

Abstract
Each class of microscope is limited to imaging specific aspects of cell structure and/or molecular organization. However, imaging the specimen by complementary microscopies and correlating the data can overcome this limitation. Whilst not a new approach, the field of correlative imaging is currently benefitting from the emergence of new microscope techniques. Here we describe the correlation of cryogenic fluorescence tomography (CFT) with soft x-ray tomography (SXT). This amalgamation of techniques integrates 3-D molecular localization data (CFT) with a high-resolution, 3-D cell reconstruction of the cell (SXT). Cells are imaged in both modalities in a near-native, cryopreserved state. Here we describe the current state of the art in correlative CFT-SXT, and discuss the future outlook for this method.
Keywords: Correlated imaging, fluorescence, microscopy, soft x-ray, tomography
Background
Cell biology takes place over an incredibly wide range of scale, from atomic-level interactions between individual molecules to large-scale movement of organelles during cell division [Alberts et al., 2008]. In an ideal world, a single microscope would be capable of imaging every type of cell and characterizing every aspect of cell structure and function [Plitzko et al., 2009]. But in our non-ideal reality, microscope characteristics such as specimen illumination place constraints on the type and depth of information contained in images of the specimen [Subramaniam, 2005]. As such, data from any one imaging modality provides a limited picture of the specimen [Caplan et al., 2011]. By imaging the specimen with a second, complementary modality and combining the data, however, the view of the specimen can be widened and made more comprehensive [Caplan et al., 2011; Hagen et al., 2012; McDermott et al., 2012b; van Rijnsoever et al., 2008; Vicidomini et al., 2008]. In general, correlative imaging produces deeper insights into the specimen than is possible using any one imaging technique alone.
Building bridges between modalities that image cell structure with those that locate specific molecules has been a longstanding goal in correlated imaging. One of the early pioneering techniques that successfully achieved this goal was the combination of light- and electron microscopy. In Correlated Light and Electron Microscopy, or CLEM, the same area is visualized by both techniques, whereas in Correlative Light and Electron Microscopy the same specimen, but not the same area, is imaged [Ellisman et al., 2012]. In CLEM the specimen is first imaged using light microscopy to locate fluorescently-tagged features within the specimen, and then by electron microscopy (EM) or tomography (ET) to obtain detailed information about the cellular ultrastructure [Rigort et al., 2012].
CLEM has a storied history spanning more than four decades, and has been the source of many fundamental discoveries [Briggs and Lakadamyali, 2012; Caplan et al., 2011; Ellisman et al., 2012; Giepmans et al., 2005; Jahn et al., 2012; Martone et al., 2000; Murphy et al., 2011; Nixon et al., 2009; Rigort et al., 2012; Sartori et al., 2005; Sartori et al., 2007; Sosinsky et al., 2007; van Driel et al., 2008; van Rijnsoever et al., 2008]. Even so – and for reasons we describe below – the development of CLEM did not write the final chapter in cellular imaging. Instrumental limitations, inherent in both the light- and electron-based techniques, continued to drive the development of new imaging modalities. One recent development was the combination of high numerical aperture cryogenic fluorescence microscopy with (CFM) with soft x-ray tomography (SXT)[Le Gros et al., 2009]. These two modalities are highly complementary – by virtue of their disparate imaging/contrast mechanisms – and address many of the shortcomings of CLEM. We will now discuss each of these techniques individually, prior to discussing practical aspects of their use in correlative studies, including the extension of CLM to allow acquisition of tomographic data, i.e. cryogenic fluorescence tomography (CFT).
Soft X-ray Microscopy
Soft x-ray microscopy (SXM) is a non-invasive method for imaging the internal structure of intact cells [Attwood, 1999; Kirz et al., 1995; Sayre et al., 1977a; Sayre et al., 1977b]. In SXM the specimen is illuminated with soft x-ray photons at energies within a spectral region termed the “water window” (i.e. 2.3 – 4.4 nm; 0.28 – 0.53 keV) [Attwood, 1999; Weiss et al., 2000]. In this region, the absorption of photons adheres to Beer’s Law and is therefore linear and a function of specimen thickness and chemical species [Larabell and Le Gros, 2004]. As the term “water window” implies, water absorbs soft x-rays weakly in comparison to carbon and nitrogen. Consequently, the differential attenuation of soft x-rays results in high contrast images of cells, without the need to dehydrate the specimen or use contrast-enhancing agents [Larabell and Nugent, 2010; Le Gros et al., 2005; McDermott et al., 2012a]. A CAD view of a soft x-ray microscope is shown in Fig. 1A and a close-up view of the sample environment is presented in Fig. 1B.
Fig. 1.
