

Abstract
Human genetics has indicated a causal role for the protein α-synuclein in the pathogenesis of familial Parkinson’s disease (PD), and the aggregation of synuclein in essentially all patients with PD suggests a central role for this protein in the sporadic disorder. Indeed, the accumulation of misfolded α-synuclein now defines multiple forms of neural degeneration. Like many of the proteins that accumulate in other neurodegenerative disorders, however, the normal function of synuclein remains poorly understood. α-Synuclein localizes specifically to the nerve terminal and inhibits neurotransmitter release when over-expressed, but the knockout has a modest effect on synaptic transmission, suggesting alternative presynaptic roles. Natively unstructured, synuclein adopts a helical conformation on membrane binding and recent work suggests a role in membrane remodeling. In neural degeneration, synuclein misfolds and aggregates as a β-sheet. Multiple observations now suggest propagation of the misfolded protein as a prion, providing a mechanism for the spread of degeneration through the neuraxis. However, the factors that trigger the original misfolding remain unknown.
α-Synuclein was independently discovered on multiple occasions, providing important but still incompletely understood clues to its normal function as well as its role in disease. It was originally identified using an antibody to purified cholinergic vesicles of the Torpedo electric organ (Maroteaux and Scheller, 1991), providing the first evidence of a presynaptic role. In addition to localization at the synapse, the antibody detected expression at the nuclear envelope, accounting for the designation ‘synuclein’ (Maroteaux et al., 1988). Subsequent work has confirmed the presence of α-synuclein in the nucleus (Goncalves and Outeiro, 2013; McLean et al., 2000; Mori et al., 2002). However, synuclein is a small protein (140 amino acid residues) that falls below the molecular weight cut-off of the nuclear pore (~40 kD). Although the distribution of synuclein may be influenced by interaction with nuclear or cytoplasmic proteins (Goers et al., 2003; Kontopoulos et al., 2006; Specht et al., 2005), untagged, endogenous synuclein would thus be expected to enter the nucleus on the basis of simple diffusion. The discovery of α-synuclein in turn led to the identification of closely related β- and γ- isoforms (Maroteaux and Scheller, 1991).
Synuclein was also identified through the biochemical characterization of senile plaques in Alzheimer’s disease (AD). Although not as abundant as the Aβ peptide, a fragment from the middle of α-synuclein (61–95) now termed the non-Aβ component (NAC) accumulates at high levels in plaques (Ueda et al., 1993). More recent work has shown that synuclein indeed contributes to the pathology of AD as well as of Dementia with Lewy bodies (DLB) (Goedert, 1999; Trojanowski et al., 1998). However, this role appears to reflect cytoplasmic deposition rather than accumulation in extracellular plaques. Nonetheless, subsequent analysis of the NAC precursor (α-synuclein) helped to establish its primarily presynaptic localization (Iwai et al., 1995).
Third, α-synuclein mRNA transcripts were found to change specifically within regions of the zebra finch brain involved in control of song. Relative to other brain regions where synuclein remains at high levels through development and maturity, specific regions implicated in bird song show large, sustained reductions in synuclein expression during song acquisition (George et al., 1995). The regulated expression of synuclein within cell populations that participate in bird song has thus suggested a specific role for the protein in synaptic plasticity, but this role remains poorly understood.
Fourth, synuclein was purified as an inhibitor of phospholipase D2 (PLD2), identifying a specific biochemical function for the protein through a presumably unbiased experimental appraoch. PLD enzymes cleave the headgroup of phosphatidylcholine (PC) to release choline and phosphatidic acid (PA), and have been implicated in membrane trafficking, particularly regulated exocytosis (Hughes et al., 2004; Humeau et al., 2001; Vitale et al., 2001; Zeniou-Meyer et al., 2007). In contrast to the PLD1 isoform, which acts downstream of an ADP ribosylating factor (ARF) GTPase (Caumont et al., 1998; Cockcroft et al., 2002; Colley et al., 1997), PLD2 has constitutive activity. To understand how PLD2 might be regulated, factors that inhibit its function were purified, resulting in the isolation of α- and β-synuclein (Jenco et al., 1998). Additional work demonstrated a requirement for multiple domains in synuclein to inhibit PLD2 (Payton et al., 2004), but the physical interaction has not been documented. Originally, genetic studies in yeast supported a role for synuclein in PLD inhibition (Outeiro and Lindquist, 2003), but subsequent work has not borne this out (Rappley et al., 2009a). Although the initial purification of synuclein as a PLD inhibitor suggested a specific biochemical function of potentially profound significance, the biological relevance of this finding has thus remained uncertain.
Fifth, point mutations in α-synuclein were found to cause an autosomal dominant form of Parkinson’s disease (PD) (Kruger et al., 1998; Polymeropoulos et al., 1997; Zarranz et al., 2004). The clinical phenotype resembles idiopathic PD, with typical tremor, rigidity and bradykinesia, and the pathology shows cytoplasmic Lewy body inclusions characteristic of PD (Golbe et al., 1996), strongly suggesting relevance for the sporadic disorder. Indeed, mutations in α-synuclein account for only a tiny fraction of PD in the general population, but the Lewy bodies and dystrophic neuritis observed in idiopathic PD label strongly for α-synuclein (Galvin et al., 1999; Spillantini et al., 1998b; Spillantini et al., 1997). Immunostaining for α-synuclein subsequently revealed abundant inclusions not previously detected using standard histological methods (Jellinger, 2011). In fact, many of the monoclonal antibodies previously raised against Lewy bodies recognize α-synuclein (Giasson et al., 2000b), supporting the impression that although other proteins may also accumulate in the inclusions of PD, α-synuclein predominates. Taken together, the genetic evidence for a causative role and the neuropathologic evidence for accumulation in essentially all patients with PD indicates a central role for synuclein in the idiopathic disorder.
The Structure of Synuclein
The N-terminus of α-synuclein contains seven 11 residue repeats that are predicted to form an amphipathic alpha-helix (Fig. 1). The repeats are very highly conserved, both across species and among the three different isoforms. The motif is also unique, with no similar sequence identified outside the synuclein family. In addition, this sequence has been detected only in vertebrates, including the lamprey (Busch and Morgan, 2012). Remarkably, all of the mutations associated with PD—A53T, A30P and E46K as well as the more recently described G51D and H50Q (Appel-Cresswell et al., 2013; Kruger et al., 1998; Lesage et al., 2013; Polymeropoulos et al., 1997; Proukakis et al., 2013; Zarranz et al., 2004)—cluster within this N-terminal domain. It is also interesting to note that rodent synuclein normally contains a threonine at position 53, which causes PD in humans. The A53T mutation thus appears pathogenic specifically within the human context. Model genetic organisms such as worms, flies and yeast do not contain identifiable homologues, indicating that the synucleins are not required for synaptic transmission or membrane trafficking more generally. On the other hand, the apolipoproteins and a set of plant proteins that accumulate during desiccation and seed formation also contain amphipathic α-helices with eleven residue repeats (George et al., 1995).
Figure 1.
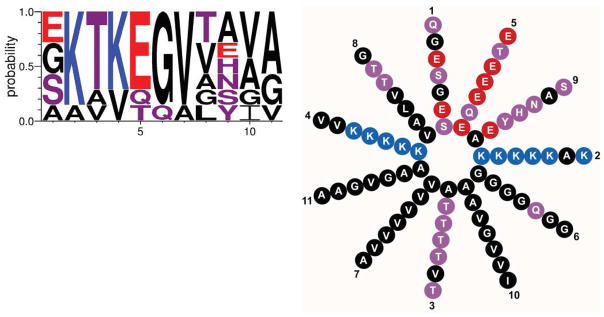
The N-terminus of synuclein contains 7 eleven residue repeats with the consensus sequence shown above and helical wheel to the right. The height of the single letter amino acid code indicates the probability of finding that particular residue in the repeats of human α-synuclein. Blue indicates basic, red acidic, purple polar uncharged and black nonpolar residues.
