

Abstract
In prokaryotes, the first three enzymes in pyrimidine biosynthesis, carbamoyl phosphate synthetase (CPS), aspartate transcarbamoylase (ATC), and dihydroorotase (DHO), are commonly expressed separately and either function independently (Escherichia coli) or associate into multifunctional complexes (Aquifex aeolicus). In mammals the enzymes are expressed as a single polypeptide chain (CAD) in the order CPS-DHO-ATC and associate into a hexamer. This study presents the three-dimensional structure of the noncovalent hexamer of DHO and ATC from the hyperthermophile A. aeolicus at 2.3 Å resolution. It is the first structure of any multienzyme complex in pyrimidine biosynthesis and is a possible model for the core of mammalian CAD. The structure has citrate, a near isosteric analogue of carbamoyl aspartate, bound to the active sites of both enzymes. Three active site loops that are intrinsically disordered in the free, inactive DHO are ordered in the complex. The reorganization also changes the peptide bond between Asp153, a ligand of the single zinc atom in DHO, and Gly154, to the rare cis conformation. In the crystal structure, six DHO and six ATC chains form a hollow dodecamer, in which the 12 active sites face an internal reaction chamber that is approximately 60 Å in diameter and connected to the cytosol by narrow tunnels. The entrances and the interior of the chamber are both electropositive, which suggests that the architecture of this nanoreactor modifies the kinetics of the bisynthase, not only by steric channeling but also by preferential escape of the product, dihydroorotase, which is less negatively charged than its precursors, carbamoyl phosphate, aspartate, or carbamoyl aspartate.
In mammals, the first three steps in pyrimidine biosynthesis are performed by a single protein, called CAD1, which combines carbamoyl phosphate synthetase (CPS), aspartate transcarbamoylase (ATC), and dihydroorotase (DHO) activities on a single polypeptide chain that self-associates into a hexamer of 1.5 MDa (reviewed in ref 1). The intermediates are channeled between the active sites of each component of the CAD complex (2–4). Other eukaryotes use CAD, or polypeptide chains containing two of the enzymes and a separately expressed version of the third enzyme, or three separately expressed enzymes. In prokaryotes, the three enzymes are commonly expressed separately and function either independently (e.g., Escherichia coli) or associate into multifunctional oligomers (e.g., Aquifex aeolicus). While resting cells can be sustained by pyrimidine salvage, the rate of de novo pyrimidine biosynthesis in proliferating cells must be accelerated to meet the increased demand for DNA precursors. Consequently, this pathway is a potential therapeutic target for cancer and for pathogenic organisms that lack the salvage pathway and depend entirely upon de novo pyrimidine synthesis for growth, such as the four plasmodia responsible for malaria.
Until this report, none of the multifunctional complexes has been visualized in three dimensions, although separate structures are available for CPS, ATC, and DHO from one, five, and four organisms, respectively (Supporting Information Table S1). Among these examples, A. aeolicus is notable because (a) it is one of the oldest species of eubacteria and has a closer phylogenetic connection to eukaryotes than other eubacteria, (b) it is a hyperthermophile that flourishes at 95 °C, and so its proteins are particularly stable, and (c) its separately expressed enzymes form a weak complex between CPS and ATC (5) and a strong complex between DHO and ATC (6). In this paper, we present the crystal structure of the DHO-ATC complex (DAC) from A. aeolicus at 2.3 Å resolution with adventitious citrate bound in the active site.
Aspartate transcarbamoylase from A. aeolicus (AaeATC) contains 291 amino acids (34 kDa) and, like all ATC enzymes, is functional only as a tightly bound trimer in which each active site is formed by residues from two subunits. The trimer alone is active, but two trimers readily associate with six AaeDHO chains to form a heteromeric dodecamer with enhanced thermal stability for both components (5, 6). This partnership identifies AaeATC as a type A1 aspartate transcarbamoylase (7, 8), in which the DHO subunits are active (e.g., A. aeolicus (6), Thermus aquaticus (9)). In type A2 complexes, the DHO domain is inactive and only fulfills a structural role (e.g., Pseudomonas aeruginosa (10)). Type B enzymes also form a heteromeric dodecamer, but six allosteric regulatory chains replace the DHO chains (e.g., E. coli (11)). Type C enzymes function as unregulated free trimers (e.g., Bacillus subtilis (12)); AaeATC functions as a type C enzyme when dihydroorotase is absent.
EcoATC, a B-type enzyme whose structure and kinetics have been analyzed in great detail (13–15), is the archetype for aspartate transcarbamoylases. The holoenzyme contains two trimers of catalytic chains linked into a dodecamer by three regulatory dimers. Each catalytic chain contains two domains joined by a hinge segment. Each active site has six proximate residues from the two domains of one monomer (Thr53, Thr55, His134, Arg167, Arg229, Gln231) and two residues from the N-terminal domain of an adjacent monomer (Ser80*, Lys84*). The native enzyme follows an ordered bi-bi mechanism: (a) carbamoyl phosphate binds first and potentiates the binding of aspartate, (b) the two domains and the loop containing Ser80 and Lys84 from the adjacent catalytic chain in the trimer close down on the two substrates, (c) nucleophilic attack by the amino group of aspartate on the polarized carbonyl carbon of carbamoyl phosphate forms a tetrahedral intermediate, (d) the intermediate collapses into carbamoyl aspartate with the release of free phosphate, and (e) carbamoyl aspartate departs followed by the phosphate ion. Histidine 134, which forms a hydrogen bond with the carbonyl oxygen of carbamoyl phosphate, is the “catalytic” residue in that it polarizes the carbonyl carbon and stabilizes the tetrahedral transition state. The regulatory chains, which are absent in type A aspartate transcarbamoylases like the A. aeolicus enzyme, increase or decrease the activity in response to allosteric effectors by altering the affinity for the substrate, aspartate.
AaeATC and EcoATC share 30% of their sequence. Although the sequence of AaeATC has eight residues (Thr46, Thr48, Ser72, Lys75, His126, Arg159, Arg213, Gln215, Ser72*, and Lys75*) that correspond to those in the active site of EcoATC, there are significant kinetic differences between the two species. At 37 °C, AaeATC turns over more slowly than EcoATC, but its apparent specificity constant (kcat/Km) is 20 times higher than that of the E. coli enzyme due to lower Km values for aspartate and carbamoyl phosphate. At 75 °C, the Km value for aspartate is unchanged at 1.1 mM, the Km value for carbamoyl phosphate increases 2-fold to 1.3 mM, and kcat increases almost 5-fold to 42 s−1.
Dihydroorotase also has interesting metabolic and kinetic properties. Flux analysis of the E. coli metabolism has recently identified DHO as a unique thermodynamic bottleneck for growth due to its having a free energy change near zero under physiological conditions (16). Based upon their sequences (17) and structures (Supporting Information Table S1), dihydroorotases segregate into two families. Type I DHO proteins, such as A. aeolicus DHO and the DHO domain in CAD, are older in evolutionary terms and larger, with molecular masses around 45 kDa. Type II DHO proteins, such as E. coli DHO, are shorter at both the N-and C-terminus, with molecular masses around 38 kDa. Consonant with these relationships, the DHO domain of human CAD is more similar to AaeDHO (26% similarity) than to EcoDHO (13% similarity). Presently, the available sequences of more than 1000 dihydroorotase/pyrC proteins (UniProt) are distributed fairly evenly between the two types of DHO. As a group, dihydroorotases belong to the superfamily of amidohydrolases, which primarily catalyze the hydrolysis of amide or ester functional groups at carbon or phosphorus centers (18). All members of the superfamily have a catalytic domain containing an eight-stranded α/β-barrel, with the catalytic residues at the C-terminal end of the barrel. The larger hydrolases, such as DHO type I, also have a composite domain formed by extensions of the sequence at the N- and C-termini of the α/β-barrel domain.
DHO catalyzes the reversible conversion of carbamoyl aspartate to dihydroorotate with an equilibrium constant of 1 at pH 6.2 (19). Because the reaction is shifted in the direction of the substrate, carbamoyl aspartate, at higher, more physiological pH values, dihydroorotase works “uphill” in metabolic terms. The active site of EcoDHO contains two zinc atoms (20, 21) that are approximately 3.6 Å apart; the more buried metal is designated Znα. A carboxylated lysine residue (PDB code: KCX) at position 102 bridges the two zinc atoms on their interior side while a water molecule bridges them on the exterior side. The latter is proposed to function as a nucleophilic hydroxide ion in the hydrolysis of dihydroorotate and as a departing water molecule in the cyclization of carbamoyl aspartate (19). An aspartate residue near Znα participates as a general acid or base as required by the reaction direction. In the active site of AaeDHO only Znα is clearly visible. It displays a distorted square-pyramidal geometry with His-61, His-63, Asp-153, Asp-305, and a water molecule as ligands (22).