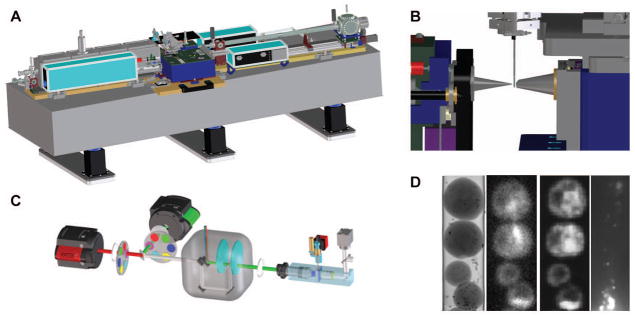
A) The overall layout of the soft x-ray microscope XM2 located at the Advanced Light Source, Lawrence Berkeley National Laboratory. All necessary instruments for SXT data collection sit on a 4m long optical bench. The synchrotron source of soft x-rays, and the associated hardware that transports photons to the microscope, are not shown for reasons of clarity.
B) Close up of a specimen capillary in position, ready for SXM data collection. Cells are mounted in a thin-walled capillary suspended from a rotation stage. Soft x-ray illumination comes via a Fresnel zone plate optic, located behind the cone on the left hand side. Soft x-ray photons transmitted by the specimen are collected and focused onto a detector by another zone plate optic situated behind the cone on the right hand side.
C) Overview of the cryolight spinning disc confocal micoscope (CLM).
D) The left hand panel shows a representative projection image from the SXM showing yeast cells aligned in single file inside a capillary. The two center panels are cryo-fluorescence data from the same yeast cells imaged in the CLM (nucleus labeled with GFP, and nucleolus labelled with mCherry. The right hand panel shows a CLM image of the far red fluorescent beads used for aligning images.
Soft x-ray microscopes operate in transmission mode and so photon absorption by the specimen determines the maximum thickness of a specimen that can be imaged. In practice, soft x-rays can penetrate cells up to 15 μm thick; as a result whole, intact eukaryotic cells can be imaged in a soft x-ray microscope [Clowney et al., 2012; Schneider et al., 2010; Spring et al., 1995; Uchida et al., 2009; Uchida et al., 2011{Schneider, 1999 #15]}. Conversely, specimens imaged by EM are restricted to a maximum thickness of 750 nm (for cryo EM, because of the strong inelastic scattering of electrons) and become thinner as observation time increases under the electron beam [Legall et al., 2012].
Cryogenic Fluorescence Microscopy
Fluorescence microscopy (FM) is so commonly used to locate and track specific molecules or proteins within a cell that it requires little introduction here. FM of cells is normally carried out on live or chemically fixed cells at room temperature. Recently, so-called “super resolution” techniques have greatly increased the precision with which fluorescent entities can be localized. Improvements in camera technology now allow detection of previously undetectable fluorescence signals, including fluorescence from a single molecule and the tracking of very quick, dynamic cellular events [Dedecker et al., 2013]. Fluorescence labeling can be achieved by a number of well-established methods, including fluorescent staining of certain cellular molecules, immuno-labeling, or the generation of a Fluorescent Protein (FP) fusion protein [Giepmans et al., 2006]. Despite the enormous utility of FM and the achievements in breaking through the diffraction limit barrier on spatial resolution, FM nonetheless has weaknesses, in particular the susceptibility of fluorescent molecules to irreversible damage by the illumination during long or repeated exposures [Shaner et al., 2008].
All high resolution imaging studies potentially cause accumulated photon damage to the specimen, which then leads to artifacts in the images [Le Gros et al., 2009; Moerner and Orrit, 1999; Prendergast, 1999]. Fixation, in most instances, can mitigate this damage to undetectable levels and is therefore an essential step in studies that require repeated or extended exposure of the specimen to harsh illumination (light, electrons or x-rays)[Caplan et al., 2011; Ellisman et al., 2012; Larabell and Le Gros, 2004; Larabell and Nugent, 2010; Weiss and 2000]. Unless data can be collected instantaneously by both modalities, the specimen must always be “fixed” prior to data acquisition in correlated imaging studies to ensure the specimen remains unchanged throughout acquisition by both modalities. Rather than chemical fixation, which has been shown to damage the structural integrity of the cell, the developers of SXM/SXT turned to cryo-preservation, established by EM as being the “gold standard” in preserving the delicate ultrastructure of a cell. In the case of SXM cryo-fixation is especially appropriate: as discussed previously, water weakly attenuates soft x-rays and therefore makes an ideal environment for this type of imaging.
Since SXM is carried out on cryo-preserved specimens, cryo-fixation was the logical choice for correlated fluorescence studies as well. At the outset this approach proved to be a major technological challenge. Low temperature microscopes are fairly common and have been available for decades. However, for biological imaging these instruments are typically equipped with low numerical aperture air lenses. The use of air lenses results in refractive index mismatches as light traverses interfaces – such as that between air and the specimen – leading to a decrease in the overall fidelity of the image and thus reducing the precision with which fluorescent molecules can be localized [Le Gros et al., 2009]. Moreover, using conventional fluorescence microscopy techniques results in anisotropic data; the effective resolution along the light path is much reduced compared to the resolution in the other two axes. The challenge was therefore to develop a cryogenic light microscope that both imaged the specimen in a refractive indexed matched fluid, and reduced anisotropy in the localization signal. Recently this challenge was met by the development of a high numerical aperture spinning disc cryogenic light microscope (CLM)[Le Gros et al., 2009], Fig. 1C. In this microscope the specimen is imaged in a refractive index matched immersion fluid held at cryogenic temperatures. In addition to locking molecules and structure in position, carrying out FM at cryogenic temperatures also greatly increases the working lifetime of fluorescent molecules [Moerner and Orrit, 1999], by a factor of 30 or more [Le Gros et al., 2009]. This feature alone makes fluorescent imaging at cryogenic temperatures a worthwhile endeavor, in general, and a virtual necessity if the goal is to collect through-focus fluorescence tomographic data. Fig. 1D shows cells imaged by SXM and CLM.