Repeats of this size enable the polypeptide to make exactly three turns of the helix and thus interact directly with the surface of a membrane through multiple repeats. However, the sequence of apolipoprotein and plant seed proteins bears little if any obvious similarity to the synucleins.
Purified, recombinant synuclein behaves like a natively unfolded protein in vitro (Bertoncini et al., 2005; Weinreb et al., 1996) but, as predicted from the sequence, forms an α-helix on binding to artificial membranes (Davidson et al., 1998). Shown initially by circular dichroism, the conformational change associated with membrane binding requires acidic phospholipid headgroups, suggesting an interaction of the membrane with lysines found on opposite sides of the helix (Fig. 1). There is minimal specificity for a particular acidic headgroup, with phosphatidylserine recognized as well as phosphatidic acid and phosphatidylinositol (Zhu and Fink, 2003). NMR of synuclein in SDS micelles also reveals an α-helix, but bent presumably due to the small size of the micelle (Eliezer et al., 2001; Ulmer et al., 2004). On membranes, which have a larger diameter than micelles, the analysis of spin-labeled protein shows that synuclein adopts the extended 11/3 helix predicted from the sequence (Jao et al., 2004). Synuclein also lies along the surface of the membrane, at least half-buried in the bilayer (Bussell et al., 2005; Jao et al., 2008; Wietek et al., 2013).
Despite the original description as a natively unfolded protein, recent work has suggested that α-synuclein may in fact remain helical in solution, with important implications for its normal function and its susceptibility to aggregation. The evidence for intrinsic disorder has depended primarily on the analysis of bacterially expressed recombinant protein, and a denaturation step used by some groups in the purification has been suggested to account for the unfolded state (Bartels et al., 2011; Wang et al., 2011). Consistent with a lack of folding, synuclein behaves like a much larger protein by size exclusion chromatography, but multimerization is another possibility. To assess the multimeric state of native synuclein, a recent study from the Selkoe lab used a combination of cross-linking and analytical ultracentrifugation to determine the molecular weight of mammalian synuclein isolated from red blood cells and cell lines. In contrast to previous studies, this work found that native α-synuclein behaves as a folded, helical tetramer (Bartels et al., 2011). EM reconstruction and NMR of protein purified under nondenaturing conditions further suggested a multimeric state that resists aggregation (Wang et al., 2011). However, subsequent work using recombinant synuclein has confirmed that even the nondenatured recombinant protein is intrinsically disordered and loses its α-helical conformation after dissociation from membranes (Fauvet et al., 2012). Loss of helicity could thus reflect the dilution inherent in preparing an extract, with the helical state maintained at higher concentrations (Dettmer et al., 2013; Wang et al., 2011), but NMR studies in E. coli have in fact suggested that macromolecular crowding maintains the disordered state of synuclein (McNulty et al., 2006). It is also possible that synuclein folds to form a multimer only in mammalian cells, but the analysis of native brain synuclein has recently confirmed its almost entirely monomeric state (Burre et al., 2013). Recently, it has also been shown that synuclein can assemble into an oligomer (possibly tetramer) on nanoparticles (Varkey et al., 2013), but this phenomenon seems to differ from the ability of a preformed tetramer to interact with membranes (Wang et al., 2011). At this point, it remains possible that α-synuclein adopts a helical tetrameric state in solution, but the evidence is not definitive. The unavoidable dilution that accompanies purification of native synuclein complicates the analysis, but it is perhaps more important to acknowledge that despite extensive biochemical studies in vitro, the conformation of synuclein in cells remains poorly understood.
In contrast to the N-terminal membrane binding domain, the C-terminus of human α-synuclein is polar, with a higher proportion of charged residues. This domain undergoes phosphorylation at multiple sites (Oueslati et al., 2010; Sato et al., 2013), suggesting a mechanism for regulation, but the function of the C-terminus remains unclear, and it is the least conserved domain across species as well as among α-, β-, and γ- isoforms. The C-terminus may affect membrane binding under particular conditions (Shvadchak et al., 2011), but phosphorylation toward the end of the N-terminal repeats, at Ser-87, more clearly affects membrane binding in vitro than phosphorylation at the other, more C-terminal sites (Paleologou et al., 2010). The observations thus suggest a potential biological role for Ser-87 phosphorylation, although this again remains to be identified in the context of the cell.
Membrane Interactions and the Presynaptic Location of Synuclein
The presynaptic location of α-synuclein has been recognized since its original identification as a protein associated with synaptic vesicles (Maroteaux et al., 1988). In contrast to many proteins involved in neurodegeneration that are distributed throughout the neuron, however, α-synuclein localizes specifically to the nerve terminal, with relatively little in the cell body, dendrites or extrasynaptic sites along the axon (George et al., 1995; Iwai et al., 1995). Several non-neural tissues including red blood cells also express α-synuclein (Barbour et al., 2008), but the protein is relatively specific to the nervous system (Iwai et al., 1995). In addition, α-synuclein is widely expressed by many neuronal populations within both central and peripheral nervous systems, suggesting a general role in neuronal function. However, α-synuclein appears to be one of the last proteins that localizes to developing synapses, arriving after integral membrane proteins of the synaptic vesicle and the peripheral membrane synapsin proteins (Withers et al., 1997). Consistent with its restriction to the vertebrate lineage, its accumulation at the synapse thus does not appear essential for synapse development or function.
Similar to α-synuclein, the β- isoform also exhibits a presynaptic location (Jakes et al., 1994; Mori et al., 2002; Quilty et al., 2003). Indeed, α- and β- isoforms colocalize at many but not all presynaptic boutons. However, γ-synuclein is expressed by glia and only specific neuronal populations, in particular dopamine neurons (Brenz Verca et al., 2003; Galvin et al., 2001). γ-Synuclein is also expressed by a variety of cancers (breast, colon, pancreas) where it apparently contributes to tumor progression through a number of potential mechanisms (Hua et al., 2009; Inaba et al., 2005; Ji et al., 1997; Pan et al., 2002).
Despite the original association with synaptic vesicles, it has been unclear how α-synuclein localizes to the nerve terminal. In the absence of an obvious transmembrane domain or lipid anchor, synuclein presumably relies on the N-terminal repeats for membrane binding in cells, similar to the observations with artificial membranes made in vitro. However, fractionation of brain extracts reveals a very weak association with synaptic vesicles, and the vast majority of synuclein behaves as a soluble protein (Fortin et al., 2004; Kahle et al., 2000). These observations suggest that the association with native synaptic vesicles is weak, or disrupted by the procedures required for biochemical fractionation: dilution alone could result in the loss of synuclein from synaptic vesicles. To examine the mobility of synuclein in intact cells, cultured hippocampal neurons were therefore transfected with GFP-tagged synuclein and individual presynaptic boutons subjected to photobleaching. The synaptic fluorescence recovered quite rapidly (within seconds) after photobleaching, indicating that the protein is highly mobile (Fortin et al., 2004). More recently, this approach has been extended in vivo, to cortical neurons of transgenic mice expressing α-synuclein-GFP (Unni et al., 2010). In this case, recovery occurred more slowly (over minutes) but this presumably reflects the altered geometry in vivo, with adjacent synapses (and unbleached synuclein-GFP) simply further away from the bleached boutons. However, synapses with higher expression of synuclein also showed less complete recovery, raising the possibility that aggregation contributes. Not surprisingly, fluorescence recovery after photobleaching in a C. elegans model of inclusion formation by synuclein in muscle has shown a substantial immobile fraction (van Ham et al., 2008). The results in neurons with more physiological levels of expression thus indicate that synuclein interacts weakly with elements of the nerve terminal.
