatment with both cytokines. Our results essentially indicate two principal differences in the regulation of IRF1, IRF2, SOCS1, and SOCS3 genes (Fig. 4) that underlie differences in the kinetics of IFNα- and IFNγ-dependent STAT1 activation (Fig. 3) in our in vitro model of non-professional APCs: (1) the upregulation of both IRF1 and IRF2 genes, especially IRF1, appeared weaker after 24 h of treatment with IFNα than after 24 h of treatment with IFNγ and, (2) SOCS1 appeared to be the only factor still showing a strong activation (of at least two order of magnitude greater to those of IRF1, IRF2 and SOCS3) after 24 h of IFNα stimulation. A number of studies [35,49,50,67,68] showed that differences in the expression of IRF1 and SOCS1 are associated with differences in the level and the extent of STAT1 activation and play an important role in differentiating the biological response to IFNs. In agreement with the model of Morris et al. [56], we found that the shorter duration of STAT1 activation detected in non professional APCs treated with IFNα relative to the duration of STAT1 activation in the same cells treated with IFNγ is accompanied by a relatively weak stimulation of IRF1 gene transcription.
SOCS1 inhibits or attenuates cytokine signal transduction pathways through binding to JAKs [69] as part of a negative feedback loop that is initiated by cytokine stimulation itself [48–50]. When we looked at the kinetics of SOCS1 upregulation after treatment with IFNα in MHCII-positive non-hematopoietic cells, we found that (1) in the absence of any IFN stimulation, both the activation of STAT1 and the constitutive expression of SOCS1 were almost undetectable in all the cell lines and, (2) consistent with the known pathway of SOCS1 regulation following cytokine-stimulation, there was a rapid IFNα-dependent upregulation of SOCS1, already evident after 15 min of treatment becoming very strong after 24 h of treatment. SOCS1 is known as an essential physiological regulator of IFN signaling in various experimental systems and as a potent inhibitor of IFN-induced immune activities [68]. Studies in vitro have shown that constitutive overexpression of SOCS1 inhibits IFNα- and IFNγ-mediated activation of STAT1 as well as the antiproliferative and antiviral activities of IFNs [70] and that ectopic expression of SOCS1 inhibits IFNγ-dependent CIITA-PIV transcription and subsequent MHCII protein expression by inhibiting the STAT1 phosphorylation and binding to the GAS element in CIITA-PIV [50]. Studies in vivo have shown that cells from mice lacking SOCS-1 exhibit a prolonged response to IFNγ and a dramatically increased sensitivity to the toxic effects of IFNγ [71]. We believe that the IFNα-mediated downregulation of MHCII molecules in our system and the block of the IFNγ-induction of MHCII expression driven by type I IFNs observed in different cell types are indeed two aspects of the same regulatory mechanism acting through the induction of SOCS1.
We hypothesized that in nonprofessional APCs, showing constitutive MHCII expression sustained by low levels of the CIITA-PIII and CIITA-PIV isoform, the IFNα-induced upregulation of CIITA is quite weak and transient due to the stimulation of the normal IFNα-initiated negative feedback mechanism that strongly represses these promoters. As matter of fact, the repression appears to be so strong that the amount of CIITA-PIII and CIITA-PIV molecules expressed remains below the level of the constitutive expression. Since SOCS1 action is crucial in supporting the IFNα-initiated negative feedback mechanism, our finding that IFNα-treatment of Me10538, M14 and U87 cells strongly induces the accumulation of SOCS1-specific RNA solidly support our hypothesis.