Gaining volume
Microscopes can only produce two-dimensional (2-D) projection images of the specimen [Natterer, 1986]. This is adequate for imaging very thin objects with little internal organization. However, biological specimens – whether they are isolated organelles, individual cells, or tissues – are all three-dimensional (3-D) objects. When imaged in 2-D the structures inside a biological specimen are superimposed on top of each other, making interpretation difficult, if not impossible [Larabell and Nugent, 2010; McDermott et al., 2012a]}[Baumeister et al., 1999; Dierksen et al., 1995; Leis et al., 2009; Plitzko and Baumeister, 2010]. However, if 2-D projection images are collected from a number of different perspectives around a rotation axis, a 3-D tomographic reconstruction of the specimen can be calculated [Baumeister et al., 1999; Natterer and Wübbeling, 2001]. This is well-established technology, and has been extensively used in both research and clinical settings (for example, Computed Tomography (CT) scans are ubiquitous in Western medicine). Consequently, the CLM and soft x-ray microscope were equipped with a similar cryogenic specimen rotation stage. This both allowed the collection of tomographic data using each modality, and built a common format for cryogenic specimen mounting. To take maximum advantage of the cryo-rotation stage the specimen is mounted in a cylindrical holder Fig. 2A, rather than a flat surface such as a glass slide (Fig. 2B) or the grids commonly used in TEM. As seen in Fig. 2C, the use of a flat specimen holder limits the field of view to approximately ± 70°; as the rotation angle increases so does the thickness of the specimen. The dramatic increase in specimen thickness as a function of tilt angle on a flat specimen mount is shown graphically in Fig. 2D.
Fig. 2.

A) Line drawing of a thin-walled glass capillary of the type used for soft x-ray tomography (SXT). The diameter of the tube is matched to the size of the cells being imaged; generally between 4 and 10μm. Cells in a capillary can be imaged at any angle around the central rotation axis.
B) Representation of a flat specimen mount, such thin-glass slides for fluorescence microscopy or grids for TEM and SXT. As the specimen is tilted its thickness increases with respect to illumination normal to the rotation axis.
C) Thickness augmentation along the rotation of a flat support when the sample is 5μm thick. At a tilt angle of 45°, the beam passes through 7.1μm of the specimen. At a 72° rotation, the beam passes through 16μm.
D) The blue curve represents the increase in sample thickness when imaging a rotating flat support. At 72°, the usual maximum tilt angle used in ET, the sample is 3.2 times thicker than it is at 0°.
The cylindrical holder used in CFT-SXT is a thin-walled (~200 nm) glass capillary. This geometry has many benefits, the most important of which is the capability to image the specimen over a full rotation of 360°. This allows the 3-D reconstruction of a specimen with isotropic spatial resolution (and avoids the systematic “missing wedge” of data that occurs when flat specimen holders are used). Glass capillary properties, such as diameter and exterior fiducial markers, can be customized to meet specific experimental needs, in particular by matching the diameter of the capillary to the size of the specimen. Placing fiducials on the outside of capillary is also superior to relying on fiducials embedded in vitreous ice together with the specimen (as is the case with flat TEM grids). As has been reported, if the ice on a flat specimen grid is too thick the fiducials fall outside the depth of field of the microscope and can’t be imaged; if the ice is too thin the specimen is susceptible to damage; and even when the ice layer has optimal thickness, fiducials on the surface can move during data collection [Hummel et al., 2012]. The capillary diameter typically used lies between 4 and 15 μm, to both fit the cells and constrain them to single file within the tube. Now that we have brought the reader up to speed on the concept of CFT and SXT as discrete modalities we will discuss the practical side of correlating these complementary data.
Specimen Mounting
Prior to rapid cryo-preservation, cells are loaded directly from their growth media into a thin glass capillary coated with fiducial markers [Parkinson et al., 2013]. A simple bright field microscope with a 40X air objective is used to check specimen loading. Ideally, the cells should be positioned near the tip of the tapered capillary. Because the field of view in the soft x-ray microscope is 15 μm, multiple cells may fit in each image; in the case of yeast, this means up to 5 cells per data set. Once loaded with cells the capillary is plunged at a speed of 2 m/s into propane cooled by liquid nitrogen. The loading and plunge-cooling of the loaded specimens usually takes no more than a few minutes to complete. The capillary is then transferred to the cryo-fluorescence microscope for data acquisition.
