

Abstract
Simple, non-invasive tests for an early detection of degenerative dementia by use of biomarkers are urgently required. However, up to the present, no validated extracerebral diagnostic markers (plasma/serum, platelets, urine, connective tissue) for the early diagnosis of Alzheimer disease (AD) are available. In disease stages with evident cognitive disturbances, the clinical diagnosis of probable AD is made with around 90% accuracy using modern clinical, neuropsychological and imaging methods. Diagnostic sensitivity and specificity even in early disease stages are improved by CSF markers, in particular combined tau and amyloid β peptides (Aβ) and plasma markers (e.g. Aβ-42/Aβ-40 ratio). Recently, a novel gene/protein – ALZAS (ALZheimer ASsociated protein) – with a 79 amino acid sequence, containing the amyloid β-42 fragment (Aβ-42), the amyloid precursor protein (APP) transmembrane signal and a 12 amino acid C-terminal, not present in any other known APP alleles, has been discovered on chromosome 21 within the APP region. Reverse transcriptase-PCR revealed the expression of the transcript of this protein in the cortex and hippocampal regions as well as in lymphocytes of human AD patients. The expression of ALZAS is mirrored by a specific autoimmune response in AD patients, directed against the ct-12 end of the ALZAS-peptide but not against the Aβ-sequence. ELISA studies of plasma dectected highest titres of ALZAS in patients with mild cognitive impairment (presymptomatic AD), but only moderately increased titres in autopsy-confirmed AD, whereas low or undetectable ct-12 titres were found in cognitively intact age-matched subjects and young controls. The antigen, ALZAS protein, was detected in plasma in later clinical stages of AD. It is suggested that ALZAS represents an indicator in a dynamic equilibrium between both peripheral and brain degenerative changes in AD and may become a useful ‘non-invasive’ diagnostic marker via a simple blood test.
Keywords: Alzheimer disease, dementia, ALZAS protein, serum ELISA test, biomarker
Introduction
Pathogenesis of AD
Alzheimer pathology and diagnosis
-
Biomarkers of AD
- CSF biomarkers
- Plasma biomarkers
- Platelet biomarkers
- Structural biomarkers
-
ALZheimer ASsociated protein (ALZAS)
- ALZAS immunohistochemistry
- ALZAS-mRNA studies
- ALZAS as a future biomarker?
Conclusions
Introduction
Alzheimer disease (AD) is the most common cause of dementia in the elderly, accounting for 65–70% of all cases [1]. The lifetime risk for AD between age 65 and 100 is 33% for men and 45% for women with an annual increase of 1–2% in the seventh decade to almost 60% in the 10th decade with doubling every 5 years. AD is very common and thus is a major public health problem. As the world's population ages, the number of people with AD is expected to increase dramatically from approximately 24 million people in 2001 up to 81 million by 2040 [2]. The total worldwide yearly costs for the treatment and care of demented patients are estimated around 250 billion US dollars [3]. Dementia, after cardiovascular disease and malignancies, is the third frequent cause of death.
From a genetic viewpoint, AD is a heterogeneous disorder with both (rare) familial and (frequent) sporadic forms. Familial AD (FAD), representing less than 2% of the total, is an autosomal dominant disorder with onset before age 65 years, caused by mutations of the APP gene on chromosome 21 [4], and, less frequently, mutations in the highly homologous presenilin 1 (PSEN1) on chromosome 14, and presenilin 2 (PSEN2) on chromosome 1 [5–7]. While these latter forms are rare with a prevalence below 0.1%[8], a number of genes, e.g. the apolipoprotein E (APOE) ɛ4 allele, have been implicated in sporadic AD (sAD), which represents the vast majority of cases. The APOE ɛ4 allele operates as a major genetic risk factor in sAD and modifies the age of onset [9–12]. The common molecular mechanism of the mutations or polymorphism of these genes associated with the pathogenesis of AD is their promoting effect of amyloid β (Aβ) generation due to an imbalance of APP metabolism [13] (Fig. 1).
Fig. 1.
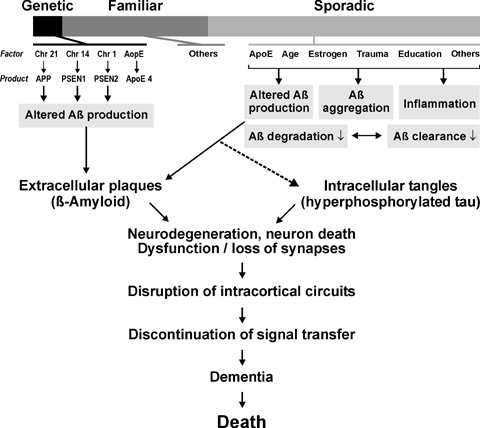
Major pathogenic factors causing Alzheimer disease.
Pathogenesis of AD
AD is a multifactorial and heterogenous disorder, the pathogenesis of which is not completely understood. A currently most popular hypothesis on its etio-pathogenesis is the ‘amyloid cascade hypothesis', according to which an imbalance between the production and clearance of Aβ in the brain is the initiating event, ultimately leading to neuronal/synaptic degeneration and dementia [14]. Altered cellular processing of the amyloid precursor protein (APP) with increased production or a reduced removal of cleavage products such as Aβ is a major factor for amyloid deposition [15]. APP is a member of a family of integral membrane proteins, which has been identified by sequentiation of Aβ deposited in AD brains [16]. On chromosome 21, 19 alternatively composed exons are coded for APP, which is up to 770 amino acids long. It is ubiquitous and expressed in various isoforms, possesses a long, extracellular/intravesicular N-terminal and a short cytoplasmic C-terminal end. All isoforms contain the full Aβ sequence. APP over-expression is the consequence of different stimuli, such as ischemia, trauma or inflammation in vitro or is achieved in transgenic (tg) animal models [17].
Consistent with this hypothesis, both intracerebral infusion of Aβ in FTDP-17 tau mutation P301L-expressing tg mice and crossing these animals with APP tg 257G mutations, showed exacerbation of neurofibrillary pathology [18, 19], and in the 3 × tg-AD mice, Aβ deposition was found to precede neurofibrillary pathology, being more severe than in double tg 2× tg-AD mice [20, 21]. However, in these mice harbouring PSEN1 APP and tau (P301L) synaptic dysfunction manifested in an age-related manner before plaque and tangle pathology [21], suggesting that plaque and tangle pathologies contribute to cognitive dysfunction at later points in time.
But, up to the present, no data from human conditions do support the amyloid cascade hypothesis (see [22]), and in the brains of AD patients there are no definite proofs for overexpression of the large type I transmembrane protein APP. Aβ is produced from APP as a result of two sequential proteolytic cleavages involving a membrane-bound aspartyl protease (β-secretase, BACE1) and two homologous membrane proteases (PSEN1 and PSEN2), corresponding to λ-secretase activites [23]. Inside raft clusters, APP is suggested to be cleaved by β-secretase and outside rafts by α-secretase [24]. This enzyme cleaves APP in the middle of the Aβ region to generate a secreted extodomain (α-APP) and a shorter α-cleaved COOH-terminal stub of APP (α-CTF) that is also cleaved by 7-secretase. Aβ-42, also suggested to be neurotoxic, is produced by proteolytic cleavage – numerous proteases are capable to cleave Aβ at multiple sites in vitro[25]– and accumulates as fibrillary aggregates in plaques and vessel walls (Fig. 2). The car-boxy-terminal fragments (CFTs), which show important neuropro-tective and neurotrophic activities, are transported by anterograde axonal transport and have functional importance for synapse formation [26]. Latest progress in understanding the role of soluble Aβ oligomers in AD has been reviewed recently [27].
Fig. 2.
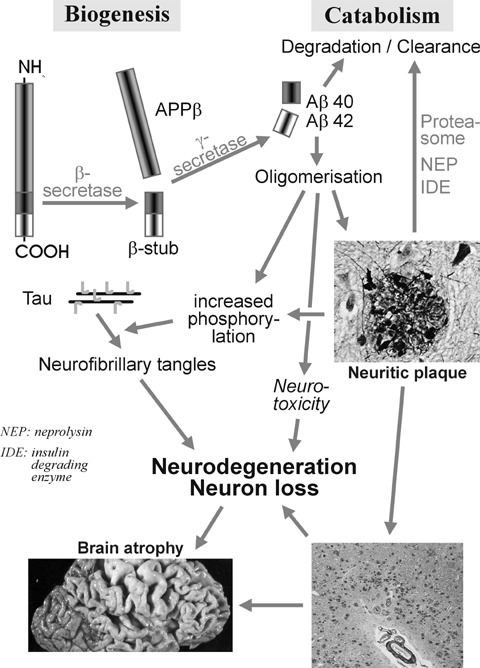
Molecular pathogenesis of Alzheimer disease. Deposition of amyloid β peptide after cleavage of amyloid precursor protein (APP) and hyperphosphorylation of tau protein induces formation of plaques, neurofibrillary tangles and neurodegeneration with neuronal and synapse loss.
The isolated role of Aβ in the pathogenesis of AD has been examined in vivo without inhibition of the complex cascade in the processing of APP tg mice carrying the construct of murine Aβ under control of the NF-L promoter showed significant pathological changes with intraneuronal expression of Aβ, widespread apoptosis and reactive gliosis finally leading to death. When Aβ peptide is transported by binding to an NCAM-signal peptide into the extracellular spaces, no pathological changes occur [21]. These data and the recent demonstration of intraneuronal accumulation of Aβ-42 as a probable product of normal neuronal metabolism with damage to synapses already before Aβ deposition in the brain tissue suggest its important role in the disease progression [17, 28] and as a source for extraneuronal aggregation of Aβ. The death of neurons, a prominent feature of AD, has been associated with the release of oligomeric intracellular Aβ-42 into the surrounding tissue, which may stimulate the production of amyloidogenic fragments of APP, amplify the levels of intracellular Aβ in neighbouring cells, and act as a nidus for the deposition of secreted Aβ[29, 30]. Recent studies, however, indicate that intraneuronal Aβ immunoreactivity, observed already in young human control brains, is not a predictor of brain amyloidosis or neurofibrillary degeneration [31]. There are only weak correlations between amyloid deposits (numbers of plaques) and neuronal dysfunction in both human brains and tg mice [32], and the rate of brain atrophy is not determined by the amount of insoluble Aβ in the grey matter [33]. However, there is a direct relationship between synaptic loss and soluble Aβ which has induced the Aβ-derived diffuse diffusible ligands (ADDL) hypothesis that suggests that soluble oligomeric are highly neurotoxic proteins and might be the culprits damaging function and plasticity of synapses, thus disrupting cognitive functions [27, 34–37]. Exogenous, soluble (blood-borne) Aβ peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons [38].
APP tg B6SJL mice immune hyporesponsiveness to Aβ suggesting that an Aβ-specific impaired adaptive immune response may contribute to neuropathology [39]; increase of autoantibodies to Aβ in AD patients suggests that a humoral immune response to Aβ may promote neuronal degeneration, a process with implications for future vaccine-based therapies of AD [40]. Short Aβ immunogens have been shown to reduce cerebral Aβ load and learning deficits in an AD mouse model in the absence of an Aβ-specific cellular immune response [41], and it is anticipated that selected Aβ human monoclonal antibodies could reduce and inhibit Aβ deposits in brain [42] in the absence of Aβ-specific T-cell reactivity [43], which requires careful antigen and antibody selection to maximize efficacy and minimize adverse events [44].
Almost in parallel with the deposition of Aβ in plaques, tangles have been shown to be composed of abnormally phosphorylated tau protein [45]. Tau is a normal axonal protein that binds microtubules, promoting their assembly and stability, its hyperphosphorylation causing disassembly of microtubules and thus impaired axonal transport, compromising neuronal and synaptic function [46, 47]. Tau protein forms abnormal fibrils in AD and other neurodegenerative diseases, called tauopathies. It is highly soluble and adopts a natively unfolded structure in solution, whereas in its phosphorylated, filamentous form, e.g. neurofibrillary tangels (NFT), small segments of tau adopt a β-conformation, and interact with other tau moledules, giving rise to a fuzzy coat of the filaments [48]. The relationship between Aβ and tau pathologies and their impact on the pathogenesis of AD are still a matter of controversy. Biochemical studies on the course and distribution pattern of their pathologies suggest independent but synergistic affects [49]. Some authors suggest that AD is a tauopathy fueled by APP dysfunction [50], and that tangles either antecede plaques or – less likely – are independently found [51]. Others believe that increased formation and deposition of Aβ caused by dysmetabolism of APP that normally occurs in neurons and synapses [52, 53] induce early pathological phosphorylation of tau via activation of caspases, the key enzymes in the final cascade of programmed cell death, inducing the formation of NFT [54]. Activity of caspase-6 that cleaves tau at amino acids 402 and 13 was detected in mild to severe AD cases [55], particularly in the entorhinal cortex in absence of plaques and tangles, indicating that it is activated as an initial site affected in AD, resulting in neurodegen-eration and inflammation. Recent developments suggest that axonal transport of APP influences Aβ-deposition and APOE ɛ4 influences both axonal tau phosphorylation and amyloid-induced pathology, thus connecting the pathogenic mechanisms of Aβ, tau and APOE ɛ[56]. An ‘alternative’ amyloid cascade hypothesis suggests that amyloid deposition does not represent the initial cause of AD, but is the consequence of other processes, such as abnormalities in proteins regulating the cell cycle, oxidative stress, mitochondrial dysfunction with disruption in neuronal energy metabolism [57–60], and may represent a neuroprotective reaction [61, 62]. Another, alternative interpretation of the amyloid hypothesis speculated that accumulations of intramembranous Aβ peptides might affect the functions of APP itself and the assembly of PSEN 1, nicastrin and other complexes [63], while another new hypothesis about Aβ toxicity gaines some support regarding its effects on ion channel functions [64]. However, due to deviating findings in tg mouse models of AD [17, 65], currently neither the exact course of the molecular cascade leading to neurodegeneration in AD (Fig. 2) nor the final pathological cascade leading to neuronal death is fully recognized. The different etiologic factors suggested to be active in the development of AD pathology may lead to some common downstream pathogenic events that ultimately precede the disease, but further extensive basic research, including the use of animal models, is warranted [66].
Alzheimer pathology and diagnosis
Sporadic AD is a slowly progressive disorder with insidious onset and duration of around 10–12 years. Its diagnosis and the differentiation from other dementing disorders is difficult and only in rather progressed stages of the disease, when memory and other cognitive disorders are severe enough to have a significant impact on the everyday life of the patient, the diagnostic accuracy when using all currently available tests is around 90%. Prior to a pathological diagnosis, only a ‘possible’ or ‘probable’ diagnosis of the disease can be made using current criteria [67–70]. A definite diagnosis of AD is only possible by the histopathological detection of its characteristic changes, the accumulation of extra-cellularly aggregated Aβ in plaques and cerebral vasculature (cerebral amyloid angiopathy / CAA) and intracellular and neurit-ic deposition of hyperphosphorylated tau protein (NFTs, neuropil threads and neuritic plaques), associated with loss of synapses and neurons in specific brain regions and loss of neuronal connectivity as a major cause of dementia (see [71, 72]). Although most of these changes are non-specific, they represent the major morphologic markers of AD, the (semi)quantitative assessment of which are the basis of current morphological criteria for the diagnosis of AD: CERAD [73], Braak & Braak, and National Institute on Aging – Reagan Institute (NIA-RI) criteria combining both CERAD and Braak stages [74], which gave good correlations between clinical data and pathological findings between AD and non-demented controls [72]. The degree of dementia usually correlates significantly with the density and extension of tau pathology, while it shows only weak correlations with the number of senile plaques but better ones to the total load of soluble Aβ (see [72, 75]).