In agreement with the hypothesis articulated by other authors on the expression of MHCII proteins on human endothelial cells and their role as non-professional APC [8,72–74], we believe the reason why non-endocrine cells populating human islets express MHCII is to aid in immune surveillance of the endocrine pancreas. Several studies have demonstrated that CIITA is a target for modulation by pathogens that are controlled by CD4+ T cells [75]. There is evidence that different viruses inhibit different steps in the IFNγ signal transduction pathway leading to induction of CIITA [76], but the effect of pathogen infection on constitutive transcription of CIITA in professional APCs still requires further investigation [77]. Our study, while it does not reveal new mechanisms involved in MHCII downregulation by pathogens is original in two fundamental aspects: (i) we provide evidence demonstrating that the action of IFNα may be an intermediate step in the effect of pathogen infection on MHCII downregulation and, (ii) we identify constitutive expression of CIITA in non-professional APCs as a target of modulation by pathogens and we describe the mechanisms responsible for downregulation.
Moreover, we propose that, in agreement with the conclusions obtained by studying CIITA-PIV knockout mice [27], the targeting of this molecule in vivo would result “in a highly selective loss of MHCII-mediated antigen presentation by nonprofessional APCs.” In the current study, we proved the feasibility of using RNA interference technology to successfully and specifically knock down CIITA-PIV in melanoma and glioma cell lines, to an extent that is definitely comparable to that obtained by IFNα treatment and, therefore, biologically relevant. Indeed, our study may contribute to the design and development of manipulations of CIITA-PIV expression in vivo, resulting in a selective reduction of MHCII-mediated antigen presentation by nonprofessional APCs, without hindering expression of MHCII molecules in professional APCs.
We believe that this system may be relevant for studies directed toward the development of novel therapies of autoimmune diseases without the unwanted side effects of systemic immunosuppression. Similar interventions may also be used to treat chronic graft rejection mediated by direct allorecognition of disparate MHCII antigens expressed by nonprofessional APCs (e.g. endothelial cells) [12].
Acknowledgements
Authors wish to thank Dr. Piergiuseppe De Berardinis for the gift of valuable reagents and Drs. Fabiola Souza and Donatella Malanga to have participated to an early phase of this project. FACS analysis data was generated in the Core Flow Cytometry Facility of the Institute of Genetics and Biophysics Adriano Buzzati-Traverso.
References
- 1.Janeway C.A., Bottomly K., Babich J., Conrad P., Conzen S., Jones B., Kaye J., Katz M., McVay L., Murphy D.B., Tite J. Quantitative variation in la antigen expression plays a central role in immune regulation. Immunology Today. 1984;5:99–105. doi: 10.1016/0167-5699(84)90043-4. [DOI] [PubMed] [Google Scholar]
- 2.Christinck E.R., Luscher M.A., Barber B.H., Williams D.B. Peptide binding to class I MHC on living cells and quantitation of complexes required for CTL lysis. Nature. 1991;352:67–70. doi: 10.1038/352067a0. [DOI] [PubMed] [Google Scholar]
- 3.DiMolfetto L., Neal H.A., Wu A., Reilly C., Lo D. The density of the class II MHC T cell receptor ligand influences IFN-Î3/IL-4 ratios in immune responsesin vivo. Cellular Immunology. 1998;183:70–79. doi: 10.1006/cimm.1997.1231. [DOI] [PubMed] [Google Scholar]
- 4.Steimle V., Siegrist C.A., Mottet A., Lisowska-Grospierre B., Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265:106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 5.Viret C., Janeway Jr C.A. MHC and T cell development. Reviews in Immunogenetics. 1999;1:91–104. [PubMed] [Google Scholar]