Despite its weak interaction with cellular membranes, synuclein nonetheless recovers more slowly after photobleaching than GFP (Fortin et al., 2004), and the N-terminal membrane-binding domain of synuclein seems likely to mediate the interaction. The A30P mutation associated with familial PD in fact disrupts both the association of synuclein with brain membranes and the presynaptic location of synuclein in cultured neurons, and accelerates the rate of recovery after photobleaching to that of GFP (Fortin et al., 2004; Jensen et al., 1998). The A30P mutation also impairs the interaction of purified, recombinant synuclein with artificial membranes (Jo et al., 2002). Although less dramatic in vitro than in cells, the effect of the A30P mutation strongly supports a role for membrane binding by the N-terminus in presynaptic localization.
How then does synuclein localize specifically to presynaptic boutons rather than other cell membranes? Acidic headgroups are found on the cytoplasmic leaflet of many intracellular membranes, but synuclein has a preference for membranes with high curvature (Jensen et al., 2011; Middleton and Rhoades, 2010), and synaptic vesicles are among the smallest biological membranes described. Consistent with this, synuclein disperses from presynaptic boutons with stimulation (Fortin et al., 2005), suggesting that it dissociates from the membrane upon delivery to the relatively flat plasma membrane by synaptic vesicle exocytosis.
What confers the specificity of synuclein for membranes with high curvature? Interestingly, the hydrophobic face of the N-terminal α-helix contains a series of threonines at position 3 in the repeat (Fig. 1). Although this polar residue might be expected to disrupt hydrophobic interactions with the membrane, threonine is in fact less polar than serine, and the precise positioning of this residue in repeats 2–5 and 7 is highly conserved among all synuclein isoforms. It is therefore possible that threonine at these positions weakens the interaction of synuclein with membranes precisely so that it can acquire specificity for high curvature. To test this possibility, the threonines were replaced by large, nonpolar residues (leucine and phenylalanine) and the recombinant mutant protein indeed loses its specificity for both acidic membranes and small vesicles (Pranke et al., 2011). When expressed in yeast, the mutant also localizes to the plasma membrane rather than to intracellular vesicles, consistent with stronger membrane interaction interfering with the preference for high curvature.
In addition to acidic phospholipid headgroups and high curvature, synuclein appears to have a preference for specific membrane microdomains. Since the large proportion of soluble synuclein makes it difficult to detect a membrane-bound fraction by morphological techniques in most cells other than neurons, digitonin was used to permeabilize selectively the plasma membrane of Hela cells expressing human α-synuclein, releasing the unbound cytosolic protein (Fortin et al., 2004). The remaining synuclein appeared punctate, but failed to colocalize with markers for many organelles. Rather, it colocalized with components of lipid rafts, a membrane microdomain with reduced fluidity that is enriched in cholesterol and saturated acyl chains (Fortin et al., 2004). The PD-associated A30P mutation abolished this localization, supporting the specificity of the interaction, and the biochemical analysis of detergent-resistant membranes by flotation gradient confirmed the localization to rafts. Importantly, the disruption of lipid rafts also prevents the accumulation of synuclein in presynaptic boutons (Fortin et al., 2004), supporting the relevance of this interaction for neurons. In addition to the requirement for acidic phospholipid, biochemical studies in vitro have indicated that synuclein requires a combination of phospholipid with oleoyl as well as polyunsaturated acyl chains (Kubo et al., 2005), suggesting that it may specifically recognize the phase boundary that arises between membranes that differ in fluidity. Remarkably, there was an apparent requirement for the acidic headgroup on the polyunsaturated acyl rather than oleoyl chain (Kubo et al., 2005), raising the possibility of a distinct and previously unknown microdomain in neurons. Further, recent work has found that synuclein can influence lipid packing within raft-like domains containing cholesterol (Leftin et al., 2013), suggesting that synuclein may not simply be recruited by these structures but also contribute to their formation, very similar to other peripheral membrane proteins such as caveolin (Parton and del Pozo, 2013).
It has also been suggested that synuclein might act as a fatty acid binding protein (Sharon et al., 2001). Synuclein promotes the uptake of polyunsaturated fatty acids into cells, and polyunsaturated fatty acids promote the oligomerization of synuclein (Assayag et al., 2007; Perrin et al., 2001; Sharon et al., 2003a; Sharon et al., 2003b). Supporting a role for this activity in vivo, the analysis of α-synuclein knockout mice has shown remarkable changes in brain cardiolipin, including acyl chain composition (Ellis et al., 2005). Fatty acid uptake and metabolism also appear affected (Golovko et al., 2005; Golovko et al., 2006; Golovko et al., 2007), although with only modest changes in other brain phospholipids (Barcelo-Coblijn et al., 2007; Rappley et al., 2009b). γ-Synuclein knockouts also show only modest brain region-specific changes in both phospholipid headgroup and acyl chain composition (Guschina et al., 2011). Nonetheless, even small changes in acyl chain composition may influence membrane fluidity and trafficking, and the expression of α-synuclein appears to promote clathrin-dependent endocytosis (Ben Gedalya et al., 2009).
The Role of Synuclein in Neurotransmitter Release
The presynaptic location of synuclein and its interaction with membranes have strongly suggested a role in transmitter release. Several of the original publications indeed reported that α-synuclein promotes release (Cabin et al., 2002; Murphy et al., 2000), but others suggested an inhibitory role. Dopamine release by α-synuclein knockout mice recovers faster from repetitive stimulation than in wild type animals, and the knockouts show a mild reduction in striatal dopamine stores consistent with increased release (Abeliovich et al., 2000). The analysis of dopamine and norepinephrine release by knockout mice in vivo also shows more rapid facilitation than in wild type and reduced depression after multiple bursts of stimulation (Yavich et al., 2006; Yavich et al., 2004). These effects on dopamine release in vivo are among the most dramatic reported for α-synuclein knockout mice, and suggest a major disturbance in the mobilization of synaptic vesicles. However, the findings at glutamate synapses are much less striking (Abeliovich et al., 2000; Chandra et al., 2004).
To mimic the increase in expression that causes PD in families with a duplication or triplication of the gene, α-synuclein has also been over-expressed both in culture and in transgenic mice. Although the over-expression of PD-associated or C-terminal truncation mutants can produce a degenerative process in vivo (Giasson et al., 2002; Gispert et al., 2003; Gomez-Isla et al., 2003; Lee et al., 2002b; Shults et al., 2005; Yazawa et al., 2005), non-viral over-expression of the wild type protein usually produces little toxicity (Matsuoka et al., 2001). Transgenic mice over-expressing the wild type human protein do show a number of behavioral abnormalities relating to olfaction, gastrointestinal motility and motor activity (Fleming et al., 2008; Kuo et al., 2010; Noorian et al., 2012; Wang et al., 2008), suggesting that these animals may reproduce the prodromal phase of PD, but there is little if any detectable degeneration.