AaeDHO is notable for being inactive unless bound to its cognate ATC, the preceding enzyme in the pathway (6). Structures of the inactive enzyme have identified three possible factors responsible for the latent state: (a) access to the active site is blocked by a bond between Znα and Cys181, which is not present in EcoDHO, (b) the enzyme lacks the second zinc atom present in EcoDHO, and (c) three loops around the periphery of the active site are intrinsically disordered (22). The relative significance of these three factors is addressed by the structure presented here, a stoichiometric complex of Aquifex DHO bound to ATC in an hollow, hetero dodecamer, in which all 12 enzyme active sites face a capacious aqueous cavity with limited access to the exterior. This striking architecture facilitates allosteric communication between the active sites and possibly increases the net production of dihydroorotate, which is thermodynamically unfavored at physiological pH, by electrostatically regulated diffusion.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were purchased from Sigma. E. coli strains DH5α and BL21(DE3) were from Invitrogen.
Preparation of the DHO-ATC Complex
The genes for A. aeolicus ATC and DHO were inserted into the BamHI/NcoI sites of plasmid pRSETC, which adds a 6×His tag fused to the N-terminal end, and expressed separately in E. coli BL21(DE3) cells (5, 6). The cells were cotransformed with the helper plasmid pSJS1240, which overexpresses tRNA species for three codons that are common in A. aeolicus but rare in E. coli. The cells were grown at 37 °C for 18 h in 500 mL of LB medium containing 100 μg/mL ampicillin and 50 μg/mL spectinomycin, harvested by centrifuge at 4000 rpm for 30 min, disrupted by sonication, and centrifuged at 12000 rpm for 30 min at 4 °C. The supernatant was applied to a column containing nickel ProBond resin (Invitrogen) equilibrated with 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, and 1 mM tris(2-carboxyethyl)phosphine. The proteins were eluted with increasing concentrations of imidazole (50–400 mM) in the same buffer.
Solutions of each protein were transferred to a “sieving buffer” of 1 mM DTT, 500 mM NaCl, and 50 mM Tris-HCl at pH 7.5 using spin columns. The solutions were adjusted to 10 mg/mL and mixed to form the complex with DHO in molar excess by 5–10% based upon Lowry measurements of the separate concentrations. The DHO-ATC dodecamer was isolated promptly by gel filtration on a Sephacryl S-300 column (170 × 1.5 cm) and equilibrated with sieving buffer at room temperature. The presence of the DTT was essential for reproducible results. The dodecamer peak, at approximately 400–500 kDa, was pooled and concentrated for crystallization using centrifugal filters (Amicon Ultra) with a MWCO of 10 kDa.
Crystallization of the DHO-ATC Complex
Before crystallization, the protein was buffer exchanged into 10 mM Hepes, 10 mM sodium citrate, and 2 mM DTT, pH 7.5, at a final protein concentration of 3.0 mg/mL. All crystals were grown at room temperature using the hanging-drop, vapor-diffusion technique: 3–6 μL of DAC solution was mixed with 1 μL of reservoir solution (25% ethylene glycol) followed by 10% of the drop volume (v/v) of 100 mM barium chloride as a crystallization additive. After mixing, the final pH was approximately 5.5. The crystals grew without barium chloride at higher concentrations of ethylene glycol but diffracted poorly. Prism-like crystals appeared within 2–7 days; the best diffracting crystals grew to 0.2 × 0.2 × 0.1 mm and were often accompanied by small needle-like crystals.
Phasing and Refinement of the DHO-ATC Complex
The X-ray diffraction data were collected in 360 frames at the Advanced Photon Source (IMCA-CAT) using a wavelength of 1.0 Å and a rotation of ½ deg per frame. The data were integrated with HKL2000 (HKL Research, Inc.) and truncated at the resolution (2.3 Å) where the average intensity was approximately twice the estimated error (Table 1). Using data between 47 and 4.0 Å, molecular replacement solutions were found by MOLREP (23) using aspartate transcarbamoylase from either E. coli (1j79A.pdb) or Pyrococcus abyssi (1ml4A.pdb) and free AaeDHO (1xrtA.pdb) as models. The electron density map was refit with ARP/wARP (24) to remove bias and refined with Refmac5 (25). The final ratio of Rfree/R was 0.204/0.164 (Table 1). Initially, the structure was refined to almost the same statistics with only water molecules in the active site. However, when the water molecules were ignored, the graphics program COOT (26) identified electron density in the active sites of DHO and ATC, respectively, as the largest and second largest “blobs” of unfit density in the crystal structure and readily fit the density in an automatic search with either the carbamoyl aspartate structure from EcoDHO (1j79B.pdb, PDB ligand: NCD) or the idealized citrate structure (PDB ligand: FLC) from the ligand database at the Protein Data Bank.
Table 1.
Crystal Structure Statistics
| statistics | DAC |
|---|---|
| PDB code | 3D6N |
| crystals | 1 |
| space group | H32 |
| unit cell dimension | |
| a (Å) | 158.43 |
| b (Å) | 158.43 |
| c (Å) | 233.55 |
| α (deg) | 90 |
| β (deg) | 90 |
| γ (deg) | 120 |
| chains per asymmetric unit | 1 DHO, 1 ATC |
| Data Collection | |
| resolution (Å) | 47.4–2.3 (2.38–2.30) |
| reflections used | 47614 |
| completeness (%) | 99.9 (99.4) |
| average (I/σ) | 23.8 (3.0) |
| Rmerge (%) | 8.0 (54.6) |
| Refinement | |
| R factor | 0.164 |
| Rfree (%) | 0.204 |
| average B factor (Å2) | 34.2 |
| atoms in asymmetric unit | 6082 |
| no. of water molecules | 423 |
| rms bond (Å) | 0.020 |
| rms angle (deg) | 1.811 |
| rms chiral | 0.136 |
Structure Analysis
The crystal structure has one DHO molecule (dho1, chain A) and one ATC molecule (atc1, chain B) in its crystallographic asymmetric unit. The biological unit was constructed from these two chains using the identity transformation [x, y, z] and the next five symmetry transformations of space group 155 in SPDBV, which generate a dodecamer with 32 symmetry. The subunit numbers and chain letters were assigned as follows: [−y, x − y, z] = dho2 (C), atc2 (D); [y − x, −x, z] = dho3 (E), atc3 (F); [y, x, −z] = dho4 (G), atc4 (H); [x − y, −y, −z] = dho5 (I), atc5 (J); [−x, y −x, −z] = dho6 (K), atc6 (L). This convention numbers the subunits in opposite order around the C3 axis in the two ATC trimers, consonant with the D3 symmetry of the dodecamer. The structure was imaged with PyMOL (27); its channels were measured and imaged with the PyMOL plug-in, CAVER, using a grid size of 0.8 Å (28). The electrostatic surfaces were calculated with APBS (29) at neutral pH and zero ionic strength using the AMBER force field (30) and visualized with VMD (31). The dielectric constant was set to 2.0 for the protein and 78.0 for the solvent. The mean radius of gyration for each ligand was calculated with GROMACS (32) after 5 ns of molecular dynamics at 298 K in a cube of water molecules containing sufficient sodium ions to give an overall net charge of zero. The relative energies of the cis and trans conformations of the D153–G154 peptide bond in free DHO and in DAC were calculated with the protein preparation wizard in the Schrodinger suite of programs using the 2005 optimized potential for liquid simulations (33). The trans conformation was generated in DAC by superposing the structure of free DHO (1xrf.pdb) onto DAC, extracting the minimal peptide around the trans-peptide bond with differences greater than 0.1 Å from DAC (T151–D159), and inserting it into the DAC structure. The reverse procedure was used to insert the cis conformation of D153–G154 from DAC into the structure of free DHO.
RESULTS
Overall Structure
The crystal structure of DAC at 2.3 Å resolution has been refined to a final R value of 0.164 and a final Rfree value of 0.204 (Table 1). The Rfree/R ratio is within the expected range for a structure with a ratio of total atoms to reflections of 0.13 (Table 1) at this resolution (34). The average temperature factors for the ATC, DHO, and solvent atoms are 26.8, 38.6, and 41.3 Å2, respectively. The estimated error calculated by Refmac5 is 0.1 Å based upon maximum likelihood and 0.2 Å based upon the R factor. The asymmetric unit contains a heterodimer of one DHO chain of 422 residues, one ATC chain of 291 residues, and two citrate molecules accompanied by 423 water molecules (Figure 1). No density was observed for the hexahistidine affinity tag on the N-terminus of either the ATC chain (31 residues) or DHO chain (34 residues). Both N-termini are on the outside surface of the dodecamer. The main chain stereochemistry of the structure is average for structures at this resolution. According to PROCHECK, 88.0% of the residues lie in the most favored regions of the Ramachandran plot, 12% in the additionally allowed region, and 0.3% of the residues in the generously allowed region. Serine 72 (B) is the only non-glycine residue in the forbidden region, and it lies in a mobile loop, as evidenced by its high B factor (78 Å), weak main chain density (1.1σ), and absent side chain density.
Figure 1.