- 6.Harris P.E., Malanga D., Liu Z., Hardy M.A., Souza F., Del Pozzo G., Winchester R.J., Maffei A. Effect of interferon alpha on MHC class II gene expression in ex vivo human islet tissue. Biochimica et Biophysica Acta. 2006;1762:627–635. doi: 10.1016/j.bbadis.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Kwak B., Mulhaupt F., Myit S., Mach F. Statins as a newly recognized type of immunomodulator. Nature Medicine. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 8.Muczynski K.A., Ekle D.M., Coder D.M., Anderson S.K. Normal human kidney HLA-DR-expressing renal microvascular endothelial cells: characterization, isolation, and regulation of MHC class II expression. Journal of the American Society of Nephrology. 2003;14:1336–1348. doi: 10.1097/01.asn.0000061778.08085.9f. [DOI] [PubMed] [Google Scholar]
- 9.Deffrennes V., Vedrenne J., Stolzenberg M.-C., Piskurich J., Barbieri G., Ting J.P., Charron D., Alcaide-Loridan C. Constitutive expression of MHC class II genes in melanoma cell lines results from the transcription of class II transactivator abnormally initiated from its B Cell-specific promoter. Journal of Immunology. 2001;167:98–106. doi: 10.4049/jimmunol.167.1.98. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin B.L., Xi H., Tejiram R., Eason D.D., Ghosh N., Wright K.L., Nagarajan U., Boss J.M., Blanck G. Varying functions of specific major histocompatibility class II transactivator promoter III and IV elements in melanoma cell lines. Cell Growth and Differentiation. 2001;12:327–335. [PubMed] [Google Scholar]
- 11.Guardiola J., Maffei A. Control of MHC class II gene expression in autoimmune, infectious, and neoplastic diseases. Critical Reviews in Immunology. 1993;13:247–268. [PubMed] [Google Scholar]
- 12.Game D.S., Lechler R.I. Pathways of allorecognition: implications for transplantation tolerance. Transplant Immunology. 2002;10:101–108. doi: 10.1016/s0966-3274(02)00055-2. [DOI] [PubMed] [Google Scholar]
- 13.Accolla R.S., Jotterand-Bellomo M., Scarpellino L., Maffei A., Carra G., Guardiola J. aIr-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. Journal of Experimental Medicine. 1986;164:369–374. doi: 10.1084/jem.164.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ting J.P., Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl.):S21–S33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 15.Muhlethaler-Mottet A., Otten L.A., Steimle V., Mach B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO Journal. 1997;16:2851–2860. doi: 10.1093/emboj/16.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masternak K., Reith W. Promoter-specific functions of CIITA and the MHC class II enhanceosome in transcriptional activation. EMBO Journal. 2002;21:1379–1388. doi: 10.1093/emboj/21.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piskurich J.F., Gilbert C.A., Ashley B.D., Zhao M., Chen H., Wu J., Bolick S.C., Wright K.L. Expression of the MHC class II transactivator (CIITA) type IV promoter in B lymphocytes and regulation by IFN-gamma. Molecular Immunology. 2006;43:519–528. doi: 10.1016/j.molimm.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honey K., Rudensky A. The pIV-otal class II transactivator promoter regulates major histocompatibility complex class II expression in the thymus. Journal of Experimental Medicine. 2001;194:F15–F18. doi: 10.1084/jem.194.4.f15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H., Gilbert C.A., Hudson J.A., Bolick S.C., Wright K.L., Piskurich J.F. Positive regulatory domain I-binding factor 1 mediates repression of the MHC class II transactivator (CIITA) type IV promoter. Molecular Immunology. 2007;44:1461–1470. doi: 10.1016/j.molimm.2006.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Y.J., Han Y., Lu H.T., Nguyen V., Qin H., Howe P.H., Hocevar B.A., Boss J.M., Ransohoff R.M., Benveniste E.N. TGF-beta suppresses IFN-gamma induction of class II MHC gene expression by inhibiting class II transactivator messenger RNA expression. Journal of Immunology. 1997;158:2065–2075. [PubMed] [Google Scholar]