To understand how over-expression of synuclein might affect behavior and cause PD, the analysis was extended to synaptic physiology and a direct analysis of the release mechanism. First studied in chromaffin cells, over-expression of the wild type human protein was found to inhibit the exocytosis of dense core vesicles as measured by direct amperometric recordings of quantal catecholamine release (Larsen et al., 2006). This did not involve a change in calcium sensitivity, quantal size or the kinetics of individual quantal events, but rather a reduction in the number of events. The PC12 cell granules also appear to accumulate at the plasma membrane, suggesting a specific defect at or close to the fusion event, and the A30P mutant had an effect surprisingly similar to wild type human synuclein considering its defect in membrane interactions (Larsen et al., 2006).
In cultured neurons, over-expression of the wild type human protein at levels that do not produce deposits or obvious toxicity causes an inhibition of synaptic vesicle exocytosis as measured by optical imaging of both hippocampal and midbrain dopamine neurons (Nemani et al., 2010) (Fig. 2). Modest over-expression in transgenic mice produced a similar defect in neurotransmission measured by postsynaptic recording at hippocampal CA1 synapses (Nemani et al., 2010). It is also important to note that there was no change in quantal size. Several reports have shown that multimeric synuclein can form pores in artificial membranes in vitro (Rochet et al., 2004; Tsigelny et al., 2007; Volles et al., 2001). This should dissipate the H+ electrochemical gradient that drives neurotransmitter uptake into vesicles, and the lack of change in quantal size argues further against pore formation by multimers, at least in these cells. Although previous work on the role of synuclein in transmitter release had identified major defects only in monoamine neurons, these findings indicated that the disturbance with over-expression is more general.
Figure 2.
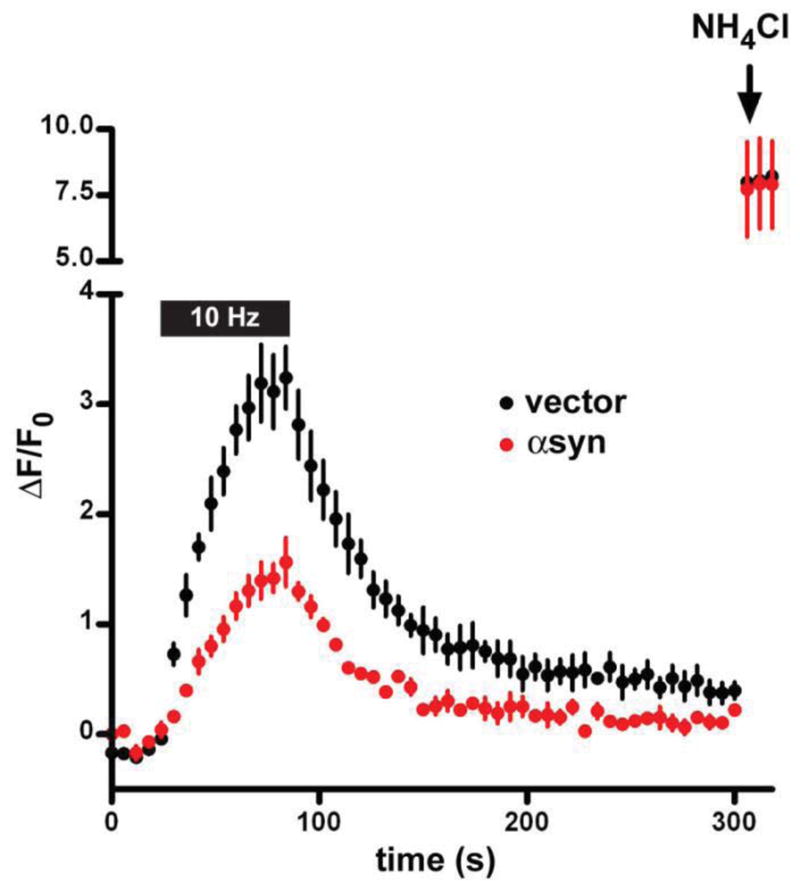
Over-expression of α-synuclein in rat hippocampal neurons inhibits synaptic vesicle exocytosis. Embryonic rat hippocampal neurons were transfected with VGLUT1-pHluorin and either wild type human α-synuclein or empty vector. After growth in culture for two weeks, the cells were stimulated at 10 Hz for 60 s and the response of VGLUT1-pHluorin monitored. Quenched at the low pH of synaptic vesicles, the pHluorin (a modified form of GFP shifted in its pH sensitivity) becomes more fluorescent when exposed to the external pH during exocytosis. The reacidification that follows endocytosis results in the quenching of fluorescence. NH4Cl is used to alkalinize all of the intracellular VGLUT1-pHluorin pool, and demonstrates that synuclein over-expression does not reduce expression of the reporter. Over-expression of synuclein impairs the exocytosis of synaptic vesicles, but has no apparent effect on endocytosis after normalization to peak stimulated fluorescence. (Reproduced from Nemani et al., 2010)
Imaging further demonstrated a specific defect in exocytosis, with no change in the endocytosis of synaptic vesicle membrane despite the effects on clathrin-dependent endocytosis observed in other cells (Ben Gedalya et al., 2009). In contrast to LDCV release by chromaffin cells (Larsen et al., 2006), the A30P mutation abolishes the effect of synuclein over-expression on synaptic vesicle exocytosis (Nemani et al., 2010). Presumably, the specific accumulation of synuclein at release sites (disrupted by the A30P mutation) is more important for neurons, with many long processes, than for small, compact chromaffin cells. However, electron microscopy in the transgenic mice over-expressing synuclein also showed a dispersion of synaptic vesicles away from the active zone and into the axon (Nemani et al., 2010), and it is more difficult to reconcile this observation with the accumulation of secretory granules at the plasma membrane in chromaffin cells that over-express synuclein (Larsen et al., 2006). Recent ultrastructural analysis of a different transgenic mouse line has shown enlargement of boutons and convoluted internal membranes connected to the cell surface (Boassa et al., 2013).
The precise nature of the defect in synaptic vesicle exocytosis remains unclear. Interestingly, the transgenic mice show a reduction in synapsins and complexin, consistent with a change in exocytosis. Subsequent work has also shown a defect in transmitter release with over-expression of synuclein in hippocampal cultures. However, this study described “vacant synapses” that were devoid of synaptic vesicle proteins and contained abnormal membranes that were not observed in other systems, suggestive of toxicity (Boassa et al., 2013; Scott et al., 2010). Importantly, defective endocytosis often accompanies toxicity, and no such “vacant synapses” were observed in another study (Nemani et al., 2010). It remains possible that even in the absence of aggregates and overt injury, toxic oligomers account for the inhibition of release by synuclein observed by multiple groups. However, the ability of the PD-associated A30P mutation to block the inhibition of release argues against this possibility. In addition, truncation of the C-terminus, which promotes aggregation of synuclein in vitro and in vivo (Crowther et al., 1998; Hoyer et al., 2004; Li et al., 2005; Wakamatsu et al., 2008), had no effect on the inhibition of release, supporting a specific effect of synuclein independent of toxicity.
Although the phenotype of the single α-synuclein knockout is relatively modest, the animals show a remarkable resistance to the parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) (Dauer et al., 2002). Exposure to MPTP results in the loss of substantia nigra dopamine neurons due to uptake of the active metabolite N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine (MPP+) by the reuptake transporters for monoamines (Javitch et al., 1985), followed by apoptosis triggered by inhibition of the respiratory chain (Krueger et al., 1990). The vesicular monoamine transporter (VMAT) protects against MPP+ toxicity by sequestering the toxin inside secretory vesicles, away from mitochondria, and selection in MPP+ was used to isolate the cDNA encoding VMAT (Liu et al., 1992a; Liu et al., 1992b). Subsequent work has confirmed the protection against MPTP toxicity conferred by loss of α-synuclein (Drolet et al., 2004; Fornai et al., 2005; Fountaine et al., 2008; Robertson et al., 2004; Thomas et al., 2011), but strains apparently differ in the magnitude of this effect (Schluter et al., 2003). In α-synuclein knockout mice, mitochondria are not affected by MPTP administration, suggesting a defect in access, but the activities of monoamine transporters known to control access of the toxin appear no different from wild type (Dauer et al., 2002). Thus, resistance to MPTP toxicity is one of the more robust aspects of the α-synuclein knockout phenotype, but the mechanism remains unknown. Although MPTP toxicity differs in important ways from PD, the ability of the α-synuclein knockout to protect against the toxin suggests a role for the normal function of synuclein in the pathogenesis of degeneration, particularly since over-expression of synuclein does not increase vulnerability to MPTP (Thomas et al., 2011).