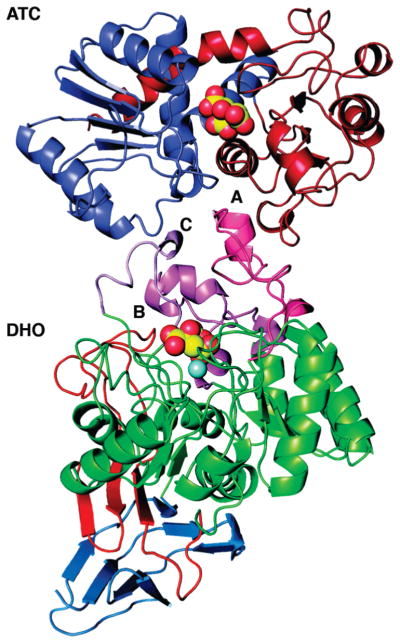
The heterodimer of AaeDHO and AaeATC. The single DHO chain (bottom) in the asymmetric unit is shown as a ribbon diagram in six color-coded segments: the N-terminal component of the composite domain (blue), (αβ)8 TIM barrel domain (green) interspersed with three neo-folded loops, A, B, and C (pink, magenta, and violet), and the C-terminal component of the composite domain (red). The zinc atom is depicted as a sphere (cyan). The single ATC chain (top) is shown as a ribbon diagram divided into the amino-terminal carbamoyl phosphate binding domain (blue) and the carboxyl-terminal aspartate-binding domain (red). The citrate molecules bound in the active sites of DHO (FLC424) and ATC (FLC292) are shown as CPK models with yellow carbon atoms.
DHO Structure
The main chain of the DHO subunit in the asymmetric unit has continuous density for all residues at the 1.0σ level and for all residues except G9, K32, D35, and E42 at the 1.5σ level. The side chains of the residues in the TIM-barrel domain (residues 56–365) have electron density for all atoms at the 1.5σ level except for A334, L337, I343, and I349, which only have complete density at the 1.0σ level. The active site in the TIM-barrel domain has strong electron density for a single zinc atom and a citrate molecule (FLC424) (Figure 2A,B). The composite domain is less well defined: 18 residues in its N-terminal half (residues 1–55) and one residue in its C-terminal half (366–422) have one or more side chain atoms without electron density at the 1.0σ level. A notable feature of this DHO structure is the well-defined electron density for three loops in the TIM-barrel domain that were disordered in the structures of free, inactive DHO (Figure 3). The DHO chain has three classical cis-peptide bonds (D66–P67, N97–P98, N278–P279) in the TIM-barrel domain and one rare cis-peptide bond (D153–G154) at the active site (Figure 4).
Figure 2.
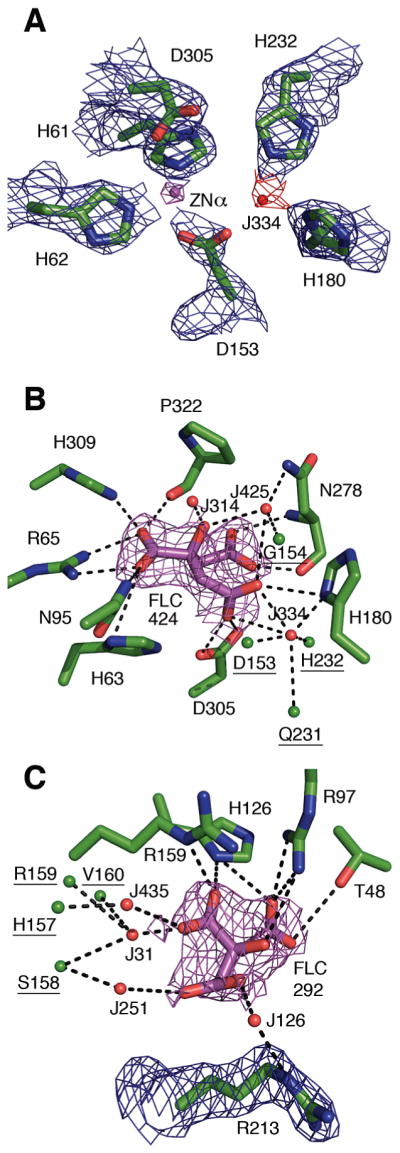
Electron density for residues and citrate (FLC) in the active sites of DHO and ATC. (A) Electron density for the residues (blue mesh), α-zinc atom (Zn423; magenta mesh), and water molecule (J334; red mesh) in the active site of AaeDHO (DAC chain A) contoured at 1σ, 5σ, and 0.75σ, respectively. The citrate molecule, FLC424, and main chain atoms were omitted to reduce clutter. (B) The 2Fo – Fc electron density for FLC424 (magenta) in the active site of DHO is shown at the 1σ level. The eight residues with 11 potential hydrogen bonds (dashed lines) to FLC424 are shown as green sticks. For clarity, uninvolved main chain atoms and Zn423 have been omitted. Water molecules (J numbers) with potential hydrogen bonds to FLC424 are shown as red spheres. The underlined residues, which bind FLC424 indirectly through a hydrogen bond to a water molecule, are represented only by the atom involved in the bond (green spheres) to avoid obscuring the central interactions. (C) The 2Fo – Fc electron density for FLC292 (magenta) and R213 (blue) in the active site of AaeATC (DAC chain B) is plotted at the 1σ contour level. An isolated blob of density (not shown) between Arg213 and FLC292 was interpreted as water molecule J126 but could also be a minor alternate orientation of Arg213. The side chains of the four active site residues that form seven hydrogen bonds with FLC292 are shown as green sticks. FLC292 is also hydrogen-bonded to four water molecules (red spheres), which are anchored by a total of six hydrogen bonds to R213 and four other residues (underlined) whose depiction is limited to the specific atom involved in the H-bond (green spheres).
Figure 3.
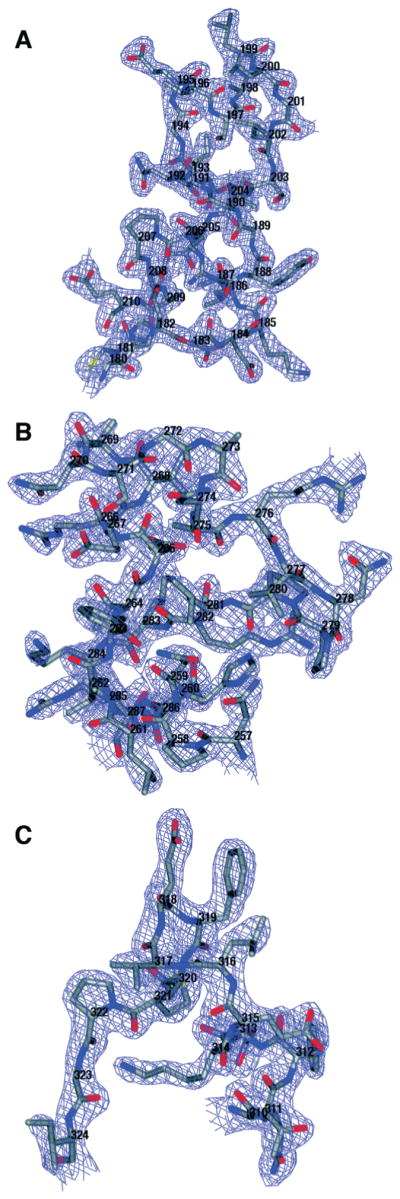
Three neo-ordered loops at the active site of DHO. Electron density contoured at 1.5σ is shown for loop A (H180–E210), loop B (N257–R287), and loop C (Q310–I324). In one structure of free AaeDHO (1xrf.pdb), the three loops, A, B, and C, have no electron density for 28/31, 23/31, and 9/15 residues, respectively. In the other structure of free DHO (1xrt.pdb), which has part of loop A bound in the active site, the respective fractions of disordered residues are 20/31, 22/31, and 11/16.
Figure 4.
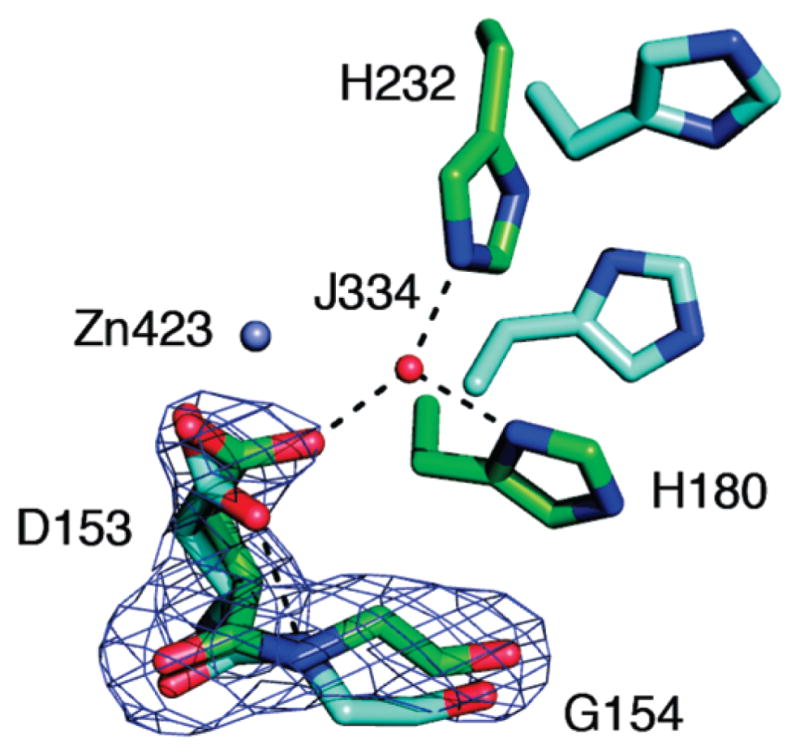
D153–G154 cis-peptide bond in AaeDHO. The “omit” difference electron density (Fo – Fc) is shown contoured at 4σ for the cis-peptide bond between Asp153 and Gly154 in the DHO chain (green) of DAC. The corresponding residues with a trans-peptide bond from the structure of free AaeDHO (1xrtA.pdb) are shown superposed onto DAC (cyan). The electron density was calculated after refinement with Refmac that did not include the two residues. Hydrogen bonds to OD2 of Asp153 are indicated with dashed lines.