- 21.Morris A.C., Spangler W.E., Boss J.M. Methylation of class II trans-activator promoter IV:a novel mechanism of MHC class II gene control. Journal of Immunology. 2000;164:4143–4149. doi: 10.4049/jimmunol.164.8.4143. [DOI] [PubMed] [Google Scholar]
- 22.Nandan D., Reiner N.E. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. Journal of Immunology. 1997;158:1095–1101. [PubMed] [Google Scholar]
- 23.Piskurich J.F., Wang Y., Linhoff M.W., White L.C., Ting J.P. Identification of distinct regions of 5’ flanking DNA that mediate constitutive, IFN-gamma, STAT1, and TGF-beta-regulated expression of the class II transactivator gene. Journal of Immunology. 1998;160:233–240. [PubMed] [Google Scholar]
- 24.Radosevich M., Jager M., Ono S.J. Inhibition of MHC class II gene expression in uveal melanoma cells is due to methylation of the CIITA gene or an upstream activator. Experimental and Molecular Pathology. 2007;82:68–76. doi: 10.1016/j.yexmp.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Satoh A., Toyota M., Ikeda H., Morimoto Y., Akino K., Mita H., Suzuki H., Sasaki Y., Kanaseki T., Takamura Y., Soejima H., Urano T., Yanagihara K., Endo T., Hinoda Y., Fujita M., Hosokawa M., Sato N., Tokino T., Imai K. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-[gamma]-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene. 2004;23:8876–8886. doi: 10.1038/sj.onc.1208144. [DOI] [PubMed] [Google Scholar]
- 26.van den Elsen P.J., van der Stoep N., Yazawa T. Class II transactivator (CIITA) deficiency in tumor cells: complicated mechanisms or not. American Journal of Pathology. 2003;163:373–376. doi: 10.1016/s0002-9440(10)63664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldburger J.-M., Suter T., Fontana A., Acha-Orbea H., Reith W. Selective abrogation of major histocompatibility complex class II expression on extrahematopoietic cells in mice lacking promoter IV of the class II transactivator gene. Journal of Experimental Medicine. 2001;194:393–406. doi: 10.1084/jem.194.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theofilopoulos A.N., Baccala R., Beutler B., Kono D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annual Review of Immunology. 2005;23:307–335. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 29.Belardelli F., Ferrantini M., Proietti E., Kirkwood J.M. Interferon-alpha in tumor immunity and immunotherapy. Cytokineand Growth Factor Reviews. 2002;13:119–134. doi: 10.1016/s1359-6101(01)00022-3. [DOI] [PubMed] [Google Scholar]
- 30.Lu H.T., Riley J.L., Babcock G.T., Huston M., Stark G.R., Boss J.M., Ransohoff R.M. Interferon (IFN) beta acts downstream of IFN-gamma-induced class II transactivator messenger RNA accumulation to block major histocompatibility complex class II gene expression and requires the 48-kD DNA-binding protein, ISGF3-gamma. Journal of Experimental Medicine. 1995;182:1517–1525. doi: 10.1084/jem.182.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang S., Bucana C.D., Van Arsdall M., Fidler I.J. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 2002;21:2504–2512. doi: 10.1038/sj.onc.1205341. [DOI] [PubMed] [Google Scholar]