The existence of three synuclein isoforms, in many cases expressed by the same cells, has raised the possibility that redundancy accounts for the modest phenotype of α-synuclein knockout mice. However, the analysis of α-/β-synuclein double knockout mice also showed no major change in synapse structure or excitatory transmission measured electrophysiologically in hippocampal region CA1 (Chandra et al., 2004). On the other hand, the α-/β- double knockout does show a modest reduction in striatal dopamine levels. In addition, α-/γ- double and synuclein triple knockouts show a substantial increase in striatal dopamine release in vivo not observed with the single knockouts (Anwar et al., 2011; Senior et al., 2008). These mutants did not exhibit a change in dopamine transporter activity or tissue dopamine levels, implicating a specific alteration of dopamine release. The mechanism remains unknown, but the α-/β- double knockout shows an increase in complexin (Chandra et al., 2004). Interestingly, synuclein over-expression reduces complexin levels (Nemani et al., 2010), suggesting that over-expression can increase the normal activity of synuclein, and that an increase in the normal function of synuclein contributes to the degeneration produced by its up-regulation.
At hippocampal synapses, the effect of the triple knockout has been controversial. According to one report from the Südhof lab, there was no change in baseline transmitter release (Burre et al., 2010). However, an independent report by a former member of the same group showed an increase in transmitter release in the triple knockout (Greten-Harrison et al., 2010). The increase was small, possibly accounting for the failure to detect a change by others, and raising the possibility that any change in release might be secondary. Indeed, the loss of all three synuclein genes results in smaller presynaptic boutons (Greten-Harrison et al., 2010), suggesting an alternative role for these proteins.
Previous work has shown a strong genetic interaction between synuclein and the degeneration produced by loss of the presynaptic chaperone cysteine string protein (CSPα) (Chandra et al., 2005). Knockout of CSPα does not affect synaptic transmission shortly after birth, but eventually results in rapidly progressive synaptic degeneration and death within two months (Fernandez-Chacon et al., 2004). CSPα thus does not itself appear required for transmitter release, but rather serves to maintain the function of the nerve terminal over a longer time frame. Work from the Südhof lab has now suggested that synuclein may have a similar role in maintenance of the nerve terminal, rather than transmitter release.
Remarkably, the over-expression of α-synuclein greatly delays the degeneration due to loss of CSPα, and the loss of synuclein exacerbates the CSPα knockout phenotype (Chandra et al., 2005), suggesting that synuclein may have a role as chaperone, very similar to CSPα. CSPα appears particularly important for the levels of t-SNARE SNAP-25 (Sharma et al., 2012; Sharma et al., 2011). As might be anticipated for a chaperone of the transmitter release machinery, the resulting perturbations of SNARE complex assembly are activity-dependent. Since synuclein over-expression inhibits transmitter release, the resulting decrease in activity might account for rescue of the CSPα phenotype. However, synuclein over-expression rescues the reduction in SNARE complex, not the decrease in SNAP-25 protein levels. The results suggest that whereas CSPα has a specific role with SNAP25 that secondarily affects SNARE complex levels, synuclein has a specific role in SNARE complex formation and can bypass the defect in SNAP-25. The original work did not detect biochemical evidence of α-synuclein associating with the presynaptic SNARE complex (Chandra et al., 2005), but a subsequent study did identify a direct biochemical interaction (Burre et al., 2010). In particular, the hydrophilic C-terminus of α-synuclein appears to interact with v-SNARE synaptobrevin 2 (Burre et al., 2010). Consistent with a requirement for the C-terminus of α-synuclein to interact with synaptobrevin, γ-synuclein, which diverges in sequence from α- at the C-terminus, does not rescue the loss of CSPα (Ninkina et al., 2012). In contrast to the role of CSPα as chaperone for SNAP-25, α-synuclein thus appears to have a role in SNARE complex formation.
How can a putative chaperone for the SNARE complex either have no effect on or inhibit transmitter release? The number of SNARE complexes may not be rate-limiting for transmitter release, and rescue of the degeneration in CSPα knockout mice does not require an increase in SNAP-25. Regardless of mechanism, SNARE complex levels correlate more closely with the degenerative process than with transmitter release. However, the levels of SNARE complex have not been studied extensively in animals with other defects in transmitter release, and may simply reflect changes in another process more directly affected by synuclein. Indeed, we do not know what comprises the total pool of SNARE complexes in the brain—cis complexes on synaptic vesicles or the plasma membrane, trans-complexes made by docked vesicles or some other pool? Recent work in vitro has also found that synuclein can inhibit membrane fusion independent of the SNARE proteins, and failed to detect an interaction of synuclein with synaptobrevin (DeWitt and Rhoades, 2013). The mechanism by which synuclein rescues the loss of CSPα thus remains uncertain.
The synuclein triple knockouts do die prematurely, but at around one year, a phenotype much milder than the CSP knockout (Fernandez-Chacon et al., 2004; Greten-Harrison et al., 2010). In addition to smaller presynaptic boutons, the synuclein triple knockout also produces an axonal defect in older animals but no obvious synapse loss. The ability to rescue loss of CSPα thus remains perhaps the most dramatic effect of synuclein observed in vivo, with a very modest degenerative phenotype in synuclein triple knockout mice alone.
Synuclein has also been reported to interact biochemically with a large number of proteins which might regulate its activity. One of the first identified, synphilin appears to promote the aggregation of synuclein (Engelender et al., 1999; McLean et al., 2001; Ribeiro et al., 2002). Synphilin also localizes to the nerve terminal in a synuclein-sensitive manner, but the physiological role of synphilin and its interaction with synuclein remain unclear. Tubulin appears to interact with a multimeric form of synuclein, and synuclein can influence the microtubule cytoskeleton (Lee et al., 2006). However, the functional ramifications of this interaction seem more relevant for the toxicity associated with synuclein than for its normal function (Alim et al., 2002; Chen et al., 2007; Kim et al., 2008; Lee et al., 2006). Since synuclein binds to membranes in an α-helical conformation, one interesting approach has been to use membrane-bound synuclein as a probe for conformation-specific interacting proteins (Woods et al., 2007). This again resulted in the isolation of tubulin, but also other proteins associated with the cytoskeleton. In addition, this approach identified one novel protein that is natively unfolded until membrane-bound (Boettcher et al., 2008). More recently, the small GTPase rab3a has been proposed to regulate the membrane association of α-synuclein in a GTP-dependent manner (Chen et al., 2013), suggesting functional integration of synuclein into the cycling of this synaptic vesicle rab and hence into the synaptic vesicle cycle. However, the role of these potential regulatory mechanisms remains unclear, largely because we do not understand the normal function of synuclein.