Autopsy studies indicate that tangle-tau pathology first appears in the medio-basal temporal lobe, particularly in the (trans)entorhinal cortex, progressing via the hippocampus to the isocortical association areas with final involvement of subcortical nuclei [76, 77]. This distribution pattern correlates with early impairment of memory due to interruption of the GABAergic ‘perforant path', which connects the hippocampus with other areas of the brain, thus isolating the hippocampus from ingoing and outgoing stimuli. The rather stereotypic development of tau pathology differs from the phases of amyloid deposition usually beginning in the neocortex with anterograde involvement of the allocortex into regions that accept neuronal projections of already involved areas [78].
Neurodegeneration in AD is estimated to start 20–30 years before clinical onset, during which preclinical phase plaque and tangle load increase and at a certain threshold the first symptoms appear [1, 72]. This clinical phase is usually designated ‘mild cognitive impairment’ (MCI), during which subjects have measurable cognitive deficits, but which are not sufficient to fulfill criteria for any specific dementing disease [79, 80]. It has been suggested to constitute a transitional stage between normal aging and AD, but data show that many patients with amnestic MCI have early neu-ropathological changes of AD including hippocampal synaptic loss and, thus, represent early AD [81–83], but many cases with CDR 0.5 show variable pathologies not restricted to AD [84–86]. While some of these patients may remain stable or even may improve, many of them convert to AD with an annual conversion rate to clinical dementia of 10–19%[87, 88]. Preclinical AD usually shows more severe neuritic pathology than ‘normal’ aging with variable intensities of tau and amyloid biochemistry. The combination of clinical, neuropsychological and imaging methods differentiated various subtypes of MCI [89–92], which, however, did not show significant differences in their annual conversion rate to dementia [88].
Biomarkers of AD
Biomarkers are required to improve the diagnostic sensitivity and specificity and to monitor the biological activity of AD in terms of the burden of neuronal involvement and the tempo of disease progression. In view of the advancing scientific knowledge regarding biomarkers for AD/MCI, it was proposed to incorporate those biomarkers in revised diagnostic criteria in the future [91–93]. Biomarkers will initially supplement our more traditional neuropsychological and imaging markers and may progress to useful surrogate clues to the pharmacological action of anti-dementia compounds [94–100].
CSF biomarkers
Senile plaques in AD brain are mainly composed of Aβ-42, while CAA mainly contains Aβ-40 [101, 102]. An inverse relation between in vivo amyloid load and CSF levels of Aβ-42 has been found in human beings [103]. CSF of AD patients even in early phases of the disease shows decreased values of Aβ-42 and, together with increased total tau (tTau) or phosphorylated tau (pTau), suggests the presence of cognitive dysfunctions even in healthy elderly subjects [104, 105]; CSF tau/Aβ-42 ratio is a prediction of cognitive decline in non-demented older adults [106–110] and with MCI [110–113]. Together with medial temporal lobe atrophy on MRI [114–118] or FDG-PET [119] and presence of the APOE ɛ4 allele, these markers predict dementia in mild cognitive impairment or higher risk for developing AD [83, 89, 120] as does rCBF-SPECT [121] and cerebral hypoperfusion induced by fMRI [122] and plasma Aβ-42, medial temporal lobe atrophy and homocysteine [123]. Antemortem CSF levels of Aβ-42, tTau and pTau-Thr231 have been reported to reflect the histopathological changes observed postmortem in the brains of AD cases [124, 125]. The CSF levels of tau are markedly increased in patients with diffuse axonal injury in head trauma, which revert on clinical improvement [126]. Thus, bulk of the evidence supports that CSF reflects the state of the brain protein metabolism; CSF levels of toxic-advanced glycation end products (TAGE) may also help in early detection of AD [127]. At present, the combination of elevated CSF tTau or pTau proteins and low CSF Aβ-42 are the only biomarkers with enough sensitivity and specificity to serve as useful diagnostic biomarkers capable of distinguishing AD from other dementias in the early stages [96, 128–136], PD patients with dementia (PDD) from those without [137] and DLB [138], while CSF serpin levels did not improve the diagnostic classification of AD versus DLB [139]. They are possible markers for severity and abundance of symptoms in AD [71, 105, 140–144] (Table 1). Other studies demonstrated that significant elevation of BACE 1 levels and activity in CSF is an indicator of MCI or other neuroimmune markers indicating AD [145, 146]. Whereas progressive increase of tTau CSF concentrations was found from early to advanced stages of AD [147], soluble Aβ-42 has been shown not to be related to the degree of cognitive impairment [140], and Aβ-42/40 ratio but not Aβ-42 alone correlates with pTau in patients with low and high CSF Aβ-40 load [148]. Recent longitudinal studies showed that levels of CSF Aβ-42 and tTau, but not pTau at threonine 181, increased over time in a memory clinic patient cohort with comparable changes in all diagnostic groups. However, the cross-sectional difference between diagnostic groups exceeded by far the longitudinal changes within individuals, suggesting that these biomarkers are not sensitive as markers of disease progression [149]. The causal relations between progress of Aβ load and plaque density in the brain and CSF Aβ reduction are not completely understood, but they were attributed to depletion of the monomeric protein into oligomeric soluble and insoluble forms in the brain and increased Aβ deposition in plaques [94]. Age and APOE ɛ4 allele accelerating pathogenic Aβ-42 brain deposition starting in late middle age in persons with normal cognition are causing decrease of CSF Aβ-42 but not the Aβ-40 concentration [150], while CSF Aβ-40 levels in frontotemporal dementia (FTD) are decreased [151] and fluctuations of CSF Aβ levels have been found in individual subjects [152]. Levels of ApoE in CSF are correlated with tau and 24S-hydroxycholesterol in patients with cognitive disorders [153, 154]. Extreme CSF Aβ levels identify familial eoAD and loAD PSEN1 mutations and, thus, can be useful endophenotypes for genetic AD [155] as well as for children with Down syndrome [156], but inverse relations between in vivo amyloid imaging load in human brain and CSF Aβ-42 have been found [103].
Table 1.
| Test | Cutoff (pg/ml) | Specificity / Sensitivity | MCI →AD |
|---|---|---|---|
| clin. AD versus controls /versus non-AD dementias | |||
| tTau | 300–500 | 0.85–1.0 / 0.40–0.89 | SP 0.70 |
| 250 | AD versus LBD SP 0.76 | ||
| 2130 | CJD versus AD 1.0 / 0.93 | ||
| Aβ-42 | 375–500 | 0.47–1.0 / 0.78–1.0 // 0.6–0.9 / 0.85 | 1.0 / 0.83 |
| Aβ-40 | Not relevant (no differences) | ||
| Aβ ratio (40 / 42) | 0.82 / 0.58 | ||
| Tau /Aβ ratio | 0.5 | 0.82 / 0.58 | AD versus LBD 0.84 / 0.79 |
| Tau × Aβ ratio | 0.88 / 0.69 | ||
| tTau + Aβ-42 | 0.81–1.0 / 0.63–0.89; 0.75–0.93 / 0.71 | 0.90 / 0.83 | |
| AD versus LBD SP 0.67; | |||
| AD versus VaD 0.58–0.83 / 0.90 | |||
| AD versus FTD SP 0.58 | |||
| pTau | 0.80–0.85 / 0.90–0.94 | MCI →AD 1.0 / 0.65 | |
| AD versus FTD 0.92 / 0.90 | |||
| pTau + Aβ-42 | 0.97 / 0.89 | 0.80 / 0.73–0.89 | |
| AD versus FTD 0.93 / 0.72 |
Formulas:
Specificity (SP) = definitely negative / (definitely negative + wrong positive)
Sensitivity (ST) = definitely positive / (definitely positive + wrong negative)
The aspects of Aβ as a biomarker for AD have been reviewed recently [25], as well as the variable patterns of CSF Aβ between synucleinopathies and tauopathies [157] or other neurodegenerative dementias [158, 159]. While initial studies suggested that CSF pTau protein correlates with neocortical neurofibrillary pathology in severely demented AD patients and may serve as in vivo surrogate marker of tangle tau pathology in AD [125], other recent studies showed no association of CSF biomarkers (Aβ-42, t- and pTau) with APOE ɛ4, plaque and tangle burden in autopsy-confirmed AD [160, 161]. Increased CSF tau is also seen in other CNS disorders with neuronal loss (degenerative, inflammatory, vascular, tumours), with highest CSF tau concentrations in Creutzfeldt-Jakob disease (CJD) and brain infarcts, the former showing a dissociation between tTau and pTau [162], while AD and AD+CVD display a similar neurochemical phenotype with increased tau and diminished Aβ-42, with some overlap between AD and VaD, the latter showing a decrease of CSF Aβ-38/40 [158, 163], as well as in plasma [164]. CSF Aβ peptides have also been suggested to distinguish between DLB, PDD, AD and FTD [158, 165]. In relatively young dementia patients, CSF neurofilament protein levels may play a role in the discrimination between FTD and early onset AD, especially in combination with Aβ-42 and pTau analysis [166]. CSF antimicroglial antibodies are also a putative marker of an ongoing inflammatory process in AD [167]. However, since most studies evaluating biomarkers are compared only with clinical rather than pathological diagnoses, the concept of in vivo surrogate markers should be further explored. Re-evaluation using immunoprecipitated CSF samples of neuropathologically defined dementing disorders will be necessary to determine, whether Aβ-40 and Aβ-42 will be applicable as neurochemical dementia markers. Comparative proteomics of CSF detecting a large number of hitherto unknown proteins in autopsy-confirmed AD and non-demented elderly subjects may provide a further means to diagnose and assess AD with 90% sensitivity and 83% specificity [129, 168–175] and to distinguish between various degenerative dementias [176]. Preliminary studies showed that redox-reactive antibodies in CSF may represent valuable biomarkers for AD diagnosis [177].
Plasma biomarkers
Plasma total amyloid or Aβ-42 is increased in cases of familial AD and trisomy 21 [178, 179], but were not consistently related to diagnosis in clinic-based cross-sectional studies of typical late-onset AD [178, 180–185].
Elevated levels of Aβ-42, low levels of Aβ-40 and a reduced Aβ-42/Aβ-40 ratio in plasma of old subjects indicate a conversion of cognitive normality to MCI or AD [186–188], and have been found in geriatric depression [189]. Others, however, found that a reduction in Aβ-42 plasma levels may be a marker for AD status, specifically a transition from normal status or MCI to AD [190], rather than a marker for neurodegenerative processes occurring in the disease [191]. Due to a high intra- and inter-personal variability of serum and plasma Aβ levels [192], plasma levels of Aβ-40 and Aβ-42 are not robust correlates of histologically or biochemically assessed amyloid burdens in brain and the origin of plasma Aβ is not completely understood [193]. Plasma Aβ levels are probably modulated by peripheral and brain metabolism and clearance as well as by transport across brain, CSF and vascular compartments. Although all forms of brain Aβ are elevated in AD, the weak correlations of the various brain Aβ measures in AD suggest that they may reflect distinct biochemical and morphological pools of Aβ[194]. The normal equilibrium between CSF and plasma Aβ may be disrupted with the initiation of Aβ deposition in the brain [195], but plasma and CSF Aβ levels are not correlated in AD [196], and Aβ-42 plasma levels are considerably influenced by concomitant medication [197], in particular by insulin treatment [198]. It is not unexpected, therefore, that plasma and brain Aβ levels are not strongly correlated. Reduction of CSF and plasma Aβ has been observed in cerebral amyloid angiopathy (CAA), amyotrophic lateral sclerosis, and dementia with Lewy bodies (DLB), and in CJD a dissociation of the Aβ peptides in CSF has been observed [199]. Measurement of CSF tau together with serum heart type fatty acid-binding protein (CH-FARP) to CSF tau may represent marker candidates for the differentiation between AD and DLB [200]. It is generally agreed that plasma Aβ-42 levels alone seem not to be reliable biomarkers for MCI and AD [123, 183, 201], while a decrease in the ratio of plasma Aβ-38/-40 peptides is considered a blood-based biomarker for vascular dementia, its diagnostic accuracy resembling that of CSF markers for AD [164]. Furthermore, blood inflammatory markers, like CRP, interleukin-6, etc., are markers for VaD [164, 202] or are increased before clinical onset of both AD and VaD [203], while other inflammatory molecules are associated with AD progression [204]. If plasma Aβ is a risk factor for AD as is suggested by several longitudinal cohort studies, the relevant Aβ levels may be those seen 5–20 years before death [187, 205]. Furthermore, changes in plasma Aβ-42 may be a biochemical predictor of rivastigmine treatment efficacy [206]. Proteomic analysis of plasma revealed higher concentrations of α-1-antitrypsin and apolipoprotein J in AD patients [207]. Recent proteomic discovery of various plasma signaling proteins may allow the development of a simple, cost-effective test for AD [208, 209].
Plasma homocysteine has been shown to be directly related to Aβ-40 levels, while the association with Aβ-42 was not significant, suggesting that homocysteine is related to aging but not specifically to AD, but it could interact to affect AD risk and cognition in PD [210, 211]. Recently, some proteome-based plasma serum biomarkers were shown to be specific for AD and to correlate with disease severity, although alternative assays will be necessary to improve sensitivity and specificity [212, 213].
Platelet biomarkers
Among changes of blood platelets, APP abnormalities have been suggested to predict conversion of MCI to dementia of the AD type [214, 215], and have been correlated with membrane fluidity and cognitive decline [216]. Increased BACE-1 activity in AD [217] and increase of monoamine oxidase B expression in blood platelets – and in brain – of demented patients with both AD and PD has been repeatedly confirmed [218–220]. Recently, increased platelet phospholipase A2 activity has been detected in patients with AD, VaD and ischemic stroke [221].
Structural biomarkers
The value of structural MRI-derived biomarkers for AD has recently reviewed [222, 223]. Medial temporal lobe atrophy (MTA) on MRI is sensitive to primary degenerative hippocampal atrophy in old subjects, but not specific for AD pathology, mild MTA score not being frequently associated with dementia [224], but for MCI [225]. The level of elevated PET amyloid ligand (11C)PIB uptake in patients with MCI / mild AD is suggestive of early AD process [226–229], even in non-demented individuals [230]. While analysis of CBF-SPECT, CMRgl-PET (glucose metabolism), proton spectroscopy (H-1 MRS), high-field strength functional MRI, voxel-based morphometry, increased activation of the mediobasal temporal lobe detected by fMRI, (R)-[(11)C]PK11195 PET to detect microglia, and other functional neuroimaging methods, in particular, combined PiB imaging and structural MRI, are used as sensitive markers for conversion of MCI into early AD [121, 231–241]. The same is true for accelerated rates of hippocampal atrophy and ApoE ɛ4 [114], FDG-PET [242], detection of subcortical hyperintensities [243–245] and MRI patterns of grey matter atrophy [246]. However, comparatively normal glucose metabolism in the presence of high frontal amyloid load suggests that amyloid plaque formation may not be directly responsible for neu-ronal dysfunction in this disorder [247].