- 32.Lesinski G.B., Anghelina M., Zimmerer J., Bakalakos T., Badgwell B., Parihar R., Hu Y., Becknell B., Abood G., Chaudhury A.R., Magro C., Durbin J., Carson 3rd W.E. The antitumor effects of IFN-alpha are abrogated in a STAT1-deficient mouse. Journal of Clinical Investigation. 2003;112:170–180. doi: 10.1172/JCI16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesinski G.B., Trefry J., Brasdovich M., Kondadasula S.V., Sackey K., Zimmerer J.M., Chaudhury A.R., Yu L., Zhang X., Crespin T.R., Walker M.J., Carson III W.E. Melanoma cells exhibit variable signal transducer and activator of transcription 1 phosphorylation and a reduced response to IFN-{alpha} compared with immune effector cells. Clinical Cancer Research. 2007;13:5010–5019. doi: 10.1158/1078-0432.CCR-06-3092. [DOI] [PubMed] [Google Scholar]
- 34.Xi H., Blanck G. The IRF-2 DNA binding domain facilitates the activation of the class II transactivator (CIITA) type IV promoter by IRF-1. Molecular Immunology. 2003;39:677–684. doi: 10.1016/s0161-5890(02)00214-6. [DOI] [PubMed] [Google Scholar]
- 35.Zhao W., Cha E.N., Lee C., Park C.Y., Schindler C. Stat2-dependent regulation of MHC class II expression. Journal of Immunology. 2007;179:463–471. doi: 10.4049/jimmunol.179.1.463. [DOI] [PubMed] [Google Scholar]
- 36.Doneda L., Ginelli E., Agresti A., Larizza L. In situ hybridization analysis of interstitial C-heterochromatin in marker chromosomes of two human melanomas. Cancer Research. 1989;49:433–438. [PubMed] [Google Scholar]
- 37.Tsuchida T., Ravindranath M.H., Saxton R.E., Irie R.F. Gangliosides of human melanoma: altered expression in vivo and in vitro. Cancer Research. 1987;47:1278–1281. [PubMed] [Google Scholar]
- 38.Carey T.E., Takahashi T., Resnick L.A., Oettgen H.F., Old L.J. Cell surface antigens of human malignant melanoma: mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1976;73:3278–3282. doi: 10.1073/pnas.73.9.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponten J., Macintyre E.H. Long term culture of normal and neoplastic human glia. Acta Pathologica Microbiologica Scandinavica. 1968;74:465–486. doi: 10.1111/j.1699-0463.1968.tb03502.x. [DOI] [PubMed] [Google Scholar]
- 40.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Winchester R.J., Wang C.-Y., Gibofsky A., Kunkel H.G., Lloyd K.O., Old L.J. Expression of Ia-like antigens on cultured human malignant melanoma cell lines. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:6235–6239. doi: 10.1073/pnas.75.12.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takamura Y., Ikeda H., Kanaseki T., Toyota M., Tokino T., Imai K., Houkin K., Sato N. Regulation of MHC class II expression in glioma cells by class II transactivator (CIITA) Glia. 2004;45:392–405. doi: 10.1002/glia.10343. [DOI] [PubMed] [Google Scholar]
- 43.Brady M.S., Lee F., Petrie H., Eckels D.D., Lee J.S. CD4(+) T cells kill HLA-class-II-antigen-positive melanoma cells presenting peptide in vitro. Cancer Immunology, Immunotherapy. 2000;48:621–626. doi: 10.1007/s002620050010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soos J.M., Krieger J.I., Stuve O., King C.L., Patarroyo J.C., Aldape K., Wosik K., Slavin A.J., Nelson P.A., Antel J.P., Zamvil S.S. Malignant glioma cells use MHC class II transactivator (CIITA) promoters III and IV to direct IFN-gamma-inducible CIITA expression and can function as nonprofessional antigen presenting cells in endocytic processing and CD4(+) T-cell activation. Glia. 2001;36:391–405. doi: 10.1002/glia.1125. [DOI] [PubMed] [Google Scholar]
- 45.Basham T.Y., Bourgeade M.F., Creasey A.A., Merigan T.C. Interferon increases HLA synthesis in melanoma cells: interferon-resistant and -sensitive cell lines. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:3265–3269. doi: 10.1073/pnas.79.10.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piskurich J.F., Linhoff M.W., Wang Y., Ting J.P. Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Molecular and Cellular Biology. 1999;19:431–440. doi: 10.1128/mcb.19.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darnell Jr J.E. Studies of IFN-induced transcriptional activation uncover the Jak-Stat pathway. Journal of Interferon, Cytokine and Mediator Research. 1998;18:549–554. doi: 10.1089/jir.1998.18.549. [DOI] [PubMed] [Google Scholar]