Acquisition of Cryo-Fluorescence and Soft X-ray Microscope data
As with CLEM, fluorescence data is always collected first in a CFT-SXT experiment [McDermott et al., 2009]. The reasoning for this in both techniques is similar: the amount of photons absorbed during SXT imaging irreversibly destroys the fluorescence signal, whereas photon accumulation during CFT image acquisition does not perturb the ultrastructure of the specimen (at least not at the level where it can be observed in SXT). To collect a CFT data set, through-focus images are recorded in the CLM at angular increments around the central axis to cover a total rotation range of 180°. Typically a through-focus data set is collected every 10°.
Once CFT data have been collected the specimen capillary is transferred in a cryogenic container to the SXM. The tip is aligned using a low magnification light microscope, and then with low doses of soft x-rays. 90 or 180 images are collected around the same rotational axis as that used for CFT data acquisition. In contrast with EM, in which the sample and sample holder progressively shrink during acquisition, the specimen and capillary usually retain their shape and volume throughout SXT data acquisition. A full SXT data set is usually collected in 4–8 minutes and later yields a reconstruction with a 15 μm3 volume. The correlative CFT-SXT acquisition workflow is presented Fig. 3.
Fig 3.
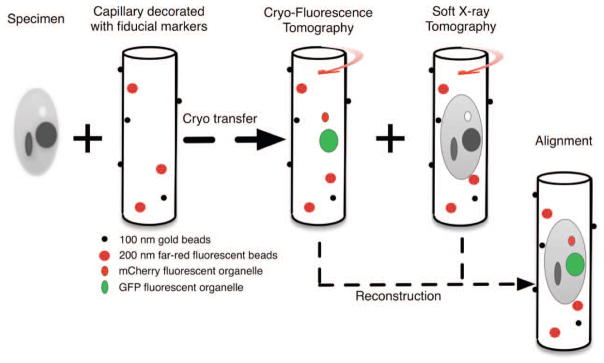
A diagrammatic representation of the workflow followed to collect correlated CFT-SXT data using cryo-light and soft x-ray microscopes.
The specimen is mounted in a capillary decorated with two kinds of fiducial markers: 100nm diameter gold beads for the soft x-ray data set reconstruction and the 200nm far red fluorescent beads for the alignment of the two data sets. The fluorescent fiducials are visible in both SXM and CLM data. The gold fiducials are only visible in SXM data. CFT data is collected prior to the specimen being cryo-transferred to the SXM for tomographic data acquisition.
Alignment of the two data sets requires two independent reconstructions. Matching of the separate data sets is then guided by the co-alignment of the fluorescence bead sets.
Correlation of CFT-SXT data
Effective and accurate correlation of datasets from two different contrast mechanisms requires every step of the workflow to be characterized and well defined. Prior to calculating a 3-D CFT tomographic reconstruction the 2-D images are aligned with respect to each other using fiducial markers as a guide. This process must also be carried out independently on the 2-D soft x-ray data set. Clearly, it is preferable if the fiducial markers that guide alignment of the two individual data sets are the same and can be viewed in both modalities. In which case, once the 2-D data from each modality have been reconstructed in 3-D, the same fiducials can be used to co-align both reconstructions.
To estimate the accuracy of the correlation between CFT and SXT data, one fiducial marker is excluded from the calculation of the transform that co-aligns the two reconstructions. The location of this fiducial is then predicted within the reconstruction. The calculated position can then be compared with the actual position to give an estimate of the error. By repeating this calculation one by one for all fiducials across all tomograms in the dataset, it is possible to obtain a direct readout of the correlation accuracy of the system [Kukulski et al., 2011].
Prior knowledge about the specimen can also be used to guide the alignment of the two reconstructions. A well-defined and fluorescently-labeled organelle (for example, a lipid droplet, the nucleus, granules or mitochondria) can function as a fiducial. Having two independent sets of fiducials – fluorescent beads coating the capillary exterior and well-characterized fluorescent structures inside the cell – is enormously powerful and produces a more precise co-alignment of the two data sets, typically with sub-pixel accuracy. An example of correlated data is shown in Fig. 4.
Fig. 4.
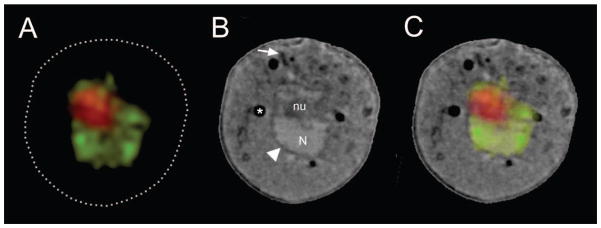
A) Virtual section (orthoslice) through CFT reconstruction of a yeast cell, nuclear volume is labeled with mCherry, the nucleolus with GFP.
B) Virtual section through the SXT reconstruction of the same yeast cell. An arrowhead points to the nuclear membrane, an arrow indicates a representative section of mitochondria. Key: N, nucleus; Nu, nucleolus.