ATC Structure
The single chain of ATC in the asymmetric unit is well ordered, as evidenced by its average temperature factor of 26.8 Å2, and has continuous electron density at 1.5σ for the main chain of all 291 residues and for the side chains of all residues except E70–K75, E143, K145, and K192, which have electron density only at the 1σ level. It has two cis-peptide bonds at F103–P104 and H249–P250. Like DHO, there is one citrate molecule (FLC292) bound to the active site (Figure 2C). Superposition by SSM onto catalytic chains of unliganded ATC (T-state) gave rmsd values of 1.97 Å for EcoATC (6at1.pdb), 1.74 Å for SacATC (1pg5.pdb), and 2.06 Å for MprATC (2be7.pdb). Superpositions onto the catalytic chains with PALA in their active sites (R-state) gave rmsd values of 1.85 Å for EcoATC (1d09.pdb) and 1.70 Å for PabATC (1ml4.pdb).
Crystal Packing and the Biological Unit
The unit cell of this crystal structure in space group 155 (hexagonal setting H32) contains 18 copies of the dho1:atc1 heterodimer. The Protein Interfaces, Surfaces, and Assemblies service (PISA) at the European Bioinformatics Institute (35) identifies seven unique interfaces within and between the heterodimers in the unit cell (Figure 5). The quantitative parameters for the seven interfaces and the color assigned to each interface, from purple to gray, based upon the relative surface area, are listed in Table 2. Interfaces 1 (purple), 2 (blue + cyan), and 3 (green), which have the greatest buried surface areas, the largest number of stabilizing interactions, and consequently the highest complexation significance scores (CSS), define a self-consistent dodecamer (Figure 6) that also includes three minor chain contacts, namely, the yellow, orange, and red interfaces (Figure 5). The seventh interface (gray), also with a negligible CSS score, represents a crystal packing contact between separate dodecamers. The six DHO chains in the dodecamer form a hollow torus (Figure 6B and table of contents figure) that is capped top and bottom by two trimers of ATC. At its simplest, the dodecamer is a hollow oblate ellipsoid with a distance of approximately 120 Å between the exterior surfaces of the ATC trimers at the poles and a distance of approximately 160 Å between the protruding tips of the DHO composite domains at the equator.
Figure 5.

Chain contact surfaces. The residues involved in the seven interfaces (Table 2) for the heterodimer of dho1 (light green surface) and atc1 (light blue surface) are shown in surface representation for dho1:dho4 (purple), atc1:atc2 (cyan), atc1:atc3 (blue), dho1:atc1 (dark green), atc1:dho4 (yellow), dho1:dho5 (orange), atc1:dho2 (red), and atc1:atc* (gray). The chain surfaces are viewed (A) from inside the chamber with the citrate ligands visible (magenta), (B) from outside the chamber (rotated 180° vertically relative to the inside view), (C) separately for atc1 and dho1 viewed vertically onto the contact surface (dark green) between the two chains, and (D) as a schematic diagram showing the seven primary interfaces and their visible symmetry equivalents for the dodecamer, the ATC trimer, and the DHO hexamer.
Table 2.
Chain Interfaces Calculated by PISAa
| interface | chain 1:chain 2 | residues | area (Å2) | H-bonds | salt bonds | CSSc | color |
|---|---|---|---|---|---|---|---|
| 1 | dho1(A):dho4(G) | 30:30 | 1082 | 10 | 26 | 0.969 | purple |
| 2 | atc1(B):atc2(D) | 26:26 | 946 | 19 | 8 | 1.00 | blue |
| (2) | atc1(B):atc3(F) | 26:26 | 946 | 19 | 8 | 1.00 | cyan |
| 3 | atc1(B):dho1(A) | 23:22 | 788 | 9 | 7 | 0.427 | green |
| 4 | atc1(B):dho4(G) | 20:22 | 768 | 12 | 0 | 0.191 | yellow |
| 5 | dho1(A):dho5(I) | 12:12 | 389 | 2 | 0 | 0.059 | orange |
| 6 | dho1(A):atc3(F) | 4:5 | 51 | 2 | 0 | 0.018 | red |
| 7b | atc1(B):atc* | 11:11 | 114 | 10 | 10 | 0.00 | gray |
The Protein Interfaces, Surfaces, and Assemblies service (PISA) at the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) identified six protein interfaces in the DAC dodecamer and one packing contact in the crystal. The six interfaces within the dodecamer are ranked in decreasing order of their buried surface area, which is an approximate measure of the strength of the interaction, and correspondingly colored in descending order of spectral energy (59). The crystal packing contact is colored gray.
Crystal packing contact between two dodecamers in the crystal.
Complexation significance score.
Figure 6.

The DAC dodecamer. (A) Top view: The “top” three ATC chains, labeled A1–A3, are depicted with solid surfaces in shades of blue. The three DHO chains attached to the “top” trimer, labeled D1–D3, are shown in shades of green. The three DHO chains associated with the bottom ATC trimer (not visible) are shown in shades of yellow. The dodecamer was generated by crystal symmetry from the DHO (chain A, “D1”) and ATC (chain B, “A1”) in the asymmetric unit. (B) Topless view: The top ATC trimer was removed to reveal the enclosed chamber and the bottom ATC trimer (A4–A6) in shades of red. The pore in the center of the bottom ATC trimer is highlighted in white. (C) Side view: The dodecamer in panel A was rotated 90° around the horizontal 2-fold axis. (D) Side pore: The side pore formed by atc1 (A1), atc3 (A3), dho1 (D1), and dho5 (D5) is highlighted in white.
Each ATC subunit interacts strongly with four other chains in the dodecamer—two other ATC chains in the same trimer (blue, cyan interfaces) and two DHO chains in the central “torus” (green, yellow interfaces)—and very slightly with a third DHO subunit (red interface). There is no contact with the ATC trimer on the opposite side of the dodecamer. Specifically, for subunit atc1 these partners are atc2 and atc3 and dho1, dho4, and dho2, respectively (Figure 5, Table 2). Each DHO chain interacts with three other chains, its ATC partner in the heterodimer (atc1) and two DHO chains attached to the opposite ATC trimer (dho4, dho5). It does not contact the other two DHO chains attached to its own ATC trimer. Finally, each ATC chain in the dodecamer has a crystal packing contact (gray interface) with an ATC chain in an adjacent dodecamer.
DISCUSSION
Although the structure of the dihydroorotase–aspartate transcarbamoylase complex (DAC) was originally solved by molecular replacement, using models of uncomplexed proteins, the bias was minimized by completely refitting the initial electron density map (R factor = 0.32) objectively with ARP/wARP. The only significant sections that required manual fitting were the cis-peptide bonds in both chains and the composite domain (residues 1–55 and 366–422) in the DHO chain (22). Unlike the crystal structures of A. aeolicus DHO alone, the composite domain, which projects out from the larger TIM-barrel domain of DHO, has no packing contacts in this DAC crystal structure to stabilize it (Figure 5) and consequently exhibits higher temperature factors. The N-terminal methionine residues of the DHO and ATC chains, to which disordered affinity tags are attached, are both on the outside surface of the dodecamer.
The PDB validation server identified seven close contacts in the DAC crystal structure that involved at least one water molecule and one close contact between the side chains of Asp179 and Asp217 in the dho1 chain. All of the water molecules in this structure had significant electron density in the 2Fo – Fc map (≥1σ) so we attribute these close contacts to water molecules that have fractional occupancies and multiple positions that alternate with their partner in the “clash”. However, the close contact between the two aspartic acid side chains merits closer inspection, because it should not occur due to the strong repulsion between two negatively charged side chains. Both side chains have excellent density with no alternate positions (not shown), so we conclude that the two side chains are correctly placed and that they share a strong hydrogen bond with one of the side chains being protonated at pH 5.5. This analysis is supported by the pKa values of 9.2 and 2.7 for Asp179 and Asp217, respectively, that are predicted by the PropKa program (36).
Oligomeric Structure
The heteromeric dimer in the asymmetric unit is not the biological unit. Separately, in solution, AaeDHO is a monomer and AaeATC is a very stable trimer that can only be dissociated with denaturants (6, 37). When mixed at room temperature and neutral pH, they form a major complex of 480 kDa and a minor complex of 240 kDa. Both complexes contain ATC and DHO in equimolar amounts. These two assemblies have been interpreted as a dodecamer formed by six DHO molecules attached to two trimers of ATC chains, and a hexamer containing three DHO molecules attached to a single ATC trimer (6). This crystallographic study provides a plausible model for the two biological units. Application of the crystal symmetry to the heterodimer in the asymmetric unit generates a hollow dodecamer with rotational 32 symmetry that contains six of the seven interfaces in the unit cell (Table 2, Figure 5). Presumably, each ATC subunit in a trimer (blue/cyan interface) binds a DHO subunit (green interface) to form a hexamer. These three DHO subunits do not interact with each other. However, each DHO chain can bind to the gap between two DHO subunits of a second hexamer through a strong interaction with one DHO chain (purple interface), a weaker interaction with the other DHO chain (orange interface), and an intermediate interaction with an ATC subunit in the second hexamer (yellow interface). The six ATC chains and six DHO chains in the dodecamer are arranged with all of their active sites facing inward toward the large internal chamber, which has restricted access to the cytosol (Figure 6).