- 48.Alexander W.S. Suppressors of cytokine signalling (SOCS) in the immune system. Nature Reviews Immunology. 2002;2:410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 49.Fenner J.E., Starr R., Cornish A.L., Zhang J.G., Metcalf D., Schreiber R.D., Sheehan K., Hilton D.J., Alexander W.S., Hertzog P.J. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nature Immunology. 2006;7:33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- 50.O’Keefe G.M., Nguyen V.T., Ping Tang L., Benveniste E.N. IFN-{{gamma}} regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. Journal of Immunology. 2001;166:2260–2269. doi: 10.4049/jimmunol.166.4.2260. [DOI] [PubMed] [Google Scholar]
- 51.Eason D.D., Blanck G. High level class II trans-activator induction does not occur with transient activation of the IFN-{{gamma}} signaling pathway. Journal of Immunology. 2001;166:1041–1048. doi: 10.4049/jimmunol.166.2.1041. [DOI] [PubMed] [Google Scholar]
- 52.Kim H., Suh J.M., Hwang E.S., Kim D.W., Chung H.K., Song J.H., Hwang J.H., Park K.C., Ro H.K., Jo E.-K., Chang J.-S., Lee T.-H., Lee M.-S., Kohn L.D., Shong M. Thyrotropin-mediated repression of class II trans-activator expression in thyroid cells: involvement of STAT3 and suppressor of cytokine signaling. Journal of Immunology. 2003;171:616–627. doi: 10.4049/jimmunol.171.2.616. [DOI] [PubMed] [Google Scholar]
- 53.Lehtonen A., Matikainen S., Julkunen I. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. Journal of Immunology. 1997;159:794–803. [PubMed] [Google Scholar]
- 54.Agarwala S. Improving survival in patients with high-risk and metastatic melanoma: immunotherapy leads the way. American Journal of Clinical Dermatology. 2003;4:333–346. doi: 10.2165/00128071-200304050-00004. [DOI] [PubMed] [Google Scholar]
- 55.Chang C.H., Fontes J.D., Peterlin M., Flavell R.A. Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. Journal of Experimental Medicine. 1994;180:1367–1374. doi: 10.1084/jem.180.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris A.C., Beresford G.W., Mooney M.R., Boss J.M. Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Molecular and Cellular Biology. 2002;22:4781–4791. doi: 10.1128/MCB.22.13.4781-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pai R.K., Askew D., Boom W.H., Harding C.V. Regulation of class II MHC expression in APCs: roles of types I, III, and IV class II transactivator. Journal of Immunology. 2002;169:1326–1333. doi: 10.4049/jimmunol.169.3.1326. [DOI] [PubMed] [Google Scholar]
- 58.Wright K.L., Ting J.P. Epigenetic regulation of MHC-II and CIITA genes. Trends inImmunology. 2006;27:405–412. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 59.Park C., Li S., Cha E., Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- 60.Leung S., Qureshi S., Kerr I., Darnell Jr J., Stark G. Role of STAT2 in the alpha interferon signaling pathway. Molecular and Cellular Biology. 1995;15:1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nature Reviews Immunology. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 62.Decker T., Lew D.J., Mirkovitch J., Darnell Jr J.E. Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO Journal. 1991;10:927–932. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shuai K., Schindler C., Prezioso V.R., Darnell Jr J.E. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science. 1992;258:1808–1812. doi: 10.1126/science.1281555. [DOI] [PubMed] [Google Scholar]