C) Virtual section through the CFT-SXT co-aligned and overlaid reconstructions.
Discussion
The correlation of complementary imaging data taken from the same specimen is a powerful technique in cell biology [Caplan et al., 2011]. The strengths of one method compensate for the weaknesses of the other, and the combination of two types of information produces insights with greater depth than is possible using any single modality. Above, we described the emergence of correlated CFT-SXT, a new tool for viewing molecular localization data directly in the context of the cell ultrastructure.
Compared to electron microscopy and conventional fluorescence microscopy, CFT and SXT are relative newcomers to cell imaging. Of course, the emergence of this new correlated modality does not signify the demise of existing techniques, such as CLEM. On the contrary there is sufficient need in cell imaging to call for the development of additional modalities above and beyond techniques such as CLEM and CFT-SXT.
CFT and SXT data are well matched in the criteria important for the creation of a new correlative modality. For example, both data sets produce 3-D reconstructions of the specimen, can sequentially image the same region of a cryo-preserved specimen, and can potentially image with similar spatial resolution. The latter is a particularly important point and will be discussed in more detail in the future outlook.
Generally speaking, imaging a single cell in isolation rarely provides adequate information to unambiguously characterize a particular aspect of cellular structures or function. Depending of the purpose of the experiment and variability of the specimens, the need to make imaging measurements from a large number of specimens is common. Consequently, specimen throughput is an important consideration in any visualization technique. CFT-SXT can achieve impressive levels of throughput because of the negligible requirement for specimen processing. For most experiments, cells are simply pipetted from the growth media into a capillary and then cryo-preserved; this process takes minutes at most. While sophisticated techniques such as FIB-SEM [Drobne et al., 2005], [Kopek et al., 2012] (which uses an abrasive Ga+ beam to remove 5 nm thickness of material between each image) have improved throughput, EM imaging is still limited to specimens with a maximum thickness of 750 nm (for cryo EM) and the generation of just one tomogram, whilst at significantly higher resolution than is possible with SXT, remains time-consuming and relatively labor intensive.
Finally, the instruments used for CFT and SXT place minimal constraints on data acquisition. For example, the specimen can be rotated through 360° and imaging at low temperature significantly increases the working lifetime of the fluorescent label. As a result highly complete data sets can be collected, eliminating the systematic errors that accompany incomplete data.
Future outlook
The correlation of SXT with CFT is very much a work in progress, with many opportunities for improvement still remaining. For example, closely matching the spatial resolution of both techniques will decrease errors in co-alignment. SXT data is currently obtained at a spatial resolution of 35 to 50 nm, while the diffraction-limited resolution of the confocal microscope used to collect CFT data is ~350 nm. As a result there is now a drive to build cryogenic microscopes that image beyond the diffraction limit, i.e. a “super resolution” instrument. This is a considerable technological challenge, but clearly achievable in the near future. A new design of microscope that incorporates lessons learned from the use of the cryo-light microscope described in [Le Gros et al., 2009] is currently under construction. This microscope is expected to localize fluorescent labels with isotropic precision very close to the current 35 to 50nm resolution of SXT.
Currently, SXM is carried out primarily at synchrotron light sources. This limits the potential growth of the technique, and means most people have to travel – often great distances – to use these instruments. However, rapid progress is being made in the development of “table top” soft x-ray sources that take up less space than an electron microscope, or a rotating anode x-ray source of the type used to collect protein crystallography diffraction data [Hertz et al., 2012; Legall et al., 2012].
The technology required to increase the spatial resolution of SXT already exists in the form of Fresnel zone plates that can image at 12 nm [Chao et al., 2009]. Installing one of these optical elements in the soft x-ray microscope is similar to routine maintenance and can be carried out in a few hours. However, as the spatial resolution increases, the depth of field decreases. At 50 nm spatial resolution, a 10 μm thick cell is fully in focus. Increasing the spatial resolution to 10 nm drops the field of view to 1–2 μm, requiring through-focus data be collected at each rotation angle in a tomographic series. Deconvolution is a well-understood imaging technique but nonetheless requires some effort before it can be applied to SXT. Consequently, in the mid-term it is likely that CFT-SXT will converge on a common spatial resolution of 35 to 50 nm for most specimens, and possibly higher for small prokaryotic cells, for example. The advantage, of course, compared to CLEM is the ability to image unstained, fully hydrated cells in a near native state.
Whilst CFT-SXT is unique, the data and findings can be combined readily with that from other modalities and utilized as part of larger studies. For example, the cryo-light microscope can easily accommodate the flat specimen grids used in cryo-EM. It would therefore require minimal work to improve the spatial resolution of cryogenic CLEM. In closing, if we were to picture our somewhat idealized world of the future it would consist of an imaging suite similar in concept to that outlined in [Caplan et al., 2011]. In our vision it consists of three core resources, i) room-temperature fluorescence microscopes for studying dynamic events, ii) a cryogenic work station for rapidly fixing, handling and storing cryo-preserving specimens, and iii) microscopes that allow the choice to be made between imaging the specimen by either CLEM (incorporating a CLM) or CFT-SXT. Such a facility would be capable of meeting a very wide range of imaging needs in cell biology and be profoundly capable of putting molecules in their cellular place.