The blue/cyan interface between each pair of catalytic chains in the ATC trimer has the maximum possible value of 1.0 for its complexation significance score (CSS), which is a function of the buried surface area and stabilizing interactions. This agrees with the observation that all known ATCs contain these trimers, which can only be separated by denaturation. However, PISI ranks the purple interface between the dho1/dho4 dimer (and its two symmetry equivalents) as being stronger than the green interface between the dho1/atc1 heterodimer (and its two symmetry equivalents) contrary to the observation that the dodecamer is in equilibrium with hexamers (6). In other words, the dho1/ dho4 interfaces (purple) and the weaker atc1/dho4 interfaces (yellow) separate before the dho1/atc1 (green) interface.
The geometrical analysis of the various interfaces by PISI is exact, but the stability predictions are only a rough guide, as exemplified by the two crystal structures of free AaeDHO. PISI correctly predicts the biological unit to be a monomer from 1xrf.pdb, which has no packing interface with a CSS score above 0.014, but the program incorrectly predicts a stable dimer from 1xrt.pdb, with a CSS value of 1.0 for its purple interface. Interestingly, the orange interface is also used in the crystal packing of the free DHO structures, but it pairs with a novel surface, not with itself, as it does in the DAC structure. The green interface is not used in the crystal structures of free AaeDHO because it contains seven residues of loop A, which is disordered in these structures. However, residues in EcoATC, which correspond to “green” residues in atc1 in DAC, are used to bind the regulatory chains. In the crystal structure of EcoDHO (1xge.pdb), which is a stable dimer in solution, the interface between chain A and chain B has a CSS score of only 0.149, which implies it plays an “auxiliary role in complexation”. The interface, which lies on a 2-fold axis, comprises 33 residues from each DHO chain; 4 residues occupy “purple” positions, 9 occupy “green” positions, and the remaining 20 residues do not correspond to any of the interface positions in the DAC structure. Comparing the three mobile loops further highlights the differences. In DAC, loop A contributes 8 residues to the purple interface with dho4 and 12 residues to the green interface with atc1 while loop B and loop C contribute only to the green interface. This distribution of contacts for loop A explains why the purple interface is weaker than the green one even though it has a greater buried surface area. With free AaeDHO, loop A is disordered, because the ATC subunit is missing. This disorder destabilizes the purple interface and makes AaeDHO a monomer in solution. In EcoDHO, all three loops are part of its purple interface between the dimers. Only three residues from EcoDHO loop A (H144, D146, and R155) and one from loop B (R193) participate in other crystal packing interfaces.
The crystal structure of free DHO from Thermus thermophilus (TthDHO) is an interesting contrast. The crystal structure has a single significant interface with a CSS of 0.599, which suggests it is a dimer in solution. It has not been characterized beyond the crystal structure but the DHO from T. aquaticus (TaqDHO), which is a close homologue (96% identical), is active as a dimer in solution and forms a complex with its cognate ATC that has a molecular mass of 480 kDa (38). TthDHO has 32 residues in its loop A, one more than AaeDHO, but the loop is stable and completely visualized in the crystal structure, unlike free AaeDHO. Like DAC, loop A in TthDHO contributes to both the purple dimer interface (11 residues) and to the equivalent of the green interface (4 residues), which is a crystal packing contact with a third TthDHO molecule. Loop B contributes only to the green interface while loop C participates in none of the interfaces in the TthDHO structure.
The interior reaction chamber and its channels to the exterior enclose 4336 water molecules when the dodecamer is immersed in a virtual cube of water generated by GROMACS. This value implies an accessible volume of approximately 1.3 × 105 Å3 for the aqueous internal chamber and an effective radius of approximately 30 Å. Access to the exterior occurs through two types of channels, two “C3” channels (Figure 7) that pass through the two ATC trimers that cap the dodecamer (Figures 6B and 8) and six “C2” channels (Figures 6D and 9) on the sides of the dodecamer, which are related to each other by 2-fold axes. Each C2 channel is divided into a major and minor channel (Figures 6D and 9A), by a limited subunit contact (Figure 9B), which forms the red interface (Table 2). The minimal radius or “gorge” radius determined by CAVER is 2.2 Å for the C3 channels, 1.8 Å for the minor C2 channel, and 2.5 Å for the major C2 channel (Figure 7). Due to the errors introduced by using a finite grid in the algorithm, these gorge radii could be underestimated by as much as 0.4 Å (28). The mean radii of gyration for carbamoyl phosphate, carbamoyl aspartate, and dihydroorotate are 1.8, 2.4, and 2.1 Å respectively. These numbers suggest that carbamoyl aspartate and, possibly, dihydroorotate are effectively excluded from the C3 channels and the minor C2 side channel. However, these calculations are based upon a static structure of DAC. The size of the side pores could dramatically increase due to local mobility and perhaps the rupture of the tenuous red interface, which divides the C2 pores, by an allosteric conformational change. In this vein, the DHO residues comprising the red interface (T311, F312) are part of loop C, which is disordered in the absence of ATC.
Figure 7.

Channel profiles. The radii of the C3 channel (black), the C2 minor channel (blue), and the C2 major side channel (red) are plotted as a function of the linear distance from the midpoint of each channel. Negative distances are toward the inside of the dodecamer; positive numbers are toward the outside. The midpoint of the C3 channel, which has a constriction or “gorge” at either end and resembles a “W”, was defined as the point between the two constrictions with the largest radius; the midpoints of the side channels, which have only one constriction and resemble a “V”, were set at the position of the single constriction.
Figure 8.

CAVER tracing of the C3 channel through the ATC trimer. The major tunnels to the outside (red) and to the inside (blue), calculated by the program CAVER, are shown as the sum of the largest spheres that can be fit at each 0.8 Å spacing along the channel. The calculation was started at the interior point in the channel with the widest radius (3.6 Å), which was at the center of the triangle defined by the carbonyl carbon of Y64 in the three ATC chains. Subunits atc2 (cyan) and atc3 (magenta) are shown as lines; subunit atc1 was removed to expose the tunnel. The minimum radius toward the exterior (2.5 Å) occurs between the side chains of the three R57 residues. The minimum radius toward the interior (2.2 Å) occurs between the side chains of the three E70 residues. In the view shown here, Y64 (atc3) and E70 (atc3) are beneath the opaque channel.
Figure 9.

The C2 side pore. (Panel A) The side pore formed by subunits dho1 (green lines), atc1 (cyan lines), atc3 (magenta lines), and dho5 (yellow lines) is depicted with red spheres marking the center of the minor channel (upper left) and the major channel (lower right) as defined by CAVER. Three residues (white rods) around each center sphere define the constriction point or “gorge” for the channel. A total of seven residues (yellow labels), two residues from subunit dho1 (green rods) and five residues from subunit atc3 (magenta rods), divide the pore into the two channels. (Panel B) The electron density (2Fo – Fc) for the seven residues that separate the two channels is shown at the 1σ level. The hydrogen bonds between T311 (dho1) and H261 (atc3) and between K238 (atc3) and D260 (atc3) are depicted with yellow dashes. The close contact between T312 (dho1) and N256 (atc3) is indicated with white dashes, and the ionic interaction between K238 (atc3) and E262 (atc3) is shown with red dashes.
DHO Active Site
Earlier structures of free AaeDHO, which is inactive, revealed four deficits in the active site compared to that of E. coli DHO (EcoDHO), which is active as a free enzyme (21): (a) the second catalytic zinc atom, Znβ, was absent; (b) two histidine residues (H180, H232) that bind Znβ were rotated out of the active site in AaeDHO; (c) in one AaeDHO structure (1xrt.pdb), Cys181 was rotated into the active site and covalently linked to the single zinc atom, Znα, and (d) three surface loops, containing 31, 31, and 15 residues, respectively (Figures 1 and 3 and Supporting Information Figure S1), were disordered in the crystal structures of free AaeDHO.
The DAC structure, which incorporates active AaeDHO, shows that complexation with AaeATC has corrected three of these deficits. The three loops are well ordered (Figure 3), Cys181 is out of the active site and the two histidines have moved in (Figure 2A), but Znβ has no significant electron density compared to Znα, which is present at the 5σ contour level (Figure 2A). Reflecting this fact, we have fit the weak electron density at the position that corresponds to Znβ in EcoDHO, which is present at 0.75σ but not at the 1σ contour level, with a water molecule (J334). We conclude that active AaeDHO requires only one zinc atom. Corroborating evidence includes chemical analyses that detect only 1 mol of zinc (6), an anomalous scattering difference density map that contained peaks for Znα and sulfur atoms but not for Znβ (not shown) and a mutant DAC with His180 and His232 replaced with alanine that was still active (R. Fernando and H. Evans, unpublished).