- 64.Adolf G.R., Pieler C., Maurer-Fogy I. Constitutive production of interferon-alpha 2 by a human B-lymphoblastoid cell line. Journal of Interferon Research. 1992;12:275–280. doi: 10.1089/jir.1992.12.275. [DOI] [PubMed] [Google Scholar]
- 65.van der Stoep N, Quinten E., Alblas G., Plancke A., van Eggermond M.C.J.A., Holling T.M., van den Elsen P.J. Constitutive and IFN[gamma]-induced activation of MHC2TA promoter type III in human melanoma cell lines is governed by separate regulatory elements within the PIII upstream regulatory region. Molecular Immunology. 2007;44:2036–2046. doi: 10.1016/j.molimm.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 66.Wesoly J., Szweykowska-Kulinska Z., Bluyssen H.A. STAT activation and differential complex formation dictate selectivity of interferon responses. Acta Biochimica Polonica. 2007;54:27–38. [PubMed] [Google Scholar]
- 67.Giroux M., Schmidt M., Descoteaux A. IFN-{gamma}-induced MHC class II expression: transactivation of class II transactivator promoter IV by IFN regulatory factor-1 is regulated by protein kinase C-{alpha} Journal ofImmunology. 2003;171:4187–4194. doi: 10.4049/jimmunol.171.8.4187. [DOI] [PubMed] [Google Scholar]
- 68.Zimmerer J.M., Lesinski G.B., Kondadasula S.V., Karpa V.I., Lehman A., RayChaudhury A., Becknell B., Carson III W.E. IFN-{alpha}-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. Journal ofImmunology. 2007;178:4832–4845. doi: 10.4049/jimmunol.178.8.4832. [DOI] [PubMed] [Google Scholar]
- 69.Kile B.T., Schulman B.A., Alexander W.S., Nicola N.A., Martin H.M.E., Hilton D.J. The SOCS box: a tale of destruction and degradation. Trends in Biochemical Sciences. 2002;27:235–241. doi: 10.1016/s0968-0004(02)02085-6. [DOI] [PubMed] [Google Scholar]
- 70.Song M.M., Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. Journal of Biological Chemistry. 1998;273:35056–35062. doi: 10.1074/jbc.273.52.35056. [DOI] [PubMed] [Google Scholar]
- 71.Brysha M., Zhang J.-G., Bertolino P., Corbin J.E., Alexander W.S., Nicola N.A., Hilton D.J., Starr R. Suppressor of cytokine signaling-1 attenuates the duration of interferon Î3 signal transduction in vitro and in vivo. Journal of Biological Chemistry. 2001;276:22086–22089. doi: 10.1074/jbc.M102737200. [DOI] [PubMed] [Google Scholar]
- 72.Al-Lamki R.S., Bradley J.R., Pober J.S. Endothelial cells in allograft rejection. Transplantation. 2008;86:1340–1348. doi: 10.1097/TP.0b013e3181891d8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nickoloff B.J., Turka L.A. Immunological functions of non-professional antigen-presenting cells: new insights from studies of T-cell interactions with keratinocytes. Immunology Today. 1994;15:464–469. doi: 10.1016/0167-5699(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 74.Taflin C, Charron D, Glotz D, Mooney N. Immunological function of the endothelial cell within the setting of organ transplantation. Immunology Letters 2011: 139, 1–6. [DOI] [PubMed]
- 75.Accolla R.S., De Lerma Barbaro A., Mazza S., Casoli C., De Maria A., Tosi G. The MHC class II transactivator: prey and hunter in infectious diseases. Trends inImmunology. 2001;22:560–563. doi: 10.1016/s1471-4906(01)02003-8. [DOI] [PubMed] [Google Scholar]
- 76.Hegde N.R., Chevalier M.S., Johnson D.C. Viral inhibition of MHC class II antigen presentation. Trends in Immunology. 2003;24:278–285. doi: 10.1016/s1471-4906(03)00099-1. [DOI] [PubMed] [Google Scholar]
- 77.Lee A.W., Wang N., Hornell T.M., Harding J.J., Deshpande C., Hertel L., Lacaille V., Pashine A., Macaubas C., Mocarski E.S., Mellins E.D. Human cytomegalovirus decreases constitutive transcription of MHC class II genes in mature Langerhans cells by reducing CIITA transcript levels. Molecular Immunology 2011;48: 1160–1167. [DOI] [PMC free article] [PubMed]