A recent review of the neuropathological basis of MR-defined cerebral lesions indicated that the presence of lacunes and WMLs provide a good signal for VaD, whereas cortical and hippocampal atrophy in aging and dementia are complex with several processes converging on similar brain structures that mediate cognitive decline [248].
ALZheimer ASsociated protein (ALZAS)
Recently, a novel, hitherto unknown Aβ protein expressed in elderly patients with the diagnosis of probable AD was discovered on chromosome 21 within the APP region. This protein, ALZAS (ALZheimer ASsociated protein), with a 79 amino acid sequence contains the Aβ-42 fragment, the AP transmembrane signal and a unique 12 amino acid c-terminal, which is not present in any known allele of the APP [249–252]. In contrast to theoretic splice variants, it has its starting codon within the exon 16 and its coding sequence ends in intron 17 (Fig. 3). Reverse transcription PCR revealed the expression of the transcript of this protein in cortical and hippocampal brain regions as well as in lymphocytes of AD patients [249–251, 253, 254]. Using cloning methods, the genetic structure of ALZAS has been clarified [255].
Fig. 3.
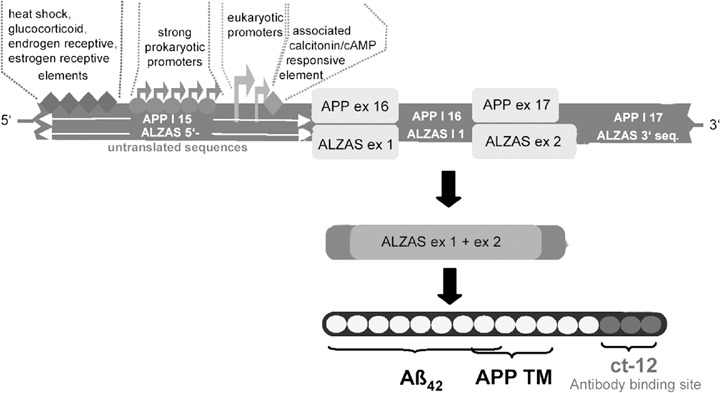
Scheme of the ALZAS gene structure (A), mRNA (B) and protein in the region of the APP gene (C) on chromosome 21. APP TM = transmembrane region.
Regulating the access of α- and β-secretases to APP and ALZAS seems to be of importance in view of recent CSF studies providing a direct evidence that Aβ-fragments (truncated Aβ 1–16, 1–33, 1–39 and 1–42) in CSF distinguish sporadic AD from non-demented controls with an overall accuracy of 86%[141]. The finding that Aβ 1–16 was the most abundant peptide may indicate that ALZAS with its predicted β-helical structure may be a substrate for α-secretase [171, 253]. Moreover, it could act as molecular chaperone that binds APP and assists in altering its conformation. ALZAS, as a specific Aβ protein, may compete or even out-compete APP for the APP transmembrane transit site in specific neurons, paralleled by intraneuronal neurotoxic accumulation of Aβ[250] and activating phospholipases, resulting in an ongoing membrane decay and synaptic disruption in AD (Fig. 4) [256]. Currently, little is known about the process in which such peptides, like Aβ, ALZAS or prions, may induce changes in neuronal phenotypes and microglial activation [257].
Fig. 4.
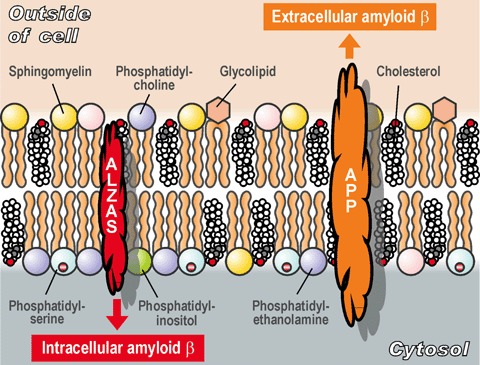
Hypothetical competing action of ALZAS with transmembrane APP.
Pilot studies in serum of patients with probable AD have detected an up to tenfold increase of the ALZAS antibody titre directed against the non-amyloidotic C-terminal of this protein. Serum ELISA studies revealed the highest titres in early stages of the disease, i.e. in patients with presymptomatic AD or MCI, but moderately increased titres in fully developed and autopsy-confirmed AD. Low or undetectable anti-ct 12 titres were found in healthy age-matched subjects or young controls (Fig. 5). Maximal values were found in the sera of patients over age 65 years who had been diagnosed as ‘depressed’ without recognizible cognition disorders. It should be emphasized that depression is common in early stages of AD (up to 87%) and is associated with significant morbidity [258–261]. Increased hippocampal tangles and plaques in patients with AD and a lifetime history of major depression suggest an interaction between both disorders [262].
Fig. 5.
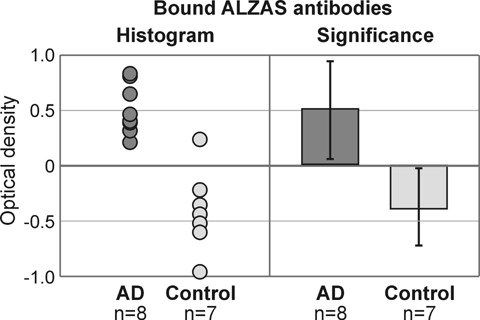
ELISA studies of EDTA plasma samples (dilution 1:20, preincubation 4 hrs, urea 1 M at 37°C). Detection of ALZAS IgG antibodies in eight AD patients and seven controls (signal: optic density /OD/ 492–620 nm).
ALZAS immunohistochemistry
Intraneuronal deposition of ALZAS protein has been demonstrated in brain slices of frontal cortex and hippocampus from autopsy-proven AD patients, using a chicken antibody, cleaned by high affinity chromatography, whereas controls were negative (Fig. 6). Wilcoxon statistics of the intraneuronal and capillary immunostain-ing of ALZAS showed significant correlations with Braak stages and NIA-Reagan classification of AD (Fig. 6A and B). Nine cases (30%) were ALZAS negative, one (3.3%) showed positive ALZAS neuronal but negative capillary decoration, while positive neuronal and capillay decoration was present in 20 cases (66.7%). In postmortem AD brain, ALZAS immunoreactivity was located intracellularly in neurons and in the inner vascular membranes (Fig. 6B and C).
Fig. 6.
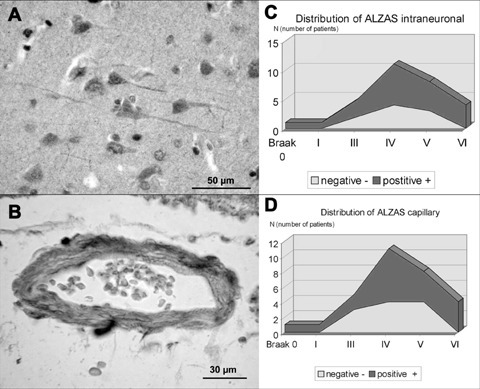
Immunostaining of ALZAS protein in human brain tissue showing intraneuronal deposits in frontal cortex (A) and capillary immunostaining (B). Distribution of ALZAS intraneuronal and intracapillary staining in frontal cortex of AD patients significantly correlates with neuritic Braak stage (C, D).
ALZAS-mRNA studies
To further verify the postulated ALZAS protein, the ALZAS-mRNA isolated from human postmortem AD brain was amplified and sequenced using primers from regions of ALZAS gene from frontal cortex of AD brains and age-matched controls. ALZAS mRNA expression was examined by qRT-PCR. The received sequence correlated with the postulated mRNS sequence of the APP-exons 16 and 17 as well as to parts of the adjacent introns APP I 15 and 17 [253].
An initial quantification of ALZAS mRNA in different tissues using RT-PCR showed that ALZAs is transcribed in all examined tissues (lymphocytes, cerebral cortex), but a preliminary assessment of the results did not show essential differences in the transcription rates between AD patients and healthy controls. Moreover, cell culture studies, using mouse embryonic stem cells that harboured an ALZAS transgene, which included the calcitonin responsive promoter, showed that ALZAS expression was initiated by adding calcitonin to the culture. In cultured transgenic mouse stem cells, ALZAS was located in neuronal plasma membranes of differentiating embryonic ES cells. Intraneuronal immunolabling of cortical sections from AD brain with Aβ specific 4G8 antibody showed similar decoration of neurons as in transgenic mouse stem cells (Fig. 7). Moreover, the ALZAS gene was cloned in an inducable expression vector for further transfection in human neu-roblastoma cells (SH-SY5Y) [253].
Fig. 7.
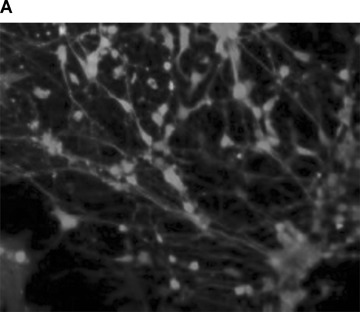
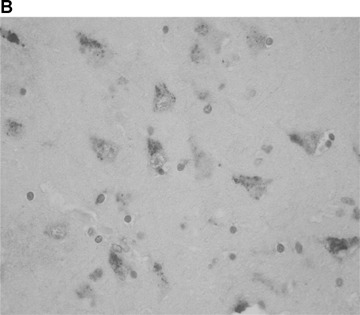
ALZAS protein is expressed in transgenic mouse stem cells (A) and intraneuronally in frontal cortex of human AD patient (B). Aβ-specific 4G8 immunostaining.
Despite increased ALZAS antibody titres in AD patients, the transcription in the examined tissues of AD patients and controls appears similar, and there are only some differences between tissues, with highest transcription and lowest transcription differences between ALZAS and APP in the blood. Up to now, the role of ALZAS in cell metabolism is highly speculative. Further studies should clarify whether ALZAS per se or in interaction with his ‘bigger brother’ Aβ-42 leads to the known disorders of metabolism and protein aggregation in AD brain. It is further unknown which type and severity of ALZAS expression is necessary, which cells are predominantly involved and by which mechanisms the pathogenic expression of ALZAS in AD patients is induced. Cloning of the ALZAS promoter region in an expression vector may enable us to find transcription factors that selectively influence the gene expression and thus probably may trigger the pathogenesis of AD. Individual findings in hitherto examined blood probes and comparison of sequences of the promoter region could indicate probable hormonal reactions.
ALZAS as a future biomarker?
The plasma of AD patients in comparison to age-matched healthy controls contains increased concentrations of ALZAS antibodies, which could be confirmed by repeated studies [255]. Using an ALZAS-Capture enzyme-linked immunosorbent assay (ELISA), specific IgG antibodies were detected that exclusively are directed against the C-terminal end, coded at the intron-region of APP. Preliminary data also revealed an increase of ALZAS peptide, possibly acting as autoantigene, in the serum of later stages of AD (Fig. 8B). However, the distinction between AD and other dementing disorders, such as vascular dementias, has to be examined by further studies. Significant elevation of serum ALZAS IgG was detected in patients with early stages of dementia, in whom neuropsychological test results were still above the threshold levels of dementia. Even in patients with MCI who later developed AD dementia, at base level increased ALZAS values were detected. This might be interesting for an early diagnosis of AD and could be a valuable sign for early treatment. However, there is considerable variability of the specificity and sensitivity of ALZAS protein depending on the compared groups, the statistical methods and the cut-off values [253, 254]. In addition, the influence of age has to be considered, which is currently under investigation.
Fig. 8.
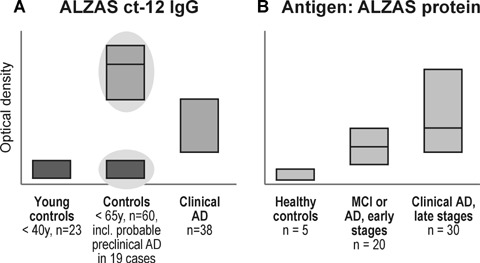
ELISA studies of EDTA plasma samples to detect anti-ALZAS IgG revealed increased levels in MCI patients (n= 19) and, less severe, in patients with AD dementia (n= 38) as compared to young and aged controls (A). There is only mild increase of ALZAS protein in plasma of MCI patients but more severe in AD dementia (B).
Conclusions
Progress in our understanding of the molecular pathology and pathogenesis of AD and its relationship to brain aging have provided clinically meaningful advances in the development of biomark-ers that are based on biochemistry and neuroimaging methods. They should have the potential to provide effective measures of early diagnosis, biological acitivty, disease outcome, and of indicating surrogate endpoints for their clinical use in diagnostic and preclinical purposes and for monitoring treatment. Because most clinical criteria lack specificity, particularly early in the course of disease, and most studies related marker values with clinical criteria and only very rarely with neuropathological diagnoses, quoted sensitivities and specificities for putative biomarkers are difficult to interprete. The currently best validated CSF biomarkers, pTau and Aβ-42, with a reported sensitivity and specificity of around 90% for the diagnosis of AD, show at least some, if inconstant, correlations with cerebral tau and Aβ pathology [125, 130, 160, 161]. As biomarkers in plasma several substances have been examined, e.g. isopostane, 3-nitrotyrosin, α-1-antichymotrypsin, interleukines, C-reactive protein, C1q complementary systems, 24S-hydrocholesterol, homocysteine, but none of these markers had enough sensitivity or specificity to diagnose AD [100, 134]. The same appears to be true for serum Aβ peptides, which do not reflect the Aβ load in the brain. Although in combination with MRI studies of the mediobasal temporal lobe they may indicate conversion of cognitive normal seniors to MCI or AD [123], but alone they are not reliable biomarkers for AD. In the future, assessment of autoantibodies in serum, serum platelets and lymphocytes against Aβ and RAGE (receptor for advanced glycation end products) appear of increasing interest [122, 123, 263, 264], and may indicate close relations between AD and autoimmune disorders [265].
Chronic inflammation with microglia activation is believed to play a central role in the pathogenesis of lesions in the central nervous system of patients with AD or multiple sclerosis (MS). In AD, abundant microglia activation is present in the affected cortex [266]. Microglia cells activated in vitro under AD-like conditions, such as by the addition of Aβ peptide to the culture medium, produce a variety of pro-inflammatory and toxic cytokines and mediators [267–269]. Such toxic factors have been suggested to augment or promote amyloid deposition and neuronal degeneration [270–272]. In contrast, active immunization or passive transfer of specific antibodies against the Aβ peptide have been shown to reduce amyloid deposits in tg models of AD and in patients [273]. Microglia cells and astrocytes are believed to play an important role in the clearing of Aβ deposits [274–277]. This may occur in experi-mental models even in a bystander fashion without the involvement of specific adaptive immune responses against Aβ[41].
Although MS has long been considered a demyelinating disease of the white matter, extensive cortical demyelination has been shown as a characteristic hallmark of the pathology of progressive MS [278, 279]. At this stage, profound inflammatory infiltrates composed by T and B lymphocytes in the meninges are associated with severe microglia activation in the cortex and the formation of widespread subpial bandlike demyelinated lesions [280–282]. Microglia cells activated in the MS brain also produce pro-inflammatory cytokines and toxic factors, which contribute to demyelination and axonal lesions [283]. To what extent chronic inflammation in the cortex of MS patients influences the development of AD lesions is so far unresolved.