The Synucleinopathies
Although the normal function of synuclein remains elusive, the protein has a central role in multiple neurodegenerative processes. Indeed, the identification of mutations in α-synuclein has shifted the focus of work on the pathogenesis of PD from a specific defect in dopamine neurons to a more widespread disturbance in the behavior of this protein. Previously, Lewy bodies had been detected by staining with hematoxylin and eosin and with somewhat more sensitivity by immunostaining for ubiquitin. However, immunostaining for α-synuclein revealed much more widespread deposits in dystrophic neurites as well as Lewy bodies of cell populations not previously known to be affected (Galvin et al., 1999; Spillantini et al., 1998b; Spillantini et al., 1997). In addition to demonstrating the relevance of synuclein for the idiopathic disorder, these observations have suggested a basis for the non-motor manifestations of PD (Ahlskog, 2007; Dickson et al., 2009; Jellinger, 2011). Constipation, hyposmia, depression and rapid eye movement (REM) behavior disorder, which involves the loss of muscle atonia during REM sleep and hence unsuppressed motor activity while dreaming, can precede the onset of characteristic parkinsonian motor symptoms by up to two decades, consistent with the deposition of α-synuclein in the enteric nervous system, olfactory bulb, dorsal motor nucleus of the vagus and glossopharyngeal nerves as well as other brainstem nuclei (Postuma et al., 2012). Additional autonomic problems (e.g., orthostatic hypotension, incontinence) become more symptomatic in advanced disease, and synuclein deposits in autonomic ganglia (Iwanaga et al., 1999; Orimo et al., 2008). Cortical deposits of synuclein that occur late in the disease presumably contribute to cognitive problems. Certain non-motor manifestations of PD can respond to dopamine replacement, raising questions about the significance of synuclein deposition outside the nigrostriatal projection. However, many symptoms do not respond, and the widespread accumulation of synuclein presumably accounts for many of the dopamine-resistant symptoms.
It is important to note that the relationship between α-synuclein deposition and neuronal dysfunction remains unclear. In the substantia nigra, substantial cell loss occurs before symptoms develop, suggesting that protein deposition is not as important as cell loss. However, cell loss may not accompany synuclein deposition elsewhere. In the enteric nervous system, Lewy pathology is indeed not associated with cell loss (Annerino et al., 2012), raising the possibility of a functional rather than anatomic disturbance. On the other hand, synuclein deposition itself may not even produce dysfunction, and pathologic investigation of many older individuals (up to 30% of centenarians) reveals extensive synucleinopathy (incidental Lewy body disease) with no clear neurological symptoms (Ding et al., 2006; Markesbery et al., 2009). Indeed, synuclein aggregation may represent a neuroprotective response, with a different species of synuclein responsible for dysfunction. Although synuclein deposition has thus superseded cell loss as evidence of degeneration, its actual role in the degenerative process remains unknown.
α-Synuclein has also been implicated in at least two other disorders, Multiple System Atrophy (MSA) and Dementia with Lewy Bodies (DLB). Interestingly, these conditions also produce clinical parkinsonism, but involve the deposition of α-synuclein in different cells from those affected by typical PD. MSA can begin with parkinsonism, autonomic failure or cerebellar ataxia, but usually progresses to involve one or both of the other components, resulting in the recognition that these initially disparate conditions reflect a common disorder. However, the parkinsonism observed in MSA does not generally respond well to dopamine replacement, presumably because the pathology affects many cell populations in addition to dopamine-producing cells of the substantia nigra, including postsynaptic medium spiny neurons in the striatum (Papp and Lantos, 1994; Sato et al., 2007). In contrast, PD affects preferentially the dopamine neurons, with spared postsynaptic cells still responsive to dopamine replacement.
In MSA, α-synuclein deposits in glial (generally oligodendroglial) cytoplasmic inclusions (GCIs) (Spillantini et al., 1998a; Tu et al., 1998), rather than in the neuronal Lewy bodies or dystrophic neurites more characteristic of PD. The expression of α-synuclein in oligodendrocytes reproduces the MSA phenotype, suggesting that the protein there is causative (Shults et al., 2005; Yazawa et al., 2005). Remarkably, α-synuclein is not normally expressed by oligodendrocytes (Miller et al., 2005), and a fundamental question remains about the origin of this protein: is it taken up from neurons, or does the pathological process activate expression by glia? In fact, the pathology shows relatively little deposition of synuclein in neurons, with only occasional nuclear and cytoplasmic inclusions (Farrer et al., 2004; Jellinger and Lantos, 2010; Nishie et al., 2004b). At the same time, oligodendrocytes do not seem to up-regulate expression of α-synuclein even in MSA (Miller et al., 2005). Regardless of its source, α-synuclein accumulates to particularly high levels in MSA, suggesting a process distinct from Lewy pathology. In addition, GCI lesions are widespread in MSA, but generally correlate with neuron loss in the substantia nigra, pons, cerebellum and intermediolateral cell columns of the spinal cord, suggesting that the glial process may be primary.
MSA is rarely familial (Soma et al., 2006) and mutations in α-synuclein have not been observed (Ozawa et al., 2006). However, polymorphisms in the synuclein gene may influence susceptibility to MSA (Al-Chalabi et al., 2009; Scholz et al., 2009). In addition, the analysis of familial MSA has recently identified mutations in COQ2, a protein required for the synthesis of coenzyme Q10 (Collaboration, 2013). The degenerative process in MSA may thus reflect a primary lesion in mitochondria.
DLB more closely resembles idiopathic PD in terms of Lewy body pathology. Although DLB appears to be a distinct syndrome, with early cognitive impairment, fluctuating alertness and visual hallucinations in addition to progressive parkinsonism, the distribution of Lewy pathology appears remarkably similar to that observed in PD, with a brainstem-predominant form and others involving the cortex as well (McKeith et al., 2005). Like PD and MSA, DLB also involves primarily the deposition of α-synuclein.
Lewy pathology was originally considered to involve only α-synuclein, but β- and γ- can deposit in both PD and DLB (Galvin et al., 1999). Similar to α-synuclein, β- accumulates presynaptically in PD, but γ- forms axonal spheroids. β-synuclein has been suggested to ameliorate the toxicity of α-synuclein, by reducing either its aggregation or its expression (Fan et al., 2006; Hashimoto et al., 2001). However, polymorphisms in β-synuclein predispose to DLB (Ohtake et al., 2004), and transgenic mice over-expressing the variant develop degeneration and behavioral abnormalities (Fujita et al., 2010). These animals do not develop typical Lewy pathology, but they do accumulate both α- and β-synuclein in axonal spheroids (Fujita et al., 2010). Indeed, β-synuclein appears as toxic as α-synuclein to cultured neurons (Taschenberger et al., 2013). Mutations in γ-synuclein have not been identified as a cause of familial PD or related disorders, and it has generally been considered nontoxic, but recent over-expression of even this isoform has produced degeneration in transgenic mice (Ninkina et al., 2009), and a polymorphism has also been linked to DLB (Nishioka et al., 2010). Rather than contribute to disease simply through a decline in their protective function (Li et al., 2004; Rockenstein et al., 2001), which nonetheless remains a possibility, β- and γ-synuclein may thus cause degeneration.
α-Synuclein also deposits in other neurodegenerative disorders. Alzheimer’s disease shows Lewy pathology in up to 60% of cases, but more often restricted to the amygdala than in PD or DLB (Hamilton, 2000; Leverenz et al., 2008; Uchikado et al., 2006). Neurodegeneration with brain iron accumulation due to mutations in pantothenate kinase type 2 also exhibit Lewy pathology labeling for α-synuclein and neuroaxonal spheroids labeling for β- and γ- (Galvin et al., 2000; Wakabayashi et al., 2000). Thus, synucleins accumulate in a variety of neurodegenerative processes, suggesting either that they are sensitive reporters for specific cellular defects, or that they participate in the response to injury.