Acknowledgments
The research reported in this publication was conducted at the National Center for X-ray Tomography (NCXT), which is supported by the National Institute of General Medical Science of the National Institutes of Health under award number P41 GM103445 and the US Department of Energy, Biological and Environmental Research (DE-AC02-05CH11231). The NCXT is located at the Advanced Light Source, a US Department of Energy supported user facility at Lawrence Berkeley National Laboratory; the authors thank the staff for providing a robust source of x-ray photons. C.A.L. and M.A.L. acknowledge generous support from the Gordon and Betty Moore Foundation. A.D.W. and O.C.F. are funded by an intramural grant from the National Institute of Diabetes and Digestive and Kidney Diseases, NIH.
Contract grant sponsor: NIH-NIGMS; Contract grant number: P41 GM103445
Contract grant sponsor: US DOE-BER; Contract grant number: DE-AC02-05CH11231
Contract grant sponsor: NIH-NIDDK; Contract grant number: Intramural
Footnotes
Competing interests
The authors declare that no competing interests exist.
References
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. New York: Garland Science; 2008. p. 1392. [Google Scholar]
- Attwood DT. Soft x-rays and extreme ultraviolet radioation: principles and applications. Cambridge, New York: Cambridge Uhiversity Press; 1999. p. 470. [Google Scholar]
- Baumeister W, Grimm R, Walz J. Electron tomography of molecules and cells. Trends in Cell Biology. 1999;9:81–85. doi: 10.1016/s0962-8924(98)01423-8. [DOI] [PubMed] [Google Scholar]
- Briggs JAG, Lakadamyali M. Imaging cellular structure across scales with correlated light, superresolution, and electron microscopy. Molecular Biology of the Cell. 2012;23:979–980. [Google Scholar]
- Caplan J, Niethammer M, Taylor RM, 2nd, Czymmek KJ. The power of correlative microscopy: multi-modal, multi-scale, multi-dimensional. Curr Opin Struct Biol. 2011;21:686–93. doi: 10.1016/j.sbi.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao W, Kim J, Rekawa S, Fischer P, Anderson EH. Demonstration of 12 nm resolution Fresnel zone plate lens based soft X-ray microscopy. Opt Express. 2009;17:17669–17677. doi: 10.1364/OE.17.017669. [DOI] [PubMed] [Google Scholar]
- Clowney EJ, Legros MA, Mosley CP, Clowney FG, Markenskoff-Papadimitriou EC, Myllys M, Barnea G, Larabell CA, Lomvardas S. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell. 2012;151:724–37. doi: 10.1016/j.cell.2012.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedecker P, De Schryver FC, Hofkens J. Fluorescent Proteins: Shine on, You Crazy Diamond. Journal of the American Chemical Society. 2013;135:2387–2402. doi: 10.1021/ja309768d. [DOI] [PubMed] [Google Scholar]
- Dierksen K, Typke D, Hegerl R, Walz J, Sackmann E, Baumeister W. Three-dimensional structure of lipid vesicles embedded in vitreous ice and investigated by automated electron tomography. Biophysical Journal. 1995;68:1416–1422. doi: 10.1016/S0006-3495(95)80314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobne D, Milani M, Zrimec A, Berden Zrimec M, Tatti F, Draslar K. Focused ion beam/scanning electron microscopy studies of Porcellio scaber (Isopoda, Crustacea) digestive gland epithelium cells. Scanning. 2005;27:30–34. doi: 10.1002/sca.4950270106. [DOI] [PubMed] [Google Scholar]
- Ellisman MH, Deerinck TJ, Shu X, Sosinsky GE. Picking faces out of a crowd: genetic labels for identification of proteins in correlated light and electron microscopy imaging. Methods Cell Biol. 2012;111:139–55. doi: 10.1016/B978-0-12-416026-2.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BN, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat Methods. 2005;2:743–9. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. The Fluorescent Toolbox for Assessing Protein Location and Function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Hagen C, Guttmann P, Klupp B, Werner S, Rehbein S, Mettenleiter TC, Schneider G, Grunewald K. Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. Journal of Structural Biology. 2012;177:193–201. doi: 10.1016/j.jsb.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz HM, von Hofsten O, Bertilson M, Vogt U, Holmberg A, Reinspach J, Martz D, Selin M, Christakou AE, Jerlström-Hultqvist J, Svärd S. Laboratory cryo soft X-ray microscopy. Journal of Structural Biology. 2012;177:267–272. doi: 10.1016/j.jsb.2011.11.015. [DOI] [PubMed] [Google Scholar]