A recent study of autopsy cases of MS, AD and age-matched controls showed profound microglia activation in both MS and AD cortices with similar patterns. But microglia activation in MS cortex, in contrast to that in AD and controls, correlated with lymphocyte and plasma cell infiltration into the meninges. Old MS patients showed AD pathology with comparable incidence as in the course of normal aging, and the density of Aβ plaques and NFTs did not differ between demyelinated and non-demyelinated cortical areas. These data suggest that the non-specific activation of microglia in MS cortex, which has been described to decrease Aβ load in experimental animals sensitized with copolymer I or adjuvant alone [41], has little or no interference with the development of cortical AD pathology [284], and does not play a major role in Aβ clearance in human beings, unless specific Aβ antibodies are present [275].
The ct-12 amino acid sequence (c-terminal immune response-elicitating sequence) that recognizes endogenous ALZAS antibodies, may convert involved cells to targets of microglial cells in brain tissue that induce an immune cascade and neuroinflammation processes as side effects of AD. In this connection, binding of ALZAS to the RAGE receptor, acting as amplification and activation factor of microglia, is under discussion. RAGE recognizes substances that are formed during chronic and age-related disorders, including Aβ[264]. Most recent studies demonstrate that the conversion of MCI to AD dementia is associated with inflammatory processes [285]. Microglia activation in AD appears to be driven by innate immunity [286–288]. A CD14-dependent inflammatory response to Aβ oligomers provides the basis for a hypothesis of a structural mimicry between aggregated, highly hydrophobic amy-loidogenic proteins and biophysically similar pathogen-associated microglial patterns, contributing to chronic neuroinflammation and neurodegeneration [289] that is suggested to be a consequence of microglial dysregulation and overactivation [290]. ALZAS peptide represents an additional source of Aβ, an auto-antigen due to its unique amino acid sequence (ct-12) and is predicted as pathogen that may induce both inflammation and neurodegeneration. The contribution of age-related loss of microglial neuroprotective functions to AD pathogenesis [286, 291] remains a plausible option. ALZAS as inducable gene could be a ‘missing link’ in the search for the causes of AD (Table 2 -[271]). Since ALZAS peptide like APP contains the transmem-brane signal and, therefore, concurs with APP for the deposition into the cell membrane, it may have relationship to membrane dysfunction and disorders of axonal transport in AD. While the clinical and pathological effects of active immunotherapy of AD with synthetic Aβ-42 are well established in animal models [273, 292], few reports have been made about the neuropathological findings in patients who received immunotherapy with Aβ-42 [274, 293–299]. These findings include various degrees of reduction in amyloid plaque burden, but persistence of tau pathology and CAA. Lymphocytic infiltration, characteristic of meningoen-cephalitis [295, 296], was not observed in all cases [297, 300], while others showed minimal effects in Aβ plaque reduction but severe CAA with brain haemorrhages [301, 302].
Table 2.
Suggested role of ALZAS in pathogenesis and possible treatment of Alzheimer disease
 |
ALZAS protein, which has been detected in plasma in both early and late stages of AD, is suggested to represent an indicator of a dynamic equilibrium between both peripheral and brain degenerative changes, thus providing a reliable and simple diagnostic marker for AD by a simple, non-invasinve blood test. Further studies are warranted in order to validate the diagnostic sensibility and sensitivity of ALZAS ELISA serum tests in comparison to other currently used and future biomarkers in prospective clinico-pathological studies.
Acknowledgments
The authors thank the medical staff of many Vienna hospitals for clinical and autopsy reports and brain material as the basis of the present study. We are grateful to Mrs. Veronika Rappelsberger and Mrs. Barbara Weidinger for their excellent laboratory work and Mr. Erich Mitter-Ferstl, PhD, for computer and graphical work. Part of the study was supported by the Society for Support of Research in Experimental Neurology, Vienna, Austria.
References
- 1.Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 2.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winblad B, Wimo A, Joönsson L. The worldwide direct costs and costs of informal care of dementia. Alzheimer's Dem. 2006;2:S19–S20. [Google Scholar]
- 4.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 5.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–7. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 7.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–70. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry. 2003;74:1206–9. doi: 10.1136/jnnp.74.9.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–50. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 10.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipopro-tein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 11.Meyer MR, Tschanz JT, Norton MC, Welsh-Bohmer KA, Steffens DC, Wyse BW, Breitner JC. APOE genotype predicts when-not whether-one is predisposed to develop Alzheimer disease. Nat Genet. 1998;19:321–2. doi: 10.1038/1206. [DOI] [PubMed] [Google Scholar]
- 12.Cedazo-Minguez A. Apolipoprotein E and Alzheimer's disease: molecular mechanisms and therapeutic opportunities. J Cell Mol Med. 2007;11:1227–38. doi: 10.1111/j.1582-4934.2007.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gandy S. The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–9. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 15.McLaurin J, Yang D, Yip CM, Fraser PE. Review: modulating factors in amyloid-β fibril formation. J Struct Biol. 2000;130:259–70. doi: 10.1006/jsbi.2000.4289. [DOI] [PubMed] [Google Scholar]
- 16.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 17.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 18.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ 42 fibrils. Science. 2001;293:1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 19.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 20.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 21.Oddo S, Caccamo A, Smith IF, Green KN, Laferla FM. A dynamic relationship between intracellular and extracellular pools of Aβ. Am J Pathol. 2006;168:184–94. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iqbal K, Grundkelqbal I. Metabolic/signal transduction hypothesis of Alzheimer's disease and other tauopathies. Acta Neuropathol. 2005;109:25–31. doi: 10.1007/s00401-004-0951-y. [DOI] [PubMed] [Google Scholar]
- 23.Zhang YW, Xu H. Molecular and cellular mechanisms for Alzheimer's disease: understanding APP metabolism. Curr Mol Med. 2007;7:687–96. doi: 10.2174/156652407782564462. [DOI] [PubMed] [Google Scholar]
- 24.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–23. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andreasson U, Portelius E, Andersson ME, Blennow K, Zetterberg H. Aspects of β-amyloid as a biomarker for Alzheimer's disease. Biomarkers in Med. 2007;1:59–78. doi: 10.2217/17520363.1.1.59. [DOI] [PubMed] [Google Scholar]
- 26.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–85. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Walsh DM, Selkoe DJ. A β Oligomers – a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 28.Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol Aging. 2007;28:1297–306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 29.Bahr BA, Hoffman KB, Yang AJ, Hess US, Glabe CG, Lynch G. Amyloid β protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J Comp Neurol. 1998;397:139–47. [PubMed] [Google Scholar]
- 30.Braak H, Del Tredici K. Alzheimer's disease: intraneuronal alterations precede insoluble amyloid-β formation. Neurobiol Aging. 2004;25:713–8. doi: 10.1016/j.neurobiolaging.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 31.Wegiel J, Kuchna I, Nowicki K, Frackowiak J, Mazur-Kolecka B, Imaki H, Mehta PD, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Pirttilla T, Frey H, Lehtimaki T, Kivimaki T, Visser FE, Kamphorst W, Potempska A, Bolton D, Currie JR, Miller DL. Intraneuronal Aβ immunoreactivity is not a predictor of brain amyloidosis-β or neurofibrillary degeneration. Acta Neuropathol. 2007;113:389–402. doi: 10.1007/s00401-006-0191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Dickson DW, Jack CR., Jr Beta-amyloid burden is not associated with rates of brain atrophy. Ann Neurol. 2008;63:204–12. doi: 10.1002/ana.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 35.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 37.Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C. Amyloid β oligomers (A β(1–42) globu-lomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–97. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clifford PM, Zarrabi S, Siu G, Kinsler KJ, Kosciuk MC, Venkataraman V, D'Andrea MR, Dinsmore S, Nagele RG. Aβ peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Res. 2007;1142:223–36. doi: 10.1016/j.brainres.2007.01.070. [DOI] [PubMed] [Google Scholar]
- 39.Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporesponsive-ness to amyloid β-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:10273–8. doi: 10.1073/pnas.191118298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nath A, Hall E, Tuzova M, Dobbs M, Jons M, Anderson C, Woodward J, Guo Z, Fu W, Kryscio R, Wekstein D, Smith C, Markesbery WR, Mattson MP. Autoantibodies to amyloid β-peptide (Aβ) are increased in Alzheimer's disease patients and Aβ antibodies can enhance Aβ neurotoxicity: implications for disease pathogenesis and vaccine development. Neuromolecular Med. 2003;3:29–39. doi: 10.1385/nmm:3:1:29. [DOI] [PubMed] [Google Scholar]
- 41.Maier M, Seabrook TJ, Lazo ND, Jiang L, Das P, Janus C, Lemere CA. Short amy-loid-β (Aβ) immunogens reduce cerebral Aβ load and learning deficits in an Alzheimer's disease mouse model in the absence of an Aβ-specific cellular immune response. J Neurosci. 2006;26:4717–28. doi: 10.1523/JNEUROSCI.0381-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geylis V, Steinitz M. Immunotherapy of Alzheimer's disease (AD): from murine models to anti-amyloid β (Aβ) human monoclonal antibodies. Autoimmun Rev. 2006;5:33–9. doi: 10.1016/j.autrev.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Lemere CA, Maier M, Peng Y, Jiang L, Seabrook TJ. Novel Aβ immunogens: is shorter better. Curr Alzheimer Res. 2007;4:427–36. doi: 10.2174/156720507781788800. [DOI] [PubMed] [Google Scholar]
- 44.Solomon B. Clinical immunologic approaches for the treatment of Alzheimer's disease. Expert Opin Investig Drugs. 2007;16:819–28. doi: 10.1517/13543784.16.6.819. [DOI] [PubMed] [Google Scholar]
- 45.Grundkelqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the micro-tubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-lqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 47.Iqbal K, Grundkelqbal I. Alzheimer neu-rofibrillary degeneration: significance etiopathogenesis, therapeutics and prevention. J Cell Mol Med. 2008;12:38–55. doi: 10.1111/j.1582-4934.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mandelkow E, von Bergen M, Biernat J, Mandelkow EM. Structural principles of tau and the paired helical filaments of Alzheimer's disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer's disease. Neurology. 2002;59:398–407. doi: 10.1212/wnl.59.3.398. [DOI] [PubMed] [Google Scholar]
- 50.Delacourte A. Alzheimer's disease: a true tauopathy fueled by amyloid precursor protein dysfunction. In: Hanin I, Cacabelos R, Fisher A, editors. Progress in Alzheimer's and Parkinson's diseases. London – New York: Taylor & Francis; 2005. pp. 301–7. [Google Scholar]
- 51.Schonheit B, Zarski R, Ohm TG. Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology. Neurobiol Aging. 2004;25:697–711. doi: 10.1016/j.neurobiolaging.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 52.Selkoe DJ. Amyloid β-peptide is produced by cultured cells during normal metabolism: a reprise. J Alzheimers Dis. 2006;9:163–8. doi: 10.3233/jad-2006-9s319. [DOI] [PubMed] [Google Scholar]
- 53.Masters CL, Beyreuther K. Pathways to the discovery of the Aβ amyloid of Alzheimer's disease. J Alzheimers Dis. 2006;9:155–61. doi: 10.3233/jad-2006-9s318. [DOI] [PubMed] [Google Scholar]
- 54.Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neu-ropathology. J Neuropathol Exp Neurol. 2005;64:104–12. doi: 10.1093/jnen/64.2.104. [DOI] [PubMed] [Google Scholar]
- 55.Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–9. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adalbert R, Gilley J, Coleman MP. Aβ, tau and ApoE4 in Alzheimer's disease: the axonal connection. Trends Mol Med. 2007;13:135–42. doi: 10.1016/j.molmed.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 57.Gibson GE, Huang HM. Oxidative stress in Alzheimer's disease. Neurobiol Aging. 2005;26:575–8. doi: 10.1016/j.neurobiolaging.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Webber KM, Raina AK, Marlatt MW, Zhu X, Prat Ml, Morelli L, Casadesus G, Perry G, Smith MA. The cell cycle in Alzheimer disease: a unique target for neuropharma-cology. Mech Ageing Dev. 2005;126:1019–25. doi: 10.1016/j.mad.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 59.Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci USA. 2007;104:11062–7. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petersen RB, Nunomura A, Lee HG, Casadesus G, Perry G, Smith MA, Zhu X. Signal transduction cascades associated with oxidative stress in Alzheimer's disease. J Alzheimers Dis. 2007;11:143–52. doi: 10.3233/jad-2007-11202. [DOI] [PubMed] [Google Scholar]
- 61.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-β in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321:823–9. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 62.Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 63.Marchesi VT. An alternative interpretation of the amyloid Aβ hypothesis with regard to the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci USA. 2005;102:9093–8. doi: 10.1073/pnas.0503181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arispe N, Diaz JC, Simakova O. Aβ ion channels. Prospects for treating Alzheimer's disease with Aβ channel blockers. Biochim Biophys Acta. 2007;1768:1952–65. doi: 10.1016/j.bbamem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 65.Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet. 2007;37:79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- 66.Games D, Buttini M, Kobayashi D, Schenk D, Seubert P. Mice as models: transgenic approaches and Alzheimer's disease. J Alzheimers Dis. 2006;9:133–49. doi: 10.3233/jad-2006-9s316. [DOI] [PubMed] [Google Scholar]