Besides the zinc content, AaeDHO also differs notably from EcoDHO in having Asp153 instead of a carboxylated lysine (KCX102) as a zinc ligand (22). This difference probably reflects the greatly reduced concentration of carbon dioxide experienced by hyperthermophiles relative to mesophiles. The carboxylation reaction is not catalyzed and depends upon adventitious carbon dioxide in the cytosol. A surprise in this study is the discovery that the peptide bond between Asp153 and Gly154 has changed from the trans conformation in inactive, free DHO to the cis conformation in DAC (Figure 4), a conformation that is seen in less than 0.05% of peptide bonds not involving proline. This transformation, which occurred spontaneously during refinement, is very robust. If forced back to the trans conformation, the peptide bond returns to the cis conformation within a few cycles of crystallographic refinement. Independent energy minimizations with the Schrodinger suite (see Experimental Procedures section) gave a free energy difference for a trans to cis conformational change of D153–G154 that was negative for the DAC structure but positive for the structure of free DHO, in agreement with the conformations actually observed in the crystal structures. Moreover, the cis conformation in DAC was 3.5 times more stable than the trans conformation in free DHO, relative to their alternative conformations.
The exact role this conformational change plays in the activation of the DHO domain in DAC remains to be determined; however, one local effect of the trans to cis transformation is the change of the hydrogen bond partner of Asp153 OD2 from the N of Gly154 to the water molecule (J334) that occupies the putative Znβ site formed by the two histidines that rotate into the active site during activation (Figure 4). In this DAC structure, J334 forms a hydrogen bond to the citrate ligand (Figure 2B). Although the functional significance of the Asp-Gly diad is unclear, it is a strongly conserved structural feature. The SAM-T2 server found more that 400 homologues to the AaeDHO sequence that possessed the Asp-Gly diad within one residue position of the diad in AaeDHO (39).
ATC Active Site
Experiments by many investigators on EcoATC, the first ATC to be analyzed in detail, have shown that its kinetic properties are well described by a two-state model. The dodecamer of six catalytic chains and six regulatory chains, in the absence of ligands, primarily occupies the T-state, which has a high Km for substrates, and switches with increasing probability to a more active R-state, which has a low Km, as more active sites become occupied (40, 41). In the catalytic chain of EcoATC, this conformational change is linked to the rotation of the carbamoyl phosphate-binding domain (residues 1–134) and aspartate-binding domain (residues 150–284) around a hinge (residues 135–149, 385–305) that closes the active site around the substrates (42, 43). The three corresponding segments in AaeATC are residues 1–126, residues 142–270, and residues 127–141 plus 271–291 (Supporting Information Figure S2). According to DynDom (44), which uses an objective algorithm to measure relative domain movements between two structures, the closure angle is 12.2° when these domains are compared in the T-state and R-state of EcoATC (6at1.pdb and 1d09.pdb respectively) and 7.4° in the T-state and R-state of the isolated catalytic trimers (1csu.pdb and 1ekx.pdb, respectively).
The corresponding movements in DAC can only be calculated definitively by comparing the structures of DAC with and without PALA bound to the active sites of the six ATC subunits. However, if the present DAC structure, with FLC bound to the active sites, is compared to the T- and R-states of EcoATC using DynDom, the rotation angles between the carbamoyl phosphate- and aspartate-binding domains are 7.4° and 1.9°, respectively, for the T- and R-states. In other words, the arrangements of the domains of the ATC subunits in DAC are closer to the R-state of EcoATC. However, citrate does not pull the DAC ATC sites fully into the R-conformation because it lacks the phosphate group present in the Michaelis complex of carbamoyl phosphate + aspartate or in the bisubstrate analogue, PALA. Instead, the phosphate binding site is occupied by three water molecules, J31, J251, and J435 (Figure 2C), which are not sufficient to pull Ser72* and Lys75*, the residues corresponding to Ser80* and Lys84* in EcoATC that hydrogen bond to the phosphate group, into the active site. When DAC atc1 is superposed onto EcoATC-R (1d09B.pdb), the Cα distance is 8.7 Å between Ser72* and Ser80* and 4.7 Å between Lys75* and Lys84* (13.8 Å between the NZ atoms).
The correct angle of domain closure for the DAC atc1 chain can only be calculated by comparing the structures of DAC with nothing and with PALA in the active sites of the ATC chains. However, the angle is likely to be smaller than the domain closure observed in EcoATC because DAC uniquely has a segment of loop A from the DHO subunit inserted between the two ATC domains (Figure 1). This interaction is part of the green interface and is a major contributor to stabilizing the loops and activating the DHO domains as evidenced by the observation that changing any of the three hydrophobic residues at the tip of loop A (A198, L199, L200) to arginine reduces the DHO activity (R. Fernando and H. Evans, unpublished).
Reactor Structure and Kinetics
Three notable features of the dodecamer that possibly influence the kinetics of the bisynthase are (a) direct contact between the DHO and ATC subunits, (b) the enclosed reaction chamber, and (c) a dramatic separation of charge between the inside and the outside of the reactor. The direct contact between the two subunits, such as loop A from DHO inserted between the two domains of the ATC chain, raises the possibility of a conformational linkage between the two sites. It can only be visualized by future structure determinations with and without bound ligands, but we have already observed, in preliminary experiments, that PALA, in sufficient amounts to just occupy the ATC active sites, reduces Vmax at the DHO site significantly (R. Fernando and H. Evans, unpublished). Offering limited access to the cytosol, the internal chamber, in effect, channels the intermediate, carbamoyl aspartate, by increasing its local concentration due to steric and electrostatic restrictions on diffusion. The small size of the chamber implies that a single molecule of carbamoyl aspartate restricted to just the volume of the chamber has a nominal concentration of approximately 12 mM.
In addition, the interior walls and the channels of the reaction chamber are electropositive while the exterior surface of the dodecamer is electronegative except at the entrances to the side pores (Figure 10). Presumably, the electropositive pores and interior preferentially attract and retain the substrates, carbamoyl phosphate and aspartate, and the intermediate, carbamoyl aspartate, which are all more negatively charged than the product, dihydroorotate. Preferential diffusion of dihydroorotate from the reaction chamber should help the bisynthase overcome the kinetic bottleneck of DHO (16), which has a greater rate for hydrolyzing dihydroorotate into carbamoyl aspartate than condensing the latter into dihydroorotate at physiological pH. The system is normally at steady state because the next synthetic step, the oxidation of dihydroorotate, is effectively irreversible. If all of the enzymes were evenly dispersed throughout the cytosol, free dihydroorotate would have two pathways similarly available by random diffusion, hydrolysis back to carbamoyl aspartate or oxidation to orotate. By sequestering the DHO active sites, DAC reduces hydrolysis of free dihydroorotate and, consequently, increases its effective concentration and the overall flux through the pyrimidine pathway. In vivo, enzymes function in an environment where the concentration of total protein is so high that free diffusion by enzymes is questionable and the concentration of most substrates is very low (45). Compartmentalization, as seen here, and aggregation into larger enzyme clusters, as seen by others (46), are two strategies that address these limitations.
Figure 10.

Exterior and interior electrostatic fields of DAC. The negative (red) and positive (blue) electrostatic field of DAC calculated by APBS, as described in the text, is shown at the 1 kT level viewed down the 2-fold axis of the dodecamer (A) from outside and (B) in cross section.
Multiprotein complexes are common in cells, and many of them have symmetric structures with the potential of enclosing substantial internal chambers (47). More than two dozen enzyme structures have been reported in PubMed to have large enclosed chambers, based upon X-ray crystallography, electron microscopy, or hydrodynamic studies. In effect, these hollow complexes are small metabolic compartments on the nanometer scale with the potential of sequestering sensitive reactions or intermediates from the cytosol, much like cellular organelles do on a larger scale. We define a complex as a reactor if it has at least one enzyme active site facing an internal chamber that is linked to the exterior of the complex by one or more channels.
Four notable reactors that have been visualized at atomic resolution and exhibit structural memes present in DAC are ferritin (48), fatty acid synthetase (49), the proteosome (50), and pyruvate dehydrogenase (51). Ferritins oxidize Fe2+ to Fe3+ and store it as a ferric hydroxyphosphate precipitate. Most ferritin structures have 24 identical or similar subunits that assemble into a truncated cube with 432 symmetry that encloses an internal chamber of approximately 80 Å diameter. Putative channels occur at the three symmetry axes and vary in relative size among species but are generally narrower than those in DAC because iron cations are smaller than the DAC metabolites. Ferritins, which bind cations, are electrostatic opposites to DAC (52). The channels, the ferrioxidase site, and nucleation sites for mineralization are all negatively charged. The electrostatic gradients calculated for human ferritin favor iron intake at the 3-fold channels, diffusion to the ferrioxidase sites, diffusion to the nucleation sites, and eventual export of the cations through the 4-fold channels (52).
Fatty acid synthase is a heteromeric dodecamer with multiple copies of six different enzyme active sites in a reaction chamber that is approximately 100 Å in diameter (49). Six α-chains form a central ring with D3 symmetry, like the DHO subunits in DAC, that is capped top and bottom by dome-shaped trimers of the β-chains. However, spokes from the α-chains extend into the center of the ring and divide the central chamber into two halves connected by large openings. Also, the acyl intermediate is tethered during the reaction so the reactor pores to the exterior are large (>20 Å) and not regulated.