Expression and Clearance
In addition to point mutations, duplication and triplication of the chromosomal region surrounding the α-synuclein gene have been found to produce dominantly inherited PD (Ahn et al., 2008; Singleton et al., 2003). The affected chromosomal region contains several other genes as well, but the neuropathology reveals deposition of synuclein (Seidel et al., 2010; Yamaguchi et al., 2005), and the phenotype most likely reflects multiplication of the α-synuclein gene. In this case, the sequence of synuclein is wild type, making the important prediction that a simple increase in the protein rather than a change in its properties suffices to produce PD. The duplication produces a form of PD similar in onset and symptoms to the sporadic disorder, but the triplication causes an exceptionally severe phenotype, with much earlier onset and prominent cognitive as well as motor impairment (Ahn et al., 2008; Ibanez et al., 2004; Ross et al., 2008).
The more global neurologic and behavioral deficits observed with gene multiplication and point mutation presumably reflect a generalized increase in synuclein by all of the neurons that normally express the gene, and α-synuclein is very widely expressed under normal conditions (Iwai et al., 1995). In contrast, the preferential involvement in sporadic PD of particular systems such as the nigrostriatal projection presumably reflects the up-regulation of synuclein within specific cells. Indeed, genome-wide association studies of risk in idiopathic PD reveal the largest contributions from the synuclein gene itself (as well as the microtubule-associated protein tau) (Simon-Sanchez et al., 2009).
Consistent with a role for the amount of α-synuclein expressed in the degenerative process, the polymorphisms implicated in PD lie outside the protein-coding region of the gene, and thus presumably affect mRNA expression. Indeed, one polymorphism has been shown to influence expression of a reporter gene in vitro (Chiba-Falek and Nussbaum, 2001). In addition, the most common inherited form of PD, due to mutations in leucine-rich repeat kinase 2 (LRRK2), generally involves Lewy pathology that may also reflect up-regulation of α-synuclein gene expression (Carballo-Carbajal et al., 2010). Further, α-synuclein has been repeatedly identified as a gene responsive to toxic insult and growth factors. Injection of the toxin quinolinic acid directly into the striatum up-regulates α-synuclein in the substantia nigra (Kholodilov et al., 1999), and oxidative stress due to insecticide or the loss of oxidant defenses also increases α-synuclein (Gillette and Bloomquist, 2003; Gohil et al., 2003). MPTP, rotenone and paraquat produce or exacerbate synuclein deposition, and synuclein can protect against some agents (paraquat) but not others (MPTP) (Fornai et al., 2005; Manning-Bog et al., 2002; Manning-Bog et al., 2003; Przedborski et al., 2001). Synuclein may thus up-regulate in response to many forms of injury, but help to alleviate only some, and exacerbate others. Perhaps consistent with a protective role, nerve growth factor induces α-synuclein expression in PC12 cells and basic fibroblast growth factor in midbrain dopamine neurons (Rideout et al., 2003; Stefanis et al., 2001). Despite these in vitro observations, however, the mechanisms that regulate synuclein expression in vivo remain poorly understood, particularly under physiological circumstances. Interestingly, microRNA-7, which down-regulates α-synuclein expression, itself decreases during MPTP toxicity, providing a mechanism for the up-regulation of synuclein in response to injury (Junn et al., 2009).
In addition to production, clearance can regulate the levels of α-synuclein. Like other natively unfolded proteins, synuclein was originally thought to be degraded by the proteasome without a requirement for ubiquitination (Bennett et al., 1999; Rideout and Stefanis, 2002; Tofaris et al., 2001). However, it was subsequently found that mono-ubiquitination apparently promotes the degradation of synuclein by the proteasome, and this modification can be bidirectionally controlled by a specific ubiquitin ligase (SIAH-2) and deubiquitinase (USP9X) (Liani et al., 2004; Rott et al., 2011). In addition, considerable evidence has also accumulated to suggest the clearance of synuclein at the lysosome. Initially thought to promote the clearance of synuclein aggregates by macroautophagy, degradation in the lysosome also contributes to the turnover of soluble oligomers and even apparently monomeric synuclein under physiological conditions (Lee et al., 2004; Mak et al., 2010; Rideout et al., 2004). Indeed, chaperone-mediated autophagy (CMA), a process that targets individual, soluble proteins to the lysosome for proteolysis, contributes to the clearance of synuclein, and synuclein can in turn disrupt CMA, altering the turnover of other CMA-dependent proteins (Cuervo et al., 2004; Vogiatzi et al., 2008). Interestingly, ubiquitination by the ligase Nedd4 has also been shown to target synuclein for degradation in the lysosome, rather than by the proteasome (Tofaris et al., 2011).
Although we do not know how changes in the expression of synuclein may actually influence the development of human PD, recent work has suggested that changes in clearance may promote degeneration. In particular, idiopathic PD has been found to associate with mutations in the glucocerebrosidase (GBA1) gene. Mutations in GBA1 are responsible for Gaucher’s disease, a recessive lysosomal storage disorder. However, the spectrum of phenotypes in Gaucher’s disease is very broad, with type 2 dying within the first two years of life, and type 1 surviving longer. Indeed, some of the so-called non-neuropathic type 1 patients eventually develop parkinsonism among other neurological problems (Alonso-Canovas et al., 2010; Neudorfer et al., 1996). In addition, it has now become clear that heterozygotes with no overt symptoms of Gaucher’s disease develop PD at higher rates than controls. GBA1 mutations have been found in ~7% patients with idiopathic PD, and up to ~30% of Ashkenazi jewish patients, with only 1.3% in the general population (Sidransky and Lopez, 2012; Sidransky et al., 2009). Mutations in GBA1 have also been reported in DLB but not MSA (Farrer et al., 2009; Segarane et al., 2009), supporting the difference in mechanism between MSA and Lewy related pathology.
GBA1 mutations presumably increase susceptibility to PD by blocking the lysosomal degradation of α-synuclein (Manning-Bog et al., 2009), but it has been difficult to understand how a modest reduction in enzyme activity could impair lysosomal function enough to produce a degenerative disorder. Recent work has indeed suggested that α-synuclein accumulates in both a mouse model of Gaucher’s disease and induced pluripotent (iPS) cells from patients with Gaucher’s disease, but attributed the increase to aggregation in the presence of increased membrane glucocerebroside (Mazzulli et al., 2011). Consistent with the localization of synuclein to lipid rafts (which are enriched in sphingolipids such as glucocerebroside) (Fortin et al., 2004), and its preference for particular lipid acyl chains as well as head groups (Davidson et al., 1998; Kubo et al., 2005), the mechanism by which GBA1 mutations confer susceptibility to PD may involve specific effects on cell membranes rather than a more general disturbance in lysosomal function that simply up-regulates the normal protein. Indeed, much of the work on synuclein has focused on its misfolding and aggregation. Since many publications have addressed the pathways to misfolding of α-synuclein, including several recent reviews (Breydo et al., 2012; Goedert et al., 2013; Lashuel et al., 2013), we will now summarize the most salient observations.
Misfolding
In contrast to its helical conformation on membranes, synuclein adopts a β-sheet structure in aggregates. Indeed, Lewy bodies and neurites contain 5–10 nm filaments that appear to be composed primarily if not exclusively of α-synuclein (Spillantini et al., 1998b). In brainstem-type Lewy bodies, the pale-staining halo, which contains filaments by electron microscopy, labels more strongly for α-synuclein than the acidophilic core (Goedert et al., 2013). Dystrophic neurites and the less discrete cortical-type Lewy bodies contain similar filaments (Marui et al., 2002). Although Lewy bodies were originally considered by some an artifact of the degenerative process, the identification of α-synuclein mutations in familial PD demonstrated a causative role for the major component of Lewy related pathology. However, it is important to remember that this is not the same as establishing a causative role for Lewy pathology in the degenerative process.