- 67.Knopman DS, DeKosky ST, Cummings JL, Chui H, Corey-Bloom J, Relkin N, Small GW, Miller B, Stevens JC. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1143–53. doi: 10.1212/wnl.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 68.Rockwood K, Bouchard RW, Camlcloll R, Léger G. Toward a revision of criteria for the dementias. Alzheimer's Dementia. 2007;3:428–40. doi: 10.1016/j.jalz.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 69.Waldemar G, Dubols B, Emre M, Georges J, McKeith IG, Rossor M, Scheltens P, Tariska P, Winblad B. Recommendations for the diagnosis and management of Alzheimer's disease and other disorders associated with dementia: EFNS guideline. Eur J Neurol. 2007;14:e1–26. doi: 10.1111/j.1468-1331.2006.01605.x. [DOI] [PubMed] [Google Scholar]
- 70.Dubols B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D, Gauthler S, Jlcha G, Meguro K, O'Brien J, Pasquler F, Robert P, Rossor M, Salloway S, Stern Y, Vlsser PJ, Scheltens P. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 71.Jelllnger KA. A view on early diagnosis of dementias from neuropathology. In: Herholz K, Morris C, Perani D, editors. The dementias: early diagnosis and evaluation. New York: Taylor & Francis; 2006. pp. 311–428. [Google Scholar]
- 72.Jelllnger KA. Alzheimer's disease. In: Gilman S, editor. Neurobiology of disease. Amsterdam: Elsevier Academic Press; 2007. pp. 69–82. [Google Scholar]
- 73.Mlrra SS, Heyman A, McKeel D, Sumi SM, Craln BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 74.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–7. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 75.Naslund J, Haroutunlan V, Mohs R, Davis KL, Davles P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–7. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 76.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 77.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredlcl K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thai DR, Rub U, Orantes M, Braak H. Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 79.Petersen RC, Smith GE, Waring SC, Ivnlk RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–8. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 80.Chertkow H, Nasreddlne Z, Joanette Y, Drolet V, Kirk J, Massoud F, Belleville S, Bergman H. Mild cognitive impairment and cognitive impairment, no dementia: Part A, concept and diagnosis. Alzheimer's Dementia. 2007;3:266–82. doi: 10.1016/j.jalz.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 81.Scheff SW, Price DA, Schmltt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–84. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 82.Markesbery WR, Schmltt FA, Krysclo RJ, Davis DG, Smith CD, Weksteln DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 83.Bouwman FH, Schoonenboom SN, Van Der Filer WM, van Elk EJ, Kok A, Barkhot F, Blankensteln MA, Scheltens P. CSF biomarkers and medial temporal lobe atrophy predict dementia in mild cognitive impairment. Neurobiol Aging. 2007;28:1070–4. doi: 10.1016/j.neurobiolaging.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 84.Saito Y, Murayama S. Neuropathology of mild cognitive impairment. Neuropathology. 2007;27:578–84. doi: 10.1111/j.1440-1789.2007.00806.x. [DOI] [PubMed] [Google Scholar]
- 85.Jlcha GA, Parlsl JE, Dlckson DW, Johnson K, Cha R, Ivnlk RJ, Tangalos EG, Boeve BF, Knopman DS, Braak H, Petersen RC. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674–81. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 86.Petersen RC, Parlsl JE, Dlckson DW, Johnson KA, Knopman DS, Boeve BF, Jlcha GA, Ivnlk RJ, Smith GE, Tangalos EG, Braak H, Kokmen E. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–72. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 87.Gauthler S, Relsberg B, Zaudlg M, Petersen RC, Ritchie K, Brolch K, Belleville S, Brodaty H, Bennett D, Chertkow H, Cummings JL, de Leon M, Feldman H, Gangull M, Hampel H, Scheltens P, Tierney MC, Whitehouse P, Winblad B. Mild cognitive impairment. Lancet. 2006;367:1262–70. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- 88.Fischer P, Jungwirth S, Zehetmayer S, Weissgram S, Hoenigschnabl S, Gelpi E, Krampla W, Tragl KH. Conversion from subtypes of mild cognitive impairment to Alzheimer dementia. Neurology. 2007;68:288–91. doi: 10.1212/01.wnl.0000252358.03285.9d. [DOI] [PubMed] [Google Scholar]
- 89.Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, Segal S, Rusinek H, Pelton GH, Honig LS, Mayeux R, Stern Y, Tabert MH, de Leon MJ. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer disease. Neurology. 2007;68:828–36. doi: 10.1212/01.wnl.0000256697.20968.d7. [DOI] [PubMed] [Google Scholar]
- 90.Rossi R, Geroldi C, Bresciani L, Testa C, Binetti G, Zanetti O, Frisoni GB. Clinical and neuropsychological features associated with structural imaging patterns in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23:175–83. doi: 10.1159/000098543. [DOI] [PubMed] [Google Scholar]
- 91.Whitwell JL, Petersen RC, Negash S, Weigand SD, Kantarci K, Ivnik RJ, Knopman DS, Boeve BF, Smith GE, Jack CR., Jr Patterns of atrophy differ among specific subtypes of mild cognitive impairment. Arch Neurol. 2007;64:1130–8. doi: 10.1001/archneur.64.8.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Panza F, Capurso C, D'Introno A, Colacicco AM, Capurso A, Solfrizzi V. Heterogeneity of mild cognitive impairment and other predementia syndromes in progression to dementia. Neurobiol Aging. 2007;28:1631–2. doi: 10.1016/j.neurobiolaging.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 93.Hyman SE. Can neuroscience be integrated into the DSM-V? Nat Rev Neurosci. 2007;8:725–32. doi: 10.1038/nrn2218. [DOI] [PubMed] [Google Scholar]
- 94.Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer's disease. NeuroRX. 2004;1:213–25. doi: 10.1602/neurorx.1.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blennow K. CSF biomarkers for Alzheimer's disease: use in early diagnosis and evaluation of drug treatment. Expert Rev Mol Diagn. 2005;5:661–72. doi: 10.1586/14737159.5.5.661. [DOI] [PubMed] [Google Scholar]
- 96.Hampel H, Buerger K. Biomarkers in blood and cerebrospinal fluid. In: Herholz K, Morris C, Perani D, editors. The dementias: early diagnosis and evaluation. New York: Taylor & Francis; 2006. pp. 73–107. [Google Scholar]
- 97.Lewczuk P, Kornhuber J, Wiltfang J. The German Competence Net Dementias: standard operating procedures for the neuro-chemical dementia diagnostics. J Neural Transm. 2006;113:1075–80. doi: 10.1007/s00702-006-0511-9. [DOI] [PubMed] [Google Scholar]
- 98.The Ronald and Nancy Reagan Research Institute of the Alzheimer's Association and the National Institute on Aging Working Group. Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer's Disease. Neurobiol Aging. 1998;19:109–16. [PubMed] [Google Scholar]
- 99.Blennow K, Zetterberg H, Minthon L, Lannfelt L, Strid S, Annas P, Basun H, Andreasen N. Longitudinal stability of CSF biomarkers in Alzheimer's disease. Neurosci Lett. 2007;419:18–22. doi: 10.1016/j.neulet.2007.03.064. [DOI] [PubMed] [Google Scholar]
- 100.Irizarry MC. Biomarkers of Alzheimer disease in plasma. NeuroRx. 2004;1:226–34. doi: 10.1602/neurorx.1.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Attems J. Sporadic cerebral amyloid angiopathy: pathology clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005;110:345–59. doi: 10.1007/s00401-005-1074-9. [DOI] [PubMed] [Google Scholar]
- 102.Mann DM, Iwatsubo T, Pickering-Brown SM, Owen F, Saido TC, Perry RH. Preferential deposition of amyloid β protein (Aβ) in the form Aβ40 in Alzheimer's disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci Lett. 1997;221:81–4. doi: 10.1016/s0304-3940(96)13294-8. [DOI] [PubMed] [Google Scholar]
- 103.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 104.Ewers M, Buerger K, Teipel SJ, Scheltens P, Schroder J, Zinkowski RP, Bouwman FH, Schonknecht P, Schoonenboom NS, Andreasen N, Wallin A, DeBernardis JF, Kerkman DJ, Heindl B, Blennow K, Hampel H. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology. 2007;69:2205–12. doi: 10.1212/01.wnl.0000286944.22262.ff. [DOI] [PubMed] [Google Scholar]
- 105.Lewczuk P, Esselmann H, Bibl M, Beck G, Maler JM, Otto M, Kornhuber J, Wiltfang J. Tau protein phosphorylated at threonine 181 in CSF as a neurochemical biomarker in Alzheimer's disease: original data and review of the literature. J Mol Neurosci. 2004;23:115–22. doi: 10.1385/JMN:23:1-2:115. [DOI] [PubMed] [Google Scholar]
- 106.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/β-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 107.Gustafson DR, Skoog I, Rosengren L, Zetterberg H, Blennow K. Cerebrospinal fluid β-amyloid 1–42 concentration may predict cognitive decline in older women. J Neurol Neurosurg Psychiatry. 2007;78:461–4. doi: 10.1136/jnnp.2006.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hansson 0, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, Blennow K. Prediction of Alzheimer's disease using the CSF Aβ42/Aβ40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23:316–20. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- 109.Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, Kaye JA, Raskind MA, Zhang J, Peskind ER, Montine TJ. CSF Aβ42/Aβ40 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69:631–9. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 110.Stomrud E, Hansson 0, Blennow K, Minthon L, Londos E. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord. 2007;24:118–24. doi: 10.1159/000105017. [DOI] [PubMed] [Google Scholar]
- 111.Hampel H, Teipel SJ, Fuchsberger T, Andreasen N, Wiltfang J, Otto M, Shen Y, Dodel R, Du Y, Farlow M, Moller HJ, Blennow K, Buerger K. Value of CSF β-amyloid1–42 and tau as predictors of Alzheimer's disease in patients with mild cognitive impairment. Mol Psychiatry. 2004;9:705–10. doi: 10.1038/sj.mp.4001473. [DOI] [PubMed] [Google Scholar]
- 112.Maccioni RB, Lavados M, Guillon M, Mujica C, Bosch R, Farias G, Fuentes P. Anomalously phosphorylated tau and Aβ fragments in the CSF correlates with cognitive impairment in MCI subjects. Neurobiol Aging. 2006;27:237–44. doi: 10.1016/j.neurobiolaging.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 113.Schonknecht P, Pantel J, Kaiser E, Thomann P, Schroder J. Increased tau protein differentiates mild cognitive impairment from geriatric depression and predicts conversion to dementia. Neurosci Lett. 2007;416:39–42. doi: 10.1016/j.neulet.2007.01.070. [DOI] [PubMed] [Google Scholar]
- 114.van de Pol LA, Van Der Flier WM, Korf ES, Fox NC, Barkhof F, Scheltens P. Baseline predictors of rates of hippocampal atrophy in mild cognitive impairment. Neurology. 2007;69:1491–7. doi: 10.1212/01.wnl.0000277458.26846.96. [DOI] [PubMed] [Google Scholar]
- 115.de Leon MJ, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Segal S, Rusinek H, Li J, Tsui W, Saint Louis LA, Clark CM, Tarshish C, Li Y, Lair L, Javier E, Rich K, Lesbre P, Mosconi L, Reisberg B, Sadowski M, DeBernadis JF, Kerkman DJ, Hampel H, Wahlund LO, Davies P. Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol Aging. 2006;27:394–401. doi: 10.1016/j.neurobiolaging.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 116.Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer's disease. Brain. 2007;130:1777–86. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van de Pol LA, Korf ES, Van Der Flier WM, Brashear HR, Fox NC, Barkhof F, Scheltens P. Magnetic resonance imaging predictors of cognition in mild cognitive impairment. Arch Neurol. 2007;64:1023–8. doi: 10.1001/archneur.64.7.1023. [DOI] [PubMed] [Google Scholar]
- 118.Tapiola T, Pennanen C, Tapiola M, Tervo S, Kivipelto M, Hanninen T, Pihlajamaki M, Laakso MP, Hallikainen M, Hamalainen A, Vanhanen M, Helkala EL, Vanninen R, Nissinen A, Rossi R, Frisoni GB, Soininen H. MRI of hippocampus and entorhinal cortex in mild cognitive impairment: a follow-up study. Neurobiol Aging. 2008;29:31–8. doi: 10.1016/j.neurobiolaging.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 119.Fellgiebel A, Scheurich A, Bartenstein P, Muller MJ. FDG-PET and CSF phospho-tau for prediction of cognitive decline in mild cognitive impairment. Psychiatry Res. 2007;155:167–71. doi: 10.1016/j.pscychresns.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 120.Herukka SK, Helisalmi S, Hallikainen M, Tervo S, Soininen H, Pirttila T. CSF Aβ42, Tau and phosphorylated Tau, APOE epsilon4 allele and MCI type in progressive MCI. Neurobiol Aging. 2007;28:507–14. doi: 10.1016/j.neurobiolaging.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 121.Huang C, Eidelberg D, Habeck C, Moeller J, Svensson L, Tarabula T, Julin P. Imaging markers of mild cognitive impairment: multivariate analysis of CBF SPECT Neurobiol Aging. 2007;28:1062–9. doi: 10.1016/j.neurobiolaging.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 122.Xu G, Antuono PG, Jones J, Xu Y, Wu G, Ward D, Li SJ. Perfusion fMRI detects deficits in regional CBF during memory-encoding tasks in MCI subjects. Neurology. 2007;69:1650–6. doi: 10.1212/01.wnl.0000296941.06685.22. [DOI] [PubMed] [Google Scholar]
- 123.Blasko I, Jellinger K, Kemmler G, Krampla W, Jungwirth S, Wichart I, Tragl KH, Fischer P. Conversion from cognitive health to mild cognitive impairment and Alzheimer's disease: prediction by plasma amyloid β 42, medial temporal lobe atrophy and homocysteine. Neurobiol Aging. 2008;29:1–11. doi: 10.1016/j.neurobiolaging.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 124.Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, Morris JC, McKeel DW, Jr, Farlow M, Weitlauf SL, Quinn J, Kaye J, Knopman D, Arai H, Doody RS, DeCarli C, Leight S, Lee VM, Trojanowski JQ. Cerebrospinal fluid tau and β-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses. Arch Neurol. 2003;60:1696–702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 125.Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain. 2006;129:3035–41. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 126.Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Welner NE, Cohen JA, Rudlck RA, Woo D. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72:741–50. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]