- Hummel E, Guttmann P, Werner S, Tarek B, Schneider G, Kunz M, Frangakis AS, Westermann B. 3D Ultrastructural organization of whole Chlamydomonas reinhardtii cells studied by nanoscale soft x-ray tomography. PLoS One. 2012;7:e53293. doi: 10.1371/journal.pone.0053293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn KA, Barton DA, Kobayashi K, Ratinac KR, Overall RL, Braet F. Correlative microscopy: providing new understanding in the biomedical and plant sciences. Micron. 2012;43:565–82. doi: 10.1016/j.micron.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Kirz J, Jacobsen C, Howells M. Soft X-ray microscopes and their biological applications. Quarterly reviews of biophysics. 1995;28:33–130. doi: 10.1017/s0033583500003139. [DOI] [PubMed] [Google Scholar]
- Kopek BG, Shtengel G, Xu CS, Clayton DA, Hess HF. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc Natl Acad Sci U S A. 2012;109:6136–41. doi: 10.1073/pnas.1121558109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukulski W, Schorb M, Welsch S, Picco A, Kaksonen M, Briggs JAG. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. Journal of Cell Biology. 2011;192:111–119. doi: 10.1083/jcb.201009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larabell C, Le Gros M. Whole cell cryo X-ray tomography and protein localization at 50 micron resolution. Biophysical Journal. 2004;86:185A–185A. [Google Scholar]
- Larabell CA, Nugent KA. Imaging cellular architecture with X-rays. Current Opinion in Structural Biology. 2010;20:623–631. doi: 10.1016/j.sbi.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gros MA, McDermott G, Larabell CA. X-ray tomography of whole cells. Current Opinion in Structural Biology. 2005;15:593–600. doi: 10.1016/j.sbi.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Le Gros MA, McDermott G, Uchida M, Knoechel CG, Larabell CA. High-aperture cryogenic light microscopy. Journal of Microscopy-Oxford. 2009;235:1–8. doi: 10.1111/j.1365-2818.2009.03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legall H, Blobel G, Stiel H, Sandner W, Seim C, Takman P, Martz DH, Selin M, Vogt U, Hertz HM, Esser D, Sipma H, Luttmann J, Hofer M, Hoffmann HD, Yulin S, Feigl T, Rehbein S, Guttmann P, Schneider G, Wiesemann U, Wirtz M, Diete W. Compact x-ray microscope for the water window based on a high brightness laser plasma source. Optics Express. 2012;20:18362–18369. doi: 10.1364/OE.20.018362. [DOI] [PubMed] [Google Scholar]
- Leis A, Rockel B, Andrees L, Baumeister W. Visualizing cells at the nanoscale. Trends in Biochemical Sciences. 2009;34:60–70. doi: 10.1016/j.tibs.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Martone ME, Deerinck TJ, Yamada N, Bushong E, Ellisman MH. Correlated 3D light and electron microscopy: Use of high voltage electron microscopy and electron tomography for imaging large biological structures. Journal of Histotechnology. 2000;23:261–270. [Google Scholar]
- McDermott G, Fox DM, Epperly L, Wetzler M, Barron AE, Le Gros MA, Larabell CA. Visualizing and quantifying cell phenotype using soft X-ray tomography. Bioessays. 2012a;34:320–327. doi: 10.1002/bies.201100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott G, Le Gros MA, Knoechel CG, Uchida M, Larabell CA. Soft X-ray tomography and cryogenic light microscopy: the cool combination in cellular imaging. Trends in Cell Biology. 2009;19:587–595. doi: 10.1016/j.tcb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott G, Le Gros MA, Larabell CA. Visualizing Cell Architecture and Molecular Location Using Soft X-Ray Tomography and Correlated Cryo-Light Microscopy. Annual Review of Physical Chemistry. 2012b;63:225–239. doi: 10.1146/annurev-physchem-032511-143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner WE, Orrit M. Illuminating single molecules in condensed matter. Science. 1999;283:1670–6. doi: 10.1126/science.283.5408.1670. [DOI] [PubMed] [Google Scholar]
- Murphy GE, Narayan K, Lowekamp BC, Hartnell LM, Heymann JAW, Fu J, Subramaniam S. Correlative 3D imaging of whole mammalian cells with light and electron microscopy. Journal of Structural Biology. 2011;176:268–278. doi: 10.1016/j.jsb.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natterer F. The Mathematics of Computerized Tomography. New York, NY: Wiley; 1986. [Google Scholar]
- Natterer F, Wübbeling F. Mathematical Methods in Image Reconstruction. Cambridge, New York: Cambridge University Press; 2001. [Google Scholar]
- Nixon SJ, Webb RI, Floetenmeyer M, Schieber N, Lo HP, Parton RG. A Single Method for Cryofixation and Correlative Light, Electron Microscopy and Tomography of Zebrafish Embryos. Traffic. 2009;10:131–136. doi: 10.1111/j.1600-0854.2008.00859.x. [DOI] [PubMed] [Google Scholar]
- Parkinson DY, Epperly LR, McDermott G, Le Gros MA, Boudreau RM, Larabell CA. Nanoimaging cells using soft x-ray tomography. Methods Mol Biol. 2013;950:457–81. doi: 10.1007/978-1-62703-137-0_25. [DOI] [PubMed] [Google Scholar]