The 20S proteosome has a very different architecture from DAC. It is a hollow barrel formed by four heptameric rings in an α7β7β7α7 arrangement. The interior cavity, which is approximately 150 Å long and 30 Å wide, is divided into a catalytic chamber that provides access to the active sites on the 14 β-chains and two antechambers, one on either end, that are formed by the α-chains. Each antechamber has an axial pore that is partially open in archaea (13 Å) and fully closed in eukaryotes. Additional protein complexes bind to these pores, open them, and allow only proteins destined for degradation to enter (50).
The pyruvate dehydrogenase complex couples glycolysis to the citric acid cycle. It contains three enzymes, E1p, E2p, and E3, where E2p forms a hollow core of 24 subunits or 60 subunits, depending upon the species, that is decorated with multiple copies of the other two subunits attached by long tethers. The smaller core has eight trimers of E2p at the apexes of a cube that encloses a chamber approximately 46 Å across with 30 Å wide channels, on each face of the cube (51). The larger core has 20 trimers at the apexes of an icosahedron that encloses a chamber approximately 118 Å across with 52 Å wide channels to the exterior (53).
Like E2p, DAC is the core of a larger noncovalent complex in A. aeolicus and possibly a model for the covalent CAD complex in mammals. Six CPS complexes, which have been shown to channel carbamoyl phosphate to the ATC trimer (5), presumably bind to the outside of the DAC dodecamer and either form an antechamber with 3-fold symmetry over a shared tunnel through the ATC trimer or individually align their active sites with one of the six C2 side pores. This overall architecture probably obtains for CAD as well, based upon the observed trend for monofunctional polypeptide chains that associate beneficially in a complex becoming fused during evolution (54). A. aeolicus, like other hyperthermophiles, retains the separate shorter chains because they are easier to synthesize successfully at high temperatures (55). This work suggests that the hollow 6-fold ring structures seen in rotary-shadowed micrographs of hamster CAD (56) arise from the association of dimers of the CAD DHO domains into a ring with D3 symmetry as seen with DAC (Figure 6 and table of contents figure). Because DHO domains isolated from hamster CAD form predominately dimers in solution, the rings must be stabilized by interactions with the other domains as seen in DAC with the green and yellow interfaces. The relative strength of the various interfaces undoubtedly varies among species; for example, the DHO domain prepared by proteolysis from frog CAD forms hexamers in solution (57). Because CAD channels its intermediates in solution with some diffusional leakage, the central hollow visible in the micrographs must be enclosed. A possible explanation is that the rings appear open in the micrographs because the trimers of ATC domains, which cap the chamber, have dissociated under the conditions used to prepare the samples. Although trimers and hexamers of hamster CAD predominate in solution, approximately 10% of the chains are monomers indicating that the blue and cyan interfaces in CAD are only moderately strong.
In CAD, the carboxy-terminal residue of the DHO domain is covalently linked to the amino-terminal residue of the ATC domain by a linker segment of 40 residues, assuming the CAD DHO domain has 422 residues as in AaeDHO. These two termini are 46 Å apart on the external surface of the DAC dodecamer, a distance that can be easily spanned by the linker, which can attain a maximal length of 140 Å if fully extended.
At the domain level, DAC and CAD have notable similarities and some differences. CAD resembles DAC by (1) having a DHO domain with extensions on the N-terminus (18 residues) and C-terminus (65 residues) relative to EcoDHO, (2) having 1 mol of zinc in the DHO domain (58), and (3) having no carboxylated lysine in its mass spectrum (H. Guy-Evans and D. R. Evans, unpublished). However, in multiple sequence alignments, CAD has a glutamic acid matched with Asp153 in the DAC DHO domain and does not have matching residues for loop A in DAC. Given our observation that none of the sequence alignment programs can match the carboxylated lysine (KCX102) in EcoDHO with its three-dimensional equivalent in AaeDHO (Thr151), the full resolution of these differences awaits further structural studies.
Supplementary Material
Acknowledgments
This project has benefited from the advice and skilled assistance of numerous staff members at the Advanced Photon Source, Argonne, IL. In particular, we thank Dr. Andrew Howard, Dr. Lisa Keefe, and Dr. Rong Huang at IMCA-CAT, Dr. Randy Alkire at SBC-CAT, and Dr. Keith Brister at LS-CAT.
Footnotes
This work was supported in part by NIH Grants HL57527, HL/ GM47399, and GM/CA60321, NSF Grant MCB9810325, and a grant for computational biology from Wayne State University. The Advanced Laboratory for Macromolecular Crystallography at Wayne State University is supported by Grant 085P1000817 from the Michigan Economic Development Corporation and Technology Tri-Corridor. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract W-31-109-Eng-38. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago.
The atomic coordinates and structure factors for the crystal structure discussed herein will be available from the Research Collaboratory for Structural Bioinformatics with accession code 3D6N upon publication.
Abbreviations: Aae, prefix for proteins from Aquifex aeolicus; Atu, prefix for Agrobacterium tumefaciens; ATC, aspartate transcarbamoylase; CAD, a polypeptide chain present in mammals and some other species that includes the first three enzymes of pyrimidine biosynthesis fused in the order CPS-DHO-ATC; CPS, carbamoyl phosphate synthetase; CSS, complexation significance score; DHO, dihydroorotase; DAC, dihydroorotase–aspartate transcarbamoylase complex; Eco, prefix for Escherichia coli; Mpr, prefix for Moritella profunda; Pab, prefix for Pyrococcus abyssi; Pgi, prefix for Porphyromonas gingivalis; Sac, prefix for Sulfolobus acidocaldarius; Sgr, prefix for Streptomyces griseus; Taq, prefix for Thermus aquaticus; Tth, prefix for Thermus thermophilus.
SUPPORTING INFORMATION AVAILABLE
A list of representative structures for carbamoyl phosphate synthetase, aspartate transcarbamoylase, and dihydroorotase (Table S1); a multiple sequence alignment derived from superpositions of DHO structures (Figure S1); and a multiple sequence alignment derived from superpositions of ATC structures (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 2.Christopherson RI, Jones ME. The overall synthesis of L-5,6-dihydroorotate by multienzymatic protein pyr1-3 from hamster cells. Kinetic studies, substrate channeling, and the effects of inhibitors. J Biol Chem. 1980;255:11381–11395. [PubMed] [Google Scholar]
- 3.Irvine HS, Shaw SM, Paton A, Carrey EA. A reciprocal allosteric mechanism for efficient transfer of labile intermediates between active sites in CAD, the mammalian pyrimidine-biosynthetic multienzyme polypeptide. Eur J Biochem. 1997;247:1063–1073. doi: 10.1111/j.1432-1033.1997.01063.x. [DOI] [PubMed] [Google Scholar]
- 4.Mally MI, Grayson DR, Evans DR. Catalytic synergy in the multifunctional protein that initiates pyrimidine biosynthesis in Syrian hamster cells. J Biol Chem. 1980;255:11372–11380. [PubMed] [Google Scholar]
- 5.Purcarea C, Ahuja A, Lu T, Kovari L, Guy HI, Evans DR. Aquifex aeolicus aspartate transcarbamoylase, an enzyme specialized for the efficient utilization of unstable car-bamoyl phosphate at elevated temperature. J Biol Chem. 2003;278:52924–52934. doi: 10.1074/jbc.M309383200. [DOI] [PubMed] [Google Scholar]
- 6.Ahuja A, Purcarea C, Ebert R, Guy H, Evans D. Aquifex aeolicus dihydroorotase: Association with aspartate transcarbamoylase switches on catalytic activity. J Biol Chem. 2004;279:53136–53144. doi: 10.1074/jbc.M403009200. [DOI] [PubMed] [Google Scholar]
- 7.Bethell MR, Jones ME. Molecular size and feedback-regulation characteristics of bacterial aspartate transcarbamoylases. Arch Biochem Biophys. 1969;134:352–365. doi: 10.1016/0003-9861(69)90294-x. [DOI] [PubMed] [Google Scholar]
- 8.Hughes LE, Hooshdaran MZ, GAOD Streptomyces aspartate transcarbamoylase is a dodecamer with dihydroorotase activity. Curr Microbiol. 1999;39:175–179. doi: 10.1007/s002849900441. [DOI] [PubMed] [Google Scholar]
- 9.Van de Casteele M, Chen P, Roovers M, Legrain C, Glansdorff N. Structure and expression of a pyrimidine gene cluster from the extreme thermophile Thermus strain ZO5. J Bacteriol. 1997;179:3470–3481. doi: 10.1128/jb.179.11.3470-3481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vickrey JF, Herve G, Evans DR. Pseudomonas aeruginosa aspartate transcarbamoylase. Characterization of its catalytic and regulatory properties. J Biol Chem. 2002;277:24490–24498. doi: 10.1074/jbc.M200009200. [DOI] [PubMed] [Google Scholar]