- 127.Takeuchl M, Sato T, Taklno J, Kobayashi Y, Furuno S, Klkuchl S, Yamaglshl S. Diagnostic utility of serum or cerebrospinal fluid levels of toxic advanced glycation end-products (TAGE) in early detection of Alzheimer's disease. Med Hypotheses. 2007;69:1358–66. doi: 10.1016/j.mehy.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 128.Bailey P. Biological markers in Alzheimer's disease. Can J Neurol Sci. 2007;34:S72–6. doi: 10.1017/s0317167100005618. [DOI] [PubMed] [Google Scholar]
- 129.Finehout EJ, Franck Z, Choe LH, Relkin N, Lee KH. Cerebrospinal fluid proteomic biomarkers for Alzheimer's disease. Ann Neurol. 2007;61:120–9. doi: 10.1002/ana.21038. [DOI] [PubMed] [Google Scholar]
- 130.Wiltfang J, Lewczuk P, Riederer P, Grunblatt E, Hock C, Scheltens P, Hampel H, Vanderstichele H, Iqbal K, Galasko D, Lannfelt L, Otto M, Esselmann H, Henkel AW, Kornhuber J, Blennow K. Consensus paper of the WFSBP Task Force on biological markers of dementia: The role of CSF and blood analysis in the early and differential diagnosis of dementia. World J Biol Psychiatry. 2005;6:69–84. doi: 10.1080/15622970510029786. [DOI] [PubMed] [Google Scholar]
- 131.Suntlerlantl T, Linker G, Mirza N, Putnam KT, Friedman DL, Kimmel LH, Bergeson J, Manetti GJ, Zimmermann M, Tang B, Bartko JJ, Cohen RM. Decreased β-amy-loid1–42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA. 2003;289:2094–103. doi: 10.1001/jama.289.16.2094. [DOI] [PubMed] [Google Scholar]
- 132.Parnetti L, Lanari A, Silvestrelli G, Saggese E, Reboldi P. Diagnosing prodromal Alzheimer's disease: role of CSF biochemical markers. Mech Ageing Dev. 2006;127:129–32. doi: 10.1016/j.mad.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 133.Galasko D. Biomarkers for Alzheimer's disease – clinical needs and application. J Alzheimers Dis. 2005;8:339–46. doi: 10.3233/jad-2005-8403. [DOI] [PubMed] [Google Scholar]
- 134.Rösier N, Wichart I, Jellinger KA. Current clinical neurochemical diagnosis of Alzheimer's disease. J Lab Med. 2002;26:139–48. [Google Scholar]
- 135.Hansson 0, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 136.Engelborghs S, De Vreese K, Van de Casteele T, Vanderstichele H, Van Everbroeck B, Cras P, Martin JJ, Vanmechelen E, De Deyn PP. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.02.016. in press. doi: 10.1016/j.neurobiolaging.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 137.Mollenhauer B, Trenkwalder C, von Ahsen N, Bibl M, Steinacker P, Brechlin P, Schindehuette J, Poser S, Wiltfang J, Otto M. β-Amlyoid 1–42 and tau-pro-tein in cerebrospinal fluid of patients with Parkinson's disease dementia. Dement Geriatr Cogn Disord. 2006;22:200–8. doi: 10.1159/000094871. [DOI] [PubMed] [Google Scholar]
- 138.Mollenhauer B, Bibl M, Wiltfang J, Steinacker P, Ciesielczyk B, Neubert K, Trenkwalder C, Otto M. Total tau protein phosphorylated tau (181p) protein, β-amyloid(1-42), and β-amyloid(1-40) in cerebrospinal fluid of patients with dementia with Lewy bodies. Clin Chem Lab Med. 2006;44:192–5. doi: 10.1515/CCLM.2006.035. [DOI] [PubMed] [Google Scholar]
- 139.Nielsen HM, Minthon L, Londos E, Blennow K, Miranda E, Perez J, Crowther DC, Lomas DA, Janciauskiene SM. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology. 2007;69:1569–79. doi: 10.1212/01.wnl.0000271077.82508.a0. [DOI] [PubMed] [Google Scholar]
- 140.Stefani A, Martorana A, Bernardini S, Panella M, Mercati F, Orlacchio A, Pierantozzi M. CSF markers in Alzheimer disease patients are not related to the different degree of cognitive impairment. J Neurol Sci. 2006;251:124–8. doi: 10.1016/j.jns.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 141.Vanderstichele H, De Meyer G, Andreasen N, Kostanjevecki V, Wallin A, Olsson A, Blennow K, Vanmechelen E. Amino-truncated β-amyloid 42 peptides in cerebrospinal fluid and prediction of progression of mild cognitive impairment. Clin Chem. 2005;51:1650–60. doi: 10.1373/clinchem.2005.051201. [DOI] [PubMed] [Google Scholar]
- 142.Wallin AK, Blennow K, Andreasen N, Minthon L. CSF biomarkers for Alzheimer's disease: levels of β-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement Geriatr Cogn Disord. 2006;21:131–8. doi: 10.1159/000090631. [DOI] [PubMed] [Google Scholar]
- 143.Speth A, Sindic C, Seron X, Ivanoiu A. Cerebrospinal fluid tau protein and amyloid β-42 correlation with the neuropsy-chological examination in mild cognitive impairment and mild Alzheimer's disease patients. 11th Europ Fed of Neurol Societies Congress. 2007 Brussels, August 25–28, 2007. [Google Scholar]
- 144.Hampel H, Mitchell A, Blennow K, Frank RA, Brettschneider S, Weller L, Moller HJ. Core biological marker candidates of Alzheimer's disease - perspectives for diagnosis, prediction of outcome and reflection of biological activity. J Neural Transm. 2004;111:247–72. doi: 10.1007/s00702-003-0065-z. [DOI] [PubMed] [Google Scholar]
- 145.Zhong Z, Ewers M, Teipel S, Burger K, Wallin A, Blennow K, He P, McAllister C, Hampel H, Shen Y. Levels of β-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry. 2007;64:718–26. doi: 10.1001/archpsyc.64.6.718. [DOI] [PubMed] [Google Scholar]
- 146.Gruden MA, Davidova TB, Malisauskas M, Sewell RD, Voskresenskaya Nl, Wilhelm K, Elistratova El, Sherstnev VV, Morozova-Roche LA. Differential neuroim-mune markers to the onset of Alzheimer's disease neurodegeneration and dementia: autoantibodies to Aβ((25–35)) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007;186:181–92. doi: 10.1016/j.jneuroim.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 147.Andersson C, Blennow K, Almkvist 0, Andreasen N, Engfeldt P, Johansson SE, Lindau M, Eriksdotter-Jonhagen M. Increasing CSF phospho-tau levels during cognitive decline and progression to dementia. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.03.027. in press. doi: 10.1016/j.neurobiolaging.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 148.Wiltfang J, Esselmann H, Bibl M, Hull M, Hampel H, Kessler H, Frolich L, Schroder J, Peters 0, Jessen F, Luckhaus C, Perneczky R, Jahn H, Fiszer M, Maler JM, Zimmermann R, Bruckmoser R, Kornhuber J, Lewczuk P. Amyloid β peptide ratio 42/40 but not A β 42 correlates with phospho-Tau in patients with low- and high-CSF A β 40 load. J Neurochem. 2007;101:1053–9. doi: 10.1111/j.1471-4159.2006.04404.x. [DOI] [PubMed] [Google Scholar]
- 149.Bouwman FH, Van Der Flier WM, Schoonenboom NS, van Elk EJ, Kok A, Rijmen F, Blankenstein MA, Scheltens P. Longitudinal changes of CSF biomarkers in memory clinic patients. Neurology. 2007;69:1006–11. doi: 10.1212/01.wnl.0000271375.37131.04. [DOI] [PubMed] [Google Scholar]
- 150.Peskind ER, Li G, Shofer J, Quinn JF, Kaye JA, Clark CM, Farlow MR, DeCarli C, Raskind MA, Schellenberg GD, Lee VM, Galasko DR. Age and apolipoprotein E epsilon4 allele effects on cerebrospinal fluid β-amyloid 42 in adults with normal cognition. Arch Neurol. 2006;63:936–9. doi: 10.1001/archneur.63.7.936. [DOI] [PubMed] [Google Scholar]
- 151.Pijnenburg YA, Schoonenboom SN, Mehta PD, Mehta SP, Mulder C, Veerhuis R, Blankenstein MA, Scheltens P. Decreased cerebrospinal fluid amyloid β (1-40) levels in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2007;78:735–7. doi: 10.1136/jnnp.2006.105064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid-β levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007;68:666–9. doi: 10.1212/01.wnl.0000256043.50901.e3. [DOI] [PubMed] [Google Scholar]
- 153.Shafaati M, Solomon A, Kivipelto M, Bjorkhem I, Leoni V. Levels of ApoE in cerebrospinal fluid are correlated with Tau and 24S-hydroxycholesterol in patients with cognitive disorders. Neurosci Lett. 2007;425:78–82. doi: 10.1016/j.neulet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 154.Cagnin A, Zambon A, Zarantonello G, Vianello D, Marchiori M, Mercurio D, Micciche F, Ermani M, Leon A, Battistin L. Serum lipoprotein profile and APOE genotype in Alzheimer's disease. J Neural Transm Suppl. 2007:175–9. doi: 10.1007/978-3-211-73574-9_22. [DOI] [PubMed] [Google Scholar]
- 155.Kauwe JS, Jacquart S, Chakraverty S, Wang J, Mayo K, Fagan AM, Holtzman DM, Morris JC, Goate AM. Extreme cerebrospinal fluid amyloid β levels identify family with late-onset Alzheimer's disease presenilin 1 mutation. Ann Neurol. 2007;61:446–53. doi: 10.1002/ana.21099. [DOI] [PubMed] [Google Scholar]
- 156.Englund H, Anneren G, Gustafsson J, Wester U, Wiltfang J, Lannfelt L, Blennow K, Hoglund K. Increase in β-amyloid levels in cerebrospinal fluid of children with Down syndrome. Dement Geriatr Cogn Disord. 2007;24:369–74. doi: 10.1159/000109215. [DOI] [PubMed] [Google Scholar]
- 157.Mollenhauer B, Bibl M, Esselmann H, Steinacker P, Trenkwalder C, Wiltfang J, Otto M. Tauopathies and synucleinopathies: do cerebrospinal fluid β-amyloid peptides reflect disease-specific pathogenesis. J Neural Transm. 2007;114:919–27. doi: 10.1007/s00702-007-0629-4. [DOI] [PubMed] [Google Scholar]
- 158.Bibl M, Mollenhauer B, Lewczuk P, Esselmann H, Wolf S, Trenkwalder C, Otto M, Stiens G, Ruther E, Kornhuber J, Wiltfang J. Validation of amyloid-β peptides in CSF diagnosis of neurodegenerative dementias. Mol Psychiatry. 2007;12:671–80. doi: 10.1038/sj.mp.4001967. [DOI] [PubMed] [Google Scholar]
- 159.Henkel AW, Dittrich PS, Groemer TW, Lemke EA, Klingauf J, Klafki HW, Lewczuk P, Esselmann H, Schwille P, Kornhuber J, Wiltfang J. Immune complexes of auto-antibodies against A β 1-42 peptides patrol cerebrospinal fluid of non-Alzheimer's patients. Mol Psychiatry. 2007;12:601–10. doi: 10.1038/sj.mp.4001947. [DOI] [PubMed] [Google Scholar]
- 160.Engelborghs S, Sleegers K, Cras P, Brouwers N, Serneels S, De Leenheir E, Martin JJ, Vanmechelen E, Van Broeckhoven C, De Deyn PP. No association of CSF biomarkers with APOE e4, plaque and tangle burden in definite Alzheimer's disease. Brain. 2007;130:2320–6. doi: 10.1093/brain/awm136. [DOI] [PubMed] [Google Scholar]
- 161.Buerger K, Alafuzoff I, Ewers M, Pirttila T, Zinkowski R, Hampel H. No correlation between CSF tau protein phosphorylated at threonine 181 with neocortical neurofibrillary pathology in Alzheimer's disease. Brain. 2007;130:e82. doi: 10.1093/brain/awm140. [DOI] [PubMed] [Google Scholar]
- 162.Buerger K, Otto M, Teipel SJ, Zinkowski R, Blennow K, DeBernardis J, Kerkman D, Schroder J, Schonknecht P, Cepek L, McCulloch C, Moller HJ, Wiltfang J, Kretzschmar H, Hampel H. Dissociation between CSF total tau and tau protein phosphorylated at threonine 231 in Creutzfeldt-Jakob disease. Neurobiol Aging. 2006;27:10–5. doi: 10.1016/j.neurobiolaging.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 163.Bibl M, Mollenhauer B, Esselmann H, Schneider M, Lewczuk P, Welge V, Falkai P, Kornhuber J, Wiltfang J. CSF neuro-chemical phenotypes in vascular dementias: original data and mini-review. Dem Ger Cogn Disord. 2008;25:256–65. doi: 10.1159/000115975. [DOI] [PubMed] [Google Scholar]
- 164.Bibl M, Esselmann H, Mollenhauer B, Weniger G, Welge V, Liess M, Lewczuk P, Otto M, Schulz JB, Trenkwalder C, Kornhuber J, Wiltfang J. Blood-based neurochemical diagnosis of vascular dementia: a pilot study. J Neurochem. 2007;103:467–74. doi: 10.1111/j.1471-4159.2007.04763.x. [DOI] [PubMed] [Google Scholar]
- 165.Bibl M, Mollenhauer B, Esselmann H, Lewczuk P, Klafki HW, Sparbier K, Smirnov A, Cepek L, Trenkwalder C, Ruther E, Kornhuber J, Otto M, Wiltfang J. CSF amyloid-β-peptides in Alzheimer's disease, dementia with Lewy bodies and Parkinson's disease dementia. Brain. 2006;129:1177–87. doi: 10.1093/brain/awl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.de Jong D, Jansen RW, Pijnenburg YA, van Geel WJ, Borm GF, Kremer HP, Verbeek MM. CSF neurofilament proteins in the differential diagnosis of dementia. J Neurol Neurosurg Psychiatry. 2007;78:936–8. doi: 10.1136/jnnp.2006.107326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.McRae A, Martins RN, Fonte J, Kraftsik R, Hirt L, Miklossy J. Cerebrospinal fluid antimicroglial antibodies in Alzheimer disease: a putative marker of an ongoing inflammatory process. Exp Gerontol. 2007;42:355–63. doi: 10.1016/j.exger.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 168.Castano EM, Roher AE, Esh CL, Kokjohn TA, Beach T. Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer's disease and non-demented elderly subjects. Neurol Res. 2006;28:155–63. doi: 10.1179/016164106X98035. [DOI] [PubMed] [Google Scholar]
- 169.Zhang J, Goodlett DR, Quinn JF, Peskind E, Kaye JA, Zhou Y, Pan C, Yi E, Eng J, Wang Q, Aebersold RH, Montine TJ. Quantitative proteomics of cerebrospinal fluid from patients with Alzheimer disease. J Alzheimers Dis. 2005;7:125–33. doi: 10.3233/jad-2005-7205. [DOI] [PubMed] [Google Scholar]
- 170.Korolainen MA, Nyman TA, Nyyssonen P, Hartikainen ES, Pirttila T. Multiplexed proteomic analysis of oxidation and concentrations of cerebrospinal fluid proteins in Alzheimer disease. Clin Chem. 2007;53:657–65. doi: 10.1373/clinchem.2006.078014. [DOI] [PubMed] [Google Scholar]
- 171.Portelius E, Zetterberg H, Andreasson U, Brinkmalm G, Andreasen N, Wallin A, Westman-Brinkmalm A, Blennow K. An Alzheimer's disease-specific β-amyloid fragment signature in cerebrospinal fluid. Neurosci Lett. 2006;409:215–9. doi: 10.1016/j.neulet.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 172.Simonsen AH, McGuire J, Hansson 0, Zetterberg H, Podust VN, Davies HA, Waldemar G, Minthon L, Blennow K. Novel panel of cerebrospinal fluid biomarkers for the prediction of progression to Alzheimer dementia in patients with mild cognitive impairment. Arch Neurol. 2007;64:366–70. doi: 10.1001/archneur.64.3.366. [DOI] [PubMed] [Google Scholar]
- 173.Lescuyer P, Allard L, Zimmermannlvol CG, Burgess JA, Hughes-Frutiger S, Burkhard PR, Sanchez JC, Hochstrasser DF. Identification of post-mortem cerebrospinal fluid proteins as potential biomarkers of ischemia and neurodegeneration. Proteomics. 2004;4:2234–41. doi: 10.1002/pmic.200300822. [DOI] [PubMed] [Google Scholar]
- 174.Puchades M, Hansson SF, Nilsson CL, Andreasen N, Blennow K, Davidsson P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer's disease. Brain Res Mol Brain Res. 2003;118:140–6. doi: 10.1016/j.molbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 175.Hu Y, Kauwe JSK, Gross J, Cairns NJ, Goate AM, Fagan AM, Townsend RR, Holtzman DM. Identification and validation of novel CSF biomarkers for early stages of Alzheimer's disease. Proteomics Clinical Applications. 2007;1:1373–84. doi: 10.1002/prca.200600999. [DOI] [PubMed] [Google Scholar]