- Plitzko J, Baumeister W. Focal issue on hybrid imaging. J Struct Biol. 2010;172:159. doi: 10.1016/j.jsb.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Plitzko JM, Rigort A, Leis A. Correlative cryo-light microscopy and cryo-electron tomography: from cellular territories to molecular landscapes. Curr Opin Biotechnol. 2009;20:83–9. doi: 10.1016/j.copbio.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Prendergast FG. Biophysics of the green fluorescent protein. Methods Cell Biol. 1999;58:1–18. doi: 10.1016/s0091-679x(08)61945-7. [DOI] [PubMed] [Google Scholar]
- Rigort A, Villa E, Bauerlein FJ, Engel BD, Plitzko JM. Integrative approaches for cellular cryo-electron tomography: correlative imaging and focused ion beam micromachining. Methods Cell Biol. 2012;111:259–81. doi: 10.1016/B978-0-12-416026-2.00014-5. [DOI] [PubMed] [Google Scholar]
- Sartori A, Gatz R, Beck F, Kossel A, Leis A, Baumeister W, Plitzko JM. Correlation microscopy: Bridging the gap between light- and cryo-electron microscopy. Microscopy and Microanalysis. 2005;11:16–17. [Google Scholar]
- Sartori A, Gatz R, Beck F, Rigort A, Baumeister W, Plitzko JM. Correlative microscopy: Bridging the gap between fluorescence light microscopy and cryo-electron tomography. Journal of Structural Biology. 2007;160:135–145. doi: 10.1016/j.jsb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Sayre D, Kirz J, Feder R, Kim DM, Spiller E. Potential operating region for ultrasoft x-ray microscopy of biological materials. Science. 1977a;196:1339–1340. doi: 10.1126/science.867033. [DOI] [PubMed] [Google Scholar]
- Sayre D, Kirz J, Feder R, Kim DM, Spiller E. Transmission microscopy of unmodified biological materials - comparative radiation dosages with electrons and ultrasoft x-ray photons. Ultramicroscopy. 1977b;2:337–349. doi: 10.1016/s0304-3991(76)91997-5. [DOI] [PubMed] [Google Scholar]
- Schneider G, Guttmann P, Heim S, Rehbein S, Mueller F, Nagashima K, Heymann JB, Muller WG, McNally JG. Three-dimensional cellular ultrastructure resolved by X-ray microscopy. Nature Methods. 2010;7:985–U116. doi: 10.1038/nmeth.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–51. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosinsky GE, Giepmans BNG, Deerinck TJ, Gaietta GM, Ellisman MH. Markers for correlated light and electron microscopy. Cellular Electron Microscopy. 2007;79:575–591. doi: 10.1016/S0091-679X(06)79023-9. [DOI] [PubMed] [Google Scholar]
- Spring H, Guttmann P, Rudolph D, Schneider G, Schmahl G, Trendelenburg MF. Applications of x-ray microscopy with hydrated specimens in biomedical research. Zoological Studies. 1995;34:214–216. [Google Scholar]
- Subramaniam S. Bridging the imaging gap: visualizing subcellular architecture with electron tomography. Current Opinion in Microbiology. 2005;8:316–322. doi: 10.1016/j.mib.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida M, McDermott G, Wetzler M, Le Gros MA, Myllys M, Knoechel C, Barron AE, Larabell CA. Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19375–19380. doi: 10.1073/pnas.0906145106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida M, Sun Y, McDermott G, Knoechel C, Le Gros MA, Parkinson D, Drubin DG, Larabell CA. Quantitative analysis of yeast internal architecture using soft X-ray tomography. Yeast. 2011;28:227–236. doi: 10.1002/yea.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Driel LF, Knoops K, Koster A, Valentijn J. Fluorescent labeling of resin-embedded sections for correlative electron microscopy using tomography-based contrast enhancement. Journal of Structural Biology. 2008;161:372–383. doi: 10.1016/j.jsb.2007.09.021. [DOI] [PubMed] [Google Scholar]
- van Rijnsoever C, Oorschot V, Klumperman J. Correlative light-electron microscopy (CLEM) combining live-cell imaging and immunolabeling of ultrathin cryosections. Nature Methods. 2008;5:973–980. doi: 10.1038/nmeth.1263. [DOI] [PubMed] [Google Scholar]
- Vicidomini G, Gagliani MC, Cortese K, Canfora M, Santangelo C, Boccacci P, Tacchetti C, Diaspro A. High throughput 3D correlative microscopy. Cytometry Part A. 2008;73A:102–103. [Google Scholar]
- Weiss D. Computed Tomography Based on Cryo X-ray Microscopic Images of Unsectioned Biological Specimenseditor. Göttingen: Georg-August University of Göttingen; 2000. [Google Scholar]
- Weiss D, Schneider G, Niemann B, Guttmann P, Rudolph D, Schmahl G. Computed tomography of cryogenic biological specimens based on X-ray microscopic images. Ultramicroscopy. 2000;84:185–197. doi: 10.1016/s0304-3991(00)00034-6. [DOI] [PubMed] [Google Scholar]