- 11.Wiley DC, Lipscomb WN. Crystallographic determination of symmetry of aspartate transcarbamylase. Nature. 1968;218:1119–1121. doi: 10.1038/2181119a0. [DOI] [PubMed] [Google Scholar]
- 12.Brabson JS, Maurizi MR, Switzer RL. Aspartate transcarbamylase from Bacillus subtilis. Methods Enzymol. 1985;113:627–635. doi: 10.1016/s0076-6879(85)13084-3. [DOI] [PubMed] [Google Scholar]
- 13.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamoylase: the molecular basis for a concerted allosteric transition. Trends Biochem Sci. 1990;15:53–59. doi: 10.1016/0968-0004(90)90176-c. [DOI] [PubMed] [Google Scholar]
- 14.Ladjimi MM, Ghellis C, Feller A, Cunin R, Glansdorff N, Pierard A, Herve G. Structure-function relationship in allosteric aspartate carbamoyltransferase from Escherichia coli II Involvement of the C-terminal region of the regulatory chain in homotropic and heterotropic interactions. J Mol Biol. 1985;186:715–724. doi: 10.1016/0022-2836(85)90391-2. [DOI] [PubMed] [Google Scholar]
- 15.Wild JR, Loughrey-Chen SJ, Corder TS. In the presence of CTP, UTP becomes an allosteric inhibitor of aspartate transcarbamoylase. Proc Natl Acad Sci USA. 1989;86:46–50. doi: 10.1073/pnas.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henry CS, Broadbelt LJ, Hatzimanikatis V. Thermodynamics-based metabolic flux analysis. Biophys J. 2007;92:1792–1805. doi: 10.1529/biophysj.106.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fields C, Brichta D, Shephardson M, Farinha M, O’Donovan G. Phylogenetic analysis and classification of dihydroorotases: a complex history for a complex enzyme. Paths Pyrimidines. 1999;7:49–63. [Google Scholar]
- 18.Seibert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 19.Porter TN, Li Y, Raushel FM. Mechanism of the dihydroorotase reaction. Biochemistry. 2004;43:16285–16292. doi: 10.1021/bi048308g. [DOI] [PubMed] [Google Scholar]
- 20.Lee M, Chan CW, Mitchell Guss J, Christopherson RI, Maher MJ. Dihydroorotase from Escherichia coli: loop movement and cooperativity between subunits. J Mol Biol. 2005;348:523–533. doi: 10.1016/j.jmb.2005.01.067. [DOI] [PubMed] [Google Scholar]
- 21.Thoden JB, Phillips GN, Jr, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry. 2001;40:6989–6997. doi: 10.1021/bi010682i. [DOI] [PubMed] [Google Scholar]
- 22.Martin PD, Purcarea C, Zhang P, Vaishnav A, Sadecki S, Guy-Evans HI, Evans DR, Edwards BF. The crystal structure of a novel, latent dihydroorotase from Aquifex aeolicus at 1.7Å resolution. J Mol Biol. 2005;348:535–547. doi: 10.1016/j.jmb.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 23.Vagin AA, Isupov MN. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr, Sect D: Biol Crystallogr. 2001;57:1451–1456. doi: 10.1107/s0907444901012409. [DOI] [PubMed] [Google Scholar]
- 24.Evrard GX, Langer GG, Perrakis A, Lamzin VS. Assessment of automatic ligand building in ARP/wARP. Acta Crystallogr, Sect D: Biol Crystallogr. 2007;63:108–117. doi: 10.1107/S0907444906023389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winn MD. An overview of the CCP4 project in protein crystallography: an example of a collaborative project. J Synchrotron Radiat. 2003;10:23–25. doi: 10.1107/s0909049502017235. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; Palo Alto, CA: 2002. [Google Scholar]
- 28.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinf. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to micro-tubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 32.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Jacobson MP, Zhu K, Zhao S, Friesner RA. Assignment of polar states for protein amino acid residues using an interaction cluster decomposition algorithm and its application to high resolution protein structure modeling. Proteins. 2007;66:824–837. doi: 10.1002/prot.21125. [DOI] [PubMed] [Google Scholar]
- 34.Tickle IJ, Laskowski RA, Moss DS. Rfree and the Rfree ratio. I Derivation of expected values of cross-validation residuals used in macromolecular least-squares refinement. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:547–557. doi: 10.1107/s0907444997013875. [DOI] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 37.Purcarea C. Aspartate transcarbamoylase from Pyrococcus abyssi. Methods Enzymol. 2001;331:248–270. doi: 10.1016/s0076-6879(01)31063-7. [DOI] [PubMed] [Google Scholar]
- 38.Van de Casteele M, Legrain C, Desmarez L, Chen PG, Pierard A, Glansdorff N. Molecular physiology of carbamoylation under extreme conditions: what can we learn from extreme thermophilic microorganisms? Comp Biochem Physiol, Part A: Mol Integr Physiol. 1997;118:463–473. doi: 10.1016/s0300-9629(97)00007-8. [DOI] [PubMed] [Google Scholar]
- 39.Karplus K, Karchin R, Draper J, Casper J, Mandel-Gutfreund Y, Diekhans M, Hughey R. Combining local-structure, fold-recognition, and new fold methods for protein structure prediction. Proteins. 2003;53:491–496. doi: 10.1002/prot.10540. [DOI] [PubMed] [Google Scholar]
- 40.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamylase: the relation between structure and function. Science. 1988;241:669–674. doi: 10.1126/science.3041592. [DOI] [PubMed] [Google Scholar]
- 41.Lee BH, Ley BW, Kantrowitz ER, O’Leary MH, Wedler FC. Domain closure in the catalytic chains of Escherichia coli aspartate transcarbamoylase influences the kinetic mechanism. J Biol Chem. 1995;270:15620–15627. doi: 10.1074/jbc.270.26.15620. [DOI] [PubMed] [Google Scholar]
- 42.Beernink PT, Endrizzi JA, Alber T, Schachman HK. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc Natl Acad Sci USA. 1999;96:5388–5393. doi: 10.1073/pnas.96.10.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanisms of catalysis and heterotropic regulation of Escherichia coli aspartate transcarbamoylase based upon a structure of the enzyme complexed with the bisubstrate analogue N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins. 1999;37:729–742. [PubMed] [Google Scholar]
- 44.Qi G, Lee R, Hayward S. A comprehensive and non-redundant database of protein domain movements. Bioinformatics. 2005;21:2832–2838. doi: 10.1093/bioinformatics/bti420. [DOI] [PubMed] [Google Scholar]
- 45.Ringe D, Petsko GA. Biochemistry. How enzymes work Science. 2008;320:1428–1429. doi: 10.1126/science.1159747. [DOI] [PubMed] [Google Scholar]
- 46.Carrey EA, Dietz C, Glubb DM, Loffler M, Lucocq JM, Watson PF. Detection and location of the enzymes of de novo pyrimidine biosynthesis in mammalian spermatozoa. Reproduction. 2002;123:757–768. [PubMed] [Google Scholar]
- 47.Devos D, Russell RB. A more complete, complexed and structured interactome. Curr Opin Struct Biol. 2007;17:370–377. doi: 10.1016/j.sbi.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Andrews SC. Iron storage in bacteria. Adv Microb Physiol. 1998;40:281–351. doi: 10.1016/s0065-2911(08)60134-4. [DOI] [PubMed] [Google Scholar]
- 49.Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Structure of fungal fatty acid synthase and implications for iterative substrate shuttling. Science. 2007;316:254–261. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- 50.Groll M, Clausen T. Molecular shredders: how proteasomes fulfill their role. Curr Opin Struct Biol. 2003;13:665–673. doi: 10.1016/j.sbi.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Mattevi A, Obmolova G, Schulze E, Kalk KH, Westphal AH, de Kok A, Hol WG. Atomic structure of the cubic core of the pyruvate dehydrogenase multienzyme complex. Science. 1992;255:1544–1550. doi: 10.1126/science.1549782. [DOI] [PubMed] [Google Scholar]
- 52.Douglas T, Ripoll DR. Calculated electrostatic gradients in recombinant human H-chain ferritin. Protein Sci. 1998;7:1083–1091. doi: 10.1002/pro.5560070502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Izard T, Aevarsson A, Allen MD, Westphal AH, Perham RN, de Kok A, Hol WG. Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA. 1999;96:1240–1245. doi: 10.1073/pnas.96.4.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 55.Snel B, Bork P, Huynen M. Genome evolution. Gene fusion versus gene fission. Trends Genet. 2000;16:9–11. doi: 10.1016/s0168-9525(99)01924-1. [DOI] [PubMed] [Google Scholar]
- 56.Lee L, Kelly RE, Pastra-Landis SC, Evans DR. Oligomeric structure of the multifunctional protein CAD that initiates pyrimidine biosynthesis in mammalian cells. Proc Natl Acad Sci USA. 1985;82:6802–6806. doi: 10.1073/pnas.82.20.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kent RJ, Lin RL, Sallach HJ, Cohen PP. Reversible dissociation of a carbamoyl phosphate synthase-aspartate transcarbamoylase-dihydroorotase complex from ovarian eggs of Rana catesbeiana: effect of uridine triphosphate and other modifiers. Proc Natl Acad Sci USA. 1975;72:1712–1716. doi: 10.1073/pnas.72.5.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelly RE, Mally MI, Evans DR. The dihydroorotase domain of the multifunctional protein CAD. Subunit structure, zinc content, and kinetics. J Biol Chem. 1986;261:6073–6083. [PubMed] [Google Scholar]
- 59.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. BI801831R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.