- 176.Abdi F, Quinn JF, Jankovic J, Mclntosh M, Leverenz JB, Peskind E, Nixon R, Nutt J, Chung K, Zabetian C, Samii A, Lin M, Hattan S, Pan C, Wang Y, Jin J, Zhu D, Li GJ, Liu Y, Waichunas D, Montine TJ, Zhang J. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neu-rodegenerative disorders. J Alzheimers Dis. 2006;9:293–348. doi: 10.3233/jad-2006-9309. [DOI] [PubMed] [Google Scholar]
- 177.Mclntyre JA, Chapman J, Shavit E, Hamilton RL, Dekosky ST. Redox-reactive autoantibodies in Alzheimer's patients' cerebrospinal fluids: preliminary studies. Autoimmunity. 2007;40:390–6. doi: 10.1080/08916930701421020. [DOI] [PubMed] [Google Scholar]
- 178.Ertekin-Taner N, Younkin LH, Yager DM, Parfitt F, Baker MC, Asthana S, Hutton ML, Younkin SG, Graff-Radford NR. Plasma amyloid beta protein is elevated in late-onset Alzheimer disease families. Neurology. 2008;70:596–606. doi: 10.1212/01.WNL.0000278386.00035.21. [DOI] [PubMed] [Google Scholar]
- 179.Schupf N, Patel B, Silverman W, Zigman WB, Zhong N, Tycko B, Mehta PD, Mayeux R. Elevated plasma amyloid β-peptide 1-42 and onset of dementia in adults with Down syndrome. Neurosci Lett. 2001;301:199–203. doi: 10.1016/s0304-3940(01)01657-3. [DOI] [PubMed] [Google Scholar]
- 180.Crystal HA, Davies P. Toward a plasma marker for Alzheimer disease: some progress, but still a long way to go. Neurology. 2008;70:586–7. doi: 10.1212/01.wnl.0000299905.05595.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tamaoka A, Fukushima T, Sawamura N, Ishikawa K, Oguni E, Komatsuzaki Y, Shoji S. Amyloid β protein in plasma from patients with sporadic Alzheimer's disease. J Neurol Sci. 1996;141:65–8. doi: 10.1016/0022-510x(96)00143-8. [DOI] [PubMed] [Google Scholar]
- 182.Vanderstichele H, Van Kerschaver E, Hesse C, Davidsson P, Buyse MA, Andreasen N, Minthon L, Wallin A, Blennow K, Vanmechelen E. Standardization of measurement of β-amyloid(1-42) in cerebrospinal fluid and plasma. Amyloid. 2000;7:245–58. doi: 10.3109/13506120009146438. [DOI] [PubMed] [Google Scholar]
- 183.Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid β proteins 1-40 and 1-42 in Alzheimer disease. Arch Neurol. 2000;57:100–5. doi: 10.1001/archneur.57.1.100. [DOI] [PubMed] [Google Scholar]
- 184.Fukumoto H, Tennis M, Locascio JJ, Hyman BT, Growdon JH, Irizarry MC. Age but not diagnosis is the main predictor of plasma amyloid β-protein levels. Arch Neurol. 2003;60:958–64. doi: 10.1001/archneur.60.7.958. [DOI] [PubMed] [Google Scholar]
- 185.Assini A, Cammarata S, Vitali A, Colucci M, Giliberto L, Borghi R, Inglese ML, Volpe S, Ratto S, Dagna-Bricarelli F, Baldo C, Argusti A, Odetti P, Piccini A, Tabaton M. Plasma levels of amyloid β-protein 42 are increased in women with mild cognitive impairment. Neurology. 2004;63:828–31. doi: 10.1212/01.wnl.0000137040.64252.ed. [DOI] [PubMed] [Google Scholar]
- 186.Graff-Radford NR, Crook JE, Lucas J, Boeve BF, Knopman DS, Ivnik RJ, Smith GE, Younkin LH, Petersen RC, Younkin SG. Association of low plasma Aβ42/Aβ40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol. 2007;64:354–62. doi: 10.1001/archneur.64.3.354. [DOI] [PubMed] [Google Scholar]
- 187.van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Aβ(1-40) and Aβ(1-42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5:655–60. doi: 10.1016/S1474-4422(06)70501-4. [DOI] [PubMed] [Google Scholar]
- 188.Sundelof J, Giedraitis V, Irizarry MC, Sundstrom J, Ingelsson E, Ronnemaa E, Arnlov J, Gunnarsson MD, Hyman BT, Basun H, Ingelsson M, Lannfelt L, Kilander L. Plasma β-amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol. 2008;65:256–63. doi: 10.1001/archneurol.2007.57. [DOI] [PubMed] [Google Scholar]
- 189.Pomara N, Doraiswamy PM, Willoughby LM, Roth AE, Mulsant BH, Sidtis JJ, Mehta PD, Reynolds CF, 3rd, Pollock BG. Elevation in plasma Aβ42 in geriatric depression: a pilot study. Neurochem Res. 2006;31:341–9. doi: 10.1007/s11064-005-9029-z. [DOI] [PubMed] [Google Scholar]
- 190.Song MS, Mook-Jung I, Lee HJ, Min JY, Park MH. Serum anti-amyloid-β antibodies and Alzheimer's disease in elderly Korean patients. J Int Med Res. 2007;35:301–6. doi: 10.1177/147323000703500303. [DOI] [PubMed] [Google Scholar]
- 191.Pesaresi M, Lovati C, Bertora P, Mailland E, Galimberti D, Scarpini E, Quadri P, Forloni G, Mariani C. Plasma levels of β-amyloid (1-42) in Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:904–5. doi: 10.1016/j.neurobiolaging.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 192.Abdullah L, Paris D, Luis C, Quadros A, Parrish J, Valdes L, Keegan AP, Mathura V, Crawford F, Mullan M. The influence of diagnosis, intra- and inter-person variability on serum and plasma Aβ levels. Neurosci Lett. 2007;428:53–8. doi: 10.1016/j.neulet.2007.09.058. [DOI] [PubMed] [Google Scholar]
- 193.Freeman SH, Raju S, Hyman BT, Frosch MP, Irizarry MC. Plasma Aβ levels do not reflect brain Aβ levels. J Neuropathol Exp Neurol. 2007;66:264–71. doi: 10.1097/NEN.0b013e31803d3ae4. [DOI] [PubMed] [Google Scholar]
- 194.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 195.Giedraitis V, Sundelof J, Irizarry MC, Garevik N, Hyman BT, Wahlund LO, Ingelsson M, Lannfelt L. The normal equilibrium between CSF and plasma amyloid β levels is disrupted in Alzheimer's disease. Neurosci Lett. 2007;427:127–31. doi: 10.1016/j.neulet.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 196.Mehta PD, Pirttila T, Patrick BA, Barshatzky M, Mehta SP. Amyloid β protein 1-40 and 1-42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci Lett. 2001;304:102–6. doi: 10.1016/s0304-3940(01)01754-2. [DOI] [PubMed] [Google Scholar]
- 197.Blasko I, Kemmler G, Krampla W, Jungwirth S, Wichart I, Jellinger K, Tragl KH, Fischer P. Plasma amyloid β protein 42 in non-demented persons aged 75 years: effects of concomitant medication and medial temporal lobe atrophy. Neurobiol Aging. 2005;26:1135–43. doi: 10.1016/j.neurobiolaging.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 198.Kulstad JJ, Green PS, Cook DG, Watson GS, Reger MA, Baker LD, Plymate SR, Asthana S, Rhoads K, Mehta PD, Craft S. Differential modulation of plasma β-amy-loid by insulin in patients with Alzheimer disease. Neurology. 2006;66:1506–10. doi: 10.1212/01.wnl.0000216274.58185.09. [DOI] [PubMed] [Google Scholar]
- 199.Wiltfang J, Esselmann H, Smirnov A, Bibl M, Cepek L, Steinacker P, Mollenhauer B, Buerger K, Hampel H, Paul S, Neumann M, Maler M, Zerr I, Kornhuber J, Kretzschmar HA, Poser S, Otto M. β-Amyloid peptides in cere-brospinal fluid of patients with Creutzfeldt-Jakob disease. Ann Neurol. 2003;54:263–7. doi: 10.1002/ana.10661. [DOI] [PubMed] [Google Scholar]
- 200.Mollenhauer B, Steinacker P, Bahn E, Bibl M, Brechlin P, Schlossmacher MG, Locascio JJ, Wiltfang J, Kretzschmar HA, Poser S, Trenkwalder C, Otto M. Serum heart-type fatty acid-binding protein and cerebrospinal fluid tau: marker candidates for dementia with Lewy bodies. Neurodegener Dis. 2007;4:366–75. doi: 10.1159/000105157. [DOI] [PubMed] [Google Scholar]
- 201.Brettschneider S, Morgenthaler NG, Teipel SJ, Fischer-Schulz C, Burger K, Dodel R, Du Y, Moller HJ, Bergmann A, Hampel H. Decreased serum amyloid β(1-42) autoantibody levels in Alzheimer's disease, determined by a newly developed immuno-precipitation assay with radiola-beled amyloid β(1-42) peptide. Biol Psychiatry. 2005;57:813–6. doi: 10.1016/j.biopsych.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 202.Ravaglia G, Forti P, Maioli F, Chiappelli M, Montesi F, Tumini E, Mariani E, Licastro F, Patterson C. Blood inflammatory markers and risk of dementia: The Conselice Study of Brain Aging. Neurobiol Aging. 2007;28:1810–20. doi: 10.1016/j.neurobiolaging.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 203.Engelhart MJ, Geerlings Ml, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, Stijnen T, Hofman A, Witteman JC, Breteler MM. Inflammatory proteins in plasma and the risk of dementia: the Rotterdam study. Arch Neurol. 2004;61:668–72. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- 204.Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in Alzheimer's disease: correlation with the disease progression. Immunol Lett. 2007;114:46–51. doi: 10.1016/j.imlet.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 205.Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, Mehta PD. Plasma A[β]40 and A[β]42 and Alzheimer's disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–90. doi: 10.1212/01.wnl.0000091890.32140.8f. [DOI] [PubMed] [Google Scholar]
- 206.Sobow T, Flirski M, Liberski P, Kloszewska I. Plasma Aβ levels as predictors of response to rivastigmine treatment in Alzheimer's disease. Acta Neurobiol Exp. 2007;67:131–9. doi: 10.55782/ane-2007-1640. [DOI] [PubMed] [Google Scholar]
- 207.Liao PC, Yu L, Kuo CC, Lin CJ, Kuo YM. Proteomics analysis of plasma for potential biomarkers in the diagnosis of Alzheimer's disease. Proteomics Clin Applic. 2007;1:506–12. doi: 10.1002/prca.200600684. [DOI] [PubMed] [Google Scholar]
- 208.German DC, Gurnani P, Nandi A, Garner HR, Fisher W, Diaz-Arrastia R, O'suilleabhain P, Rosenblatt KP. Serum biomarkers for Alzheimer's disease: proteomic discovery. Biomed Pharmacother. 2007;61:383–9. doi: 10.1016/j.biopha.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 209.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 210.Irizarry MC, Gurol ME, Raju S, Diaz-Arrastia R, Locascio JJ, Tennis M, Hyman BT, Growdon JH, Greenberg SM, Bottiglieri T. Association of homocysteine with plasma amyloid β protein in aging and neurodegenerative disease. Neurology. 2005;65:1402–8. doi: 10.1212/01.wnl.0000183063.99107.5c. [DOI] [PubMed] [Google Scholar]
- 211.Luchsinger JA, Tang MX, Miller J, Green R, Mehta PD, Mayeux R. Relation of plasma homocysteine to plasma amyloid β levels. Neurochem Res. 2007;32:775–81. doi: 10.1007/s11064-006-9207-7. [DOI] [PubMed] [Google Scholar]
- 212.Hye A, Lynham S, Thambisetty M, Causevic M, Campbell J, Byers HL, Hooper C, Rijsdijk F, Tabrizi SJ, Banner S, Shaw CE, Foy C, Poppe M, Archer N, Hamilton G, Powell J, Brown RG, Sham P, Ward M, Lovestone S. Proteome-based plasma biomarkers for Alzheimer's disease. Brain. 2006;129:3042–50. doi: 10.1093/brain/awl279. [DOI] [PubMed] [Google Scholar]
- 213.Lopez MF, Mikulskis A, Kuzdzal S, Bennett DA, Kelly J, Golenko E, DiCesare J, Denoyer E, Patton WF, Ediger R, Sapp L, Ziegert T, Lynch C, Kramer S, Whiteley GR, Wall MR, Mannion DP, Delia Cioppa G, Rakitan JS, Wolfe GM. High-resolution serum proteomic profiling of Alzheimer disease samples reveals disease-specific carrier-protein-bound mass signatures. Clin Chem. 2005;51:1946–54. doi: 10.1373/clinchem.2005.053090. [DOI] [PubMed] [Google Scholar]
- 214.Borroni B, Colciaghi F, Caltagirone C, Rozzini L, Broglio L, Cattabeni F, Di Luca M, Padovani A. Platelet amyloid precursor protein abnormalities in mild cognitive impairment predict conversion to dementia of Alzheimer type: a 2-year follow-up study. Arch Neurol. 2003;60:1740–4. doi: 10.1001/archneur.60.12.1740. [DOI] [PubMed] [Google Scholar]
- 215.Tang K, Hynan LS, Baskin F, Rosenberg RN. Platelet amyloid precursor protein processing: a bio-marker for Alzheimer's disease. J Neurol Sci. 2006;240:53–8. doi: 10.1016/j.jns.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zainaghi IA, Forlenza OV, Gattaz WF. Abnormal APP processing in platelets of patients with Alzheimer's disease: correlations with membrane fluidity and cognitive decline. Psychopharmacology. 2007;192:547–53. doi: 10.1007/s00213-007-0748-5. [DOI] [PubMed] [Google Scholar]
- 217.Johnston JA, Liu WW, Coulson DT, Todd S, Murphy S, Brennan S, Foy CJ, Craig D, Irvine GB, Passmore AP. Platelet β-secretase activity is increased in Alzheimer's disease. Neurobiol Aging. 2008;29:661–8. doi: 10.1016/j.neurobiolaging.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 218.Adolfsson R, Gottfries CG, Oreland L, Wiberg A, Winblad B. Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 1980;27:1029–34. doi: 10.1016/0024-3205(80)90025-9. [DOI] [PubMed] [Google Scholar]
- 219.Jarman J, Glover V, Sandler M, Turjanski N, Stern G. Platelet monoamine oxidase B activity in Parkinson's disease: a re-evaluation. J Neural Transm Park Dis Dement Sect. 1993;5:1–4. doi: 10.1007/BF02260909. [DOI] [PubMed] [Google Scholar]
- 220.Zellner M, Diestinger M, Rappold E, Kotzailias N, Sycha T, Winkler W, Pirchegger P, Attems J, Gerner C, Oehler R, Jellinger KA, Roth E. Monoamine oxidase B expression in blood platelets as biomarker for dementia. Neurobiol Aging. 2007 (submitted) [Google Scholar]
- 221.Krzystanek E, Krzystanek M, Opala G, Trzeciak HI, Siuda J, Malecki A. Platelet phospholipase A(2) activity in patients with Alzheimer's disease, vascular dementia and ischemic stroke. J Neural Transm. 2007;114:1033–9. doi: 10.1007/s00702-007-0669-9. [DOI] [PubMed] [Google Scholar]
- 222.Barnes J, Foster J, Fox NC. Structural magnetic resonance imaging-derived bio-markers for Alzheimer's disease. Biomarkers Med. 2007;1:79–92. doi: 10.2217/17520363.1.1.79. [DOI] [PubMed] [Google Scholar]
- 223.Vemuri P, Gunter JL, Senjem ML, Whitwell JL, Kantarci K, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr Alzheimer's disease diagnosis in individual subjects using structural MR images: validation studies. Neuroimage. 2008;39:1186–97. doi: 10.1016/j.neuroimage.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Barkhof F, Polvikoski TM, van Straaten EC, Kalaria RN, Sulkava R, Aronen HJ, Niinisto L, Rastas S, Oinas M, Scheltens P, Erkinjuntti T. The significance of medial temporal lobe atrophy: a postmortem MRI study in the very old. Neurology. 2007;69:1521–7. doi: 10.1212/01.wnl.0000277459.83543.99. [DOI] [PubMed] [Google Scholar]
- 225.Mevel K, Desgranges B, Baron JC, Landeau B, De la Sayette V, Viader F, Eustache F, Chetelat G. Detecting hip-pocampal hypometabolism in Mild Cognitive Impairment using automatic voxel-based approaches. Neuroimage. 2007;37:18–25. doi: 10.1016/j.neuroimage.2007.04.048. [DOI] [PubMed] [Google Scholar]
- 226.Kemppainen NM, Aalto S, Wilson IA, Nagren K, Helin S, Bruck A, Oikonen V, Kailajarvi M, Scheinin M, Viitanen M, Parkkola R, Rinne JO. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology. 2007;68:1603–6. doi: 10.1212/01.wnl.0000260969.94695.56. [DOI] [PubMed] [Google Scholar]
- 227.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 228.Klunk WE, Nebes RD, Tsopelas N, Lopresti BJ, Price JC, DeKosky ST, Mathis CA. Development and application of β-amyloid imaging agents in Alzheimer's disease. In: Herholz K, Perani D, Morris JC, editors. The dementias: early diagnosis and evaluation. New York -London: Taylor & Francis; 2007. pp. 279–309. [Google Scholar]
- 229.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O'Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging β-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 230.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. β-Amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 231.Apostolova LG, Steiner CA, Akopyan GG, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Three-dimensional gray matter atrophy mapping in mild cognitive impairment and mild Alzheimer disease. Arch Neurol. 2007;64:1489–95. doi: 10.1001/archneur.64.10.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bracco L, Bessi V, Piccini C, Mosconi L, Pupi A, Sorbi S. Metabolic correlates of executive dysfunction. J Neurol. 2007;254:1052–65. doi: 10.1007/s00415-006-0488-1. Different patterns in mild and very mild Alzheimer's disease. [DOI] [PubMed] [Google Scholar]
- 233.Caroli A, Testa C, Geroldi C, Nobili F, Guerra UP, Bonetti M, Frisoni GB. Brain perfusion correlates of medial temporal lobe atrophy and white matter hyperintensities in mild cognitive impairment. J Neurol. 2007;254:1000–8. doi: 10.1007/s00415-006-0498-z. [DOI] [PubMed] [Google Scholar]
- 234.Hamalainen A, Tervo S, Grau-Olivares M, Niskanen E, Pennanen C, Huuskonen J, Kivipelto M, Hanninen T, Tapiola M, Vanhanen M, Hallikainen M, Helkala EL, Nissinen A, Vanninen R, Soininen H. Voxel-based morphometry to detect brain atrophy in progressive mild cognitive impairment. Neuroimage. 2007;37:1122–31. doi: 10.1016/j.neuroimage.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 235.Kantarci K, Weigand SD, Petersen RC, Boeve BF, Knopman DS, Gunter J, Reyes D, Shiung M, O'Brien PC, Smith GE, Ivnik RJ, Tangalos EG, Jack CR., Jr Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2007;28:1330–9. doi: 10.1016/j.neurobiolaging.2006.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kircher TT, Weis S, Freymann K, Erb M, Jessen F, Grodd W, Heun R, Leube DT. Hippocampal activation in patients with mild cognitive impairment is necessary for successful memory encoding. J Neurol Neurosurg Psychiatry. 2007;78:812–8. doi: 10.1136/jnnp.2006.104877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kropholler MA, Boellaard R, van Berckel BN, Schuitemaker A, Kloet RW, Lubberink MJ, Jonker C, Scheltens P, Lammertsma AA. Evaluation of reference regions for (R)-[(11)C]PK11195 studies in Alzheimer's disease and mild cognitive impairment. J Cereb Blood Flow Metab. 2007;27:1965–74. doi: 10.1038/sj.jcbfm.9600488. [DOI] [PubMed] [Google Scholar]
- 238.Petrella JR, Wang L, Krishnan S, Slavin MJ, Prince SE, Tran TT, Doraiswamy PM. Cortical deactivation in mild cognitive impairment: high-field-strength functional MR imaging. Radiology. 2007;245:224–35. doi: 10.1148/radiol.2451061847. [DOI] [PubMed] [Google Scholar]
- 239.Rami L, Gomez-Anson B, Bosch B, Sanchez-Valle R, Monte GC, Villar A, Molinuevo JL. Cortical brain metabolism as measured by proton spectroscopy is related to memory performance in patients with amnestic mild cognitive impairment and Alzheimer's disease. Dement Geriatr Cogn Disord. 2007;24:274–9. doi: 10.1159/000107487. [DOI] [PubMed] [Google Scholar]
- 240.Prince SE, Woo S, Doraiswamy PM, Petrella JR. Functional MRI in the early diagnosis of Alzheimer's disease: is it time to refocus. Expert Rev Neurother. 2008;8:169–75. doi: 10.1586/14737175.8.2.169. [DOI] [PubMed] [Google Scholar]
- 241.Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Samuraki M, Matsunari I, Chen WP, Yajima K, Yanase D, Fujikawa A, Takeda N, Nishimura S, Matsuda H, Yamada M. Partial volume effect-corrected FDG PET and grey matter volume loss in patients with mild Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2007;34:1658–69. doi: 10.1007/s00259-007-0454-x. [DOI] [PubMed] [Google Scholar]
- 243.Debette S, Bombois S, Bruandet A, Delbeuck X, Lepoittevin S, Delmaire C, Leys D, Pasquier F. Subcortical hyperin-tensities are associated with cognitive decline in patients with mild cognitive impairment. Stroke. 2007;38:2924–30. doi: 10.1161/STROKEAHA.107.488403. [DOI] [PubMed] [Google Scholar]
- 244.Huang J, Friedland RP, Auchus AP. Diffusion tensor imaging of normal-appearing white matter in mild cognitive impairment and early Alzheimer disease: preliminary evidence of axonal degeneration in the temporal lobe. AJNR Am J Neuroradiol. 2007;28:1943–8. doi: 10.3174/ajnr.A0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Smith EE, Egorova S, Blacker D, Killiany RJ, Muzikansky A, Dickerson BC, Tanzi RE, Albert MS, Greenberg SM, Guttmann CR. Magnetic resonance imaging white matter hyperintensities and brain volume in the prediction of mild cognitive impairment and dementia. Arch Neurol. 2008;65:94–100. doi: 10.1001/archneurol.2007.23. [DOI] [PubMed] [Google Scholar]
- 246.Whitwell JL, Shiung MM, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology. 2008;70:512–20. doi: 10.1212/01.wnl.0000280575.77437.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–8. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- 248.Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, Weiner MW, Ellis WG, Zarow C, Mungas D, Reed BR, Kramer JH, Schuff N, DeCarli C, Chui HC. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol. 2008;63:72–80. doi: 10.1002/ana.21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kienzl E, Jellinger K, Janetzky B, Steindl H, Bergmann J. A broader horizon of Alzheimer pathogenesis: ALZAS - an early serum biomarker. J Neural Transm Suppl. 2002:87–95. doi: 10.1007/978-3-7091-6139-5_9. [DOI] [PubMed] [Google Scholar]
- 250.Kienzl E, Jellinger KA, Janetzky B, Attems J, Dal-Bianco P, Stachelberger H. Nested gene and autoimmune response in Alzheimer disease. Alzheimer's Dementia. 2006;2:S205. [Google Scholar]
- 251.Kienzl E, Attems J, Bergmann J, Janetzky B, Dal-Bianco P, Steindl F, Rattay F, Preddie E, Jellinger KA. A proteomic and immunohistochemical approach to the study of Alzheimer disease (Poster) Neurobiol Aging. 2004;25:S401. [Google Scholar]
- 252.Jellinger KA, Kienzl E, Janetzky B, Attems J, Dal-Bianco P, Preddie E, Bergmann J, Rattay F. Autoantibody against an abnormal protein as biomarker for pre-symptomatic and stage diagnosis of Alzheimer disease (Poster) 5th Leonard Berg Symposium. 2005 St. Louis, USA, Oct. 07–08, 2005. [Google Scholar]
- 253.Kienzl E, Jellinger KA, Attems J, Janetzky B, Rattay F, Stachelberger H, Dal-Bianco P. Neue Wege zur Früherkennung der Alzheimer Krankheit: “Alzheimer ASsociated Gene” und Biomarker im Blut. NeuroGeriatrie. 2007;4:61–7. [Google Scholar]
- 254.Jellinger KA. New possibilites for early diagnosis of Alzheimer disease: “ALZheimer ASsociated gene” and blood biomarkers. Psychoger Polska. 2007;4:159–77. [Google Scholar]
- 255.Bergmann J, Janetzky B, Kienzl E, Preddie E. Two pathogenic AD-specific transmembrane proteins and their related promotor provide credible targets for passive vaccination and siRNA silencing to prevent and stop AD. 2008. 10th International Hong Kong / Springfield Symposium on Advances in Alzheimer Therapy. Hong Kong, 28, 29 Feb, 1 March 2008.
- 256.Jellinger KA, Kienzl E, Janetzky B, Attems J, Dai-Bianco P, Bergmann J, Preddie E, Rattay F. En-coding an abnormal protein for Alzheimer early diagnosis (poster) 20th Bienn Meeting of the Internatl Soc Neurochemistry (ISN) & Europ Soc Neurochemistry (ESN) 2005 Innsbruck, Austria, Aug. 21–26, 2005. [Google Scholar]
- 257.Bate C, Kempster S, Williams A. Platelet-activating factor antagonists protect amy-loid-β damaged neurons from microglia-mediated death. Neuropharmacology. 2006;51:173–81. doi: 10.1016/j.neuropharm.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 258.Amore M, Tagariello P, Laterza C, Savoia EM. Subtypes of depression in dementia. Arch Gerontol Geriatr. 2007;44:23–33. doi: 10.1016/j.archger.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 259.Potter GG, Steffens DC. Contribution of depression to cognitive impairment and dementia in older adults. Neurologist. 2007;13:105–17. doi: 10.1097/01.nrl.0000252947.15389.a9. [DOI] [PubMed] [Google Scholar]
- 260.Starkstein SE, Mizrahi R. Depression in Alzheimer's disease. Expert Rev Neurother. 2006;6:887–95. doi: 10.1586/14737175.6.6.887. [DOI] [PubMed] [Google Scholar]
- 261.Thompson S, Herrmann N, Rapoport MJ, Lanctot KL. Efficacy and safety of antide-pressants for treatment of depression in Alzheimer's disease: a metaanalysis. Can J Psychiatry. 2007;52:248–55. doi: 10.1177/070674370705200407. [DOI] [PubMed] [Google Scholar]
- 262.Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, Gorman JM, Haroutunian V. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry. 2006;63:161–7. doi: 10.1001/archpsyc.63.2.161. [DOI] [PubMed] [Google Scholar]
- 263.Leszek J, Malyszczak K, Bartys A, Staniszewska M, Gamian A. Analysis of serum of patients with Alzheimer's disease for the level of advanced glycation end products. Am J Alzheimers Dis Other Demen. 2006;21:360–5. doi: 10.1177/1533317506291075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mruthinti S, Buccafusco JJ, Hill WD, Waller JL, Jackson TW, Zamrini EY, Schade RF. Autoimmunity in Alzheimer's disease: increased levels of circulating IgGs binding Aβ and RAGE peptides. Neurobiol Aging. 2004;25:1023–32. doi: 10.1016/j.neurobiolaging.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 265.D'Andrea MR. Add Alzheimer's disease to the list of autoimmune diseases. Med Hypotheses. 2005;64:458–63. doi: 10.1016/j.mehy.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 266.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–56. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 268.Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid β peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 269.Tuppo EE, Arias HR. The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005;37:289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 270.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 271.McGeer EG, McGeer PL. Inflammatory processes in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–9. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 272.Weisman D, Hakimian E, Ho GJ. Interleukins, inflammation, and mechanisms of Alzheimer's disease. Vitam Horm. 2006;74:505–30. doi: 10.1016/S0083-6729(06)74020-1. [DOI] [PubMed] [Google Scholar]
- 273.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 274.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuro-pathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 275.Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Aβ plaque removal following intracranial anti-Aβ antibody administration. Neurobiol Dis. 2004;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 276.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimerlike disease. Nat Med. 2007;13:432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 277.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-βin vitro and in situ. Nat Med. 2003;9:453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 278.Kidd D, Barkhof F, McConnell R, Algra PR, Allen IV, Revesz T. Cortical lesions in multiple sclerosis. Brain. 1999;122:17–26. doi: 10.1093/brain/122.1.17. [DOI] [PubMed] [Google Scholar]
- 279.Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 280.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bo L, Vedeler CA, Nyland H, Trapp BD, Mork SJ. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 2003;9:323–31. doi: 10.1191/1352458503ms917oa. [DOI] [PubMed] [Google Scholar]
- 282.Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 283.Wekerle H, Lassmann H. The immunology of inflammatory demyelinating disease. In: Compston A, Confavreux C, Lassmann H, editors. McAlpine's multiple sclerosis. Philadelphia: Churchill Livingstone; 2006. pp. 491–555. [Google Scholar]
- 284.Dal Bianco A, Bradl M, Frischer J, Kutzelnigg A, Jellinger K, Lassmann H. Multiple sclerosis and Alzheimer's disease. Ann Neurol. 2008;63:174–83. doi: 10.1002/ana.21240. [DOI] [PubMed] [Google Scholar]
- 285.Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, Cotman CW. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–33. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-β and inflammation: two faces of innate immunity in Alzheimer's disease. J Alzheimers Dis. 2007;11:457–63. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- 288.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Fassbender K, Walter S, Kuhl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, Rivest S, Lentschat A, Gulbins E, Jucker M, Staufenbiel M, Brechtel K, Walter J, Multhaup G, Penke B, Adachi Y, Hartmann T, Beyreuther K. The LPS receptor (CD14) links innate immunity with Alzheimer's disease. FASEB J. 2004;18:203–5. doi: 10.1096/fj.03-0364fje. [DOI] [PubMed] [Google Scholar]
- 290.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 291.Streit WJ. Microglial senescence: does the brain's immune system have an expiration date. Trends Neurosci. 2006;29:506–10. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 292.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Home P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A β peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–82. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 293.Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-β immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Bridges LR, Boche D, Neal J, Love S, Esiri E, Wilkinson D, Holmes C, Nicoll J. Immunization with Aβ42 results in variable clearance of Aβ plaques in Alzheimer's disease. Brain Pathol. 2006;16:S81. [Google Scholar]
- 295.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Aβ species removal after aβ42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 296.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 297.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–31. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 298.Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickie GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Amyloid-β peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–63. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, Lavielle R, Delacourte A, Pasquier F. Absence of β-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. 2007;64:583–7. doi: 10.1001/archneur.64.4.583. [DOI] [PubMed] [Google Scholar]
- 300.Delisle MB, Sastre N, Uro-Coste E, Musso-Rigal C, Gillette S, Vellas B. Neuropathology of human Alzheimer disease, in the absence of encephalitis, after Aβ vaccination: a case report. Brain Pathol. 2006;16:S80. [Google Scholar]
- 301.Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-β vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–60. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Aβ immunothera-py. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]