Recombinant synuclein also forms filaments after incubation in vitro for a protracted period (Conway et al., 1998). By X-ray diffraction, these filaments adopt a cross-beta structure characteristic of amyloid (Sawaya et al., 2007; Serpell et al., 2000). Recent solid-state NMR has also begun to analyze fibrils at high resolution, identifying the repeated units that underlie this structure (Comellas et al., 2011). Since aggregation has been considered the critical event in the pathogenesis of PD, the in vitro assay has received considerable attention. The point mutations originally identified in familial PD (A53T, A30P and E46K) were originally proposed to accelerate aggregation, but the A30P mutant appears to form fibrils more slowly than the wild type although oligomerization may be enhanced (Conway et al., 2000; Giasson et al., 1999; Li et al., 2001; Narhi et al., 1999). β-synuclein does not fibrillize and both β- and γ- can inhibit the aggregation of α-synuclein in vitro and in vivo (Hashimoto et al., 2001; Uversky et al., 2002), but as noted above, β- and γ- can still contribute to disease (see Synucleinopathies above), suggesting that tendency to aggregate may not correlate closely with potential to cause degeneration.
Many other putative pathogenic factors have also been tested for their ability to influence the aggregation of synuclein, either through direct modification of the protein, or indirectly, through effects on its environment. α-Synuclein does not contain any cysteines, but can undergo nitration and methionine oxidation in response to oxidative stress (Breydo et al., 2012; Giasson et al., 2000a). However, these modifications do not appear to promote aggregation. Similarly, the α-synuclein that deposits in Lewy bodies appears more heavily phosphorylated at Ser-129 than the soluble protein (Fujiwara et al., 2002; Nishie et al., 2004a). Phosphorylation indeed appears to promote synuclein aggregation (Smith et al., 2005b), but it remains unclear whether phosphorylation at this site actually promotes degeneration, with divergent effects in different models (Azeredo da Silveira et al., 2009; Chen and Feany, 2005). Deletion of the C-terminus promotes both aggregation of synuclein in vitro and pathological changes in vivo, suggesting an important role for proteolysis in cells (Li et al., 2005; Murray et al., 2003; Periquet et al., 2007; Tofaris et al., 2006; Ulusoy et al., 2010). Environmental factors may also predispose to synuclein aggregation, and heavy metals appear to promote deposition of the protein in cells as well as in vitro (Breydo et al., 2012; Paik et al., 1999).
It also remains unclear whether synuclein fibrils promote toxicity. First, as noted above, the A30P mutation causes familial PD but does not promote fibrillization (Conway et al., 2000). Second, protein aggregation is not always accompanied by cell loss in a viral model of PD (Lo Bianco et al., 2002). In a Drosophila model, aggregates can occur in the absence of toxicity—the chaperone hsp70 can ameliorate the toxicity of α-synuclein without affecting inclusions (Auluck et al., 2002). The recently identified PD-associated α-synuclein mutant G51D also oligomerizes more slowly than wild type α-synuclein but produces a severe form of degeneration, with early onset and pyramidal as well as extrapyramidal deficits (Lesage et al., 2013). In addition, dopamine has been suggested to promote the aggregation of α-synuclein, but not the formation of amyloid (Bisaglia et al., 2010; Herrera et al., 2008; Rekas et al., 2010). Indeed, dopamine appears to stabilize synuclein aggregation at the stage of protofibrils, and oligomers of synuclein appear more toxic than fibrils (Norris et al., 2005; Rochet et al., 2004). There are multiple cellular mechanisms that regulate the cytosolic concentration of monoamines, from vesicular monoamine transport to feedback inhibition of tyrosine hydroxylase (Fon et al., 1997; Mosharov et al., 2003; Mosharov et al., 2009), and a change in any of these may thus increase the interaction with synuclein to produce degeneration.
Taken together, these results suggest that soluble, oligomeric forms of α-synuclein rather than fibrils may be responsible for toxicity. However, it is even possible that the monomeric form contributes. Indeed, gene multiplication causes a substantially more severe form of PD than the point mutations, and the amount of synuclein rather than its altered properties may be the principal factor that increases susceptibility to degeneration. It is also important to note that although many publications report the formation of aggregates in transfected cells, often in response to toxic insult, α-synuclein in fact rarely forms aggregates detectable by immunofluorescence in transfected cells (R.H.E., unpublished observations). The principal form of synuclein in cells thus appears to be monomer or soluble oligomer.
Although the normal function of synuclein has often been considered irrelevant for its role in degeneration, the interaction of synuclein with membranes, which presumably involves a helical conformation and reflects its normal function, has been reported to influence misfolding and aggregation. As anticipated from the structural studies, one report has shown that membrane association inhibits synuclein oligomerization (Zhu and Fink, 2003), but others have suggested that oligomerization occurs on membranes (Jo et al., 2000; Lee et al., 2002a) and can be promoted by polyunsaturated fatty acids (Perrin et al., 2001). It is important to recognize that the oligomers formed on membranes may be helical, as suggested by the recent work using nanoparticles (Varkey et al., 2013); however, recent NMR and EM have shown directly that anionic phospholipid membranes can convert helical α-synuclein into fibrils (Comellas et al., 2012). It will now be important to determine how membranes influence the conformation and oligomerization of synuclein in cells.
Propagation and Prions
Careful neuropathologic examination of synuclein deposition in brains with Lewy pathology (from incidental Lewy body disease to end-stage PD) has suggested that the degenerative process advances through the nervous system along specific anatomic pathways (Braak et al., 2003). The first synuclein deposits arise in either the dorsal motor nucleus of the glossopharyngeal and vagal nerves or the olfactory bulb (stage 1). In stage 2, the medulla and pontine tegmentum develop Lewy pathology and only in stage 3 does synuclein deposition occur in the midbrain as well as amygdala. At this point, the typical motor manifestations of PD generally appear. In stage 4, α-synuclein deposits in temporal cortex, and in stages 5 and 6 in neocortex, presumably contributing to the cognitive deficits observed in LBD and advanced PD. A minority of cases do not fit this pattern, and isolated Lewy pathology can arise in the amygdala of patients with AD, but the progression otherwise appears quite stereotyped (Dickson et al., 2010). The Braak staging of Lewy pathology presumably accounts for the development of symptoms such as hyposmia and REM behavior disorder up to decades before the onset of typical parkinsonism. It has also suggested a portal of entry for the disease in either the olfactory mucosa or the gastrointestinal tract. Indeed, the retrospective analysis of routine colon biopsies has recently shown synuclein deposits in the enteric nervous system years before the clinical onset of PD, suggesting a useful and accessible biomarker (Shannon et al., 2012). However, it remains unclear whether the process originates in the gut, spreading to the central nervous system through the vagal nerves rather than the spinal cord, or arises independently at multiple sites in sympathetic as well as parasympathetic nerves (Bloch et al., 2006; Iwanaga et al., 1999; Orimo et al., 2008). How then does misfolded synuclein spread between neurons—directly, or through effects on the cell that indirectly promote misfolding?
Several observations have suggested that misfolded synuclein may propagate through a prion-like mechanism (Lee et al., 2010b; Olanow and Prusiner, 2009). First, fetal dopamine cells transplanted into the striatum of patients with PD were found to develop Lewy pathology when examined neuropathologically 1–2 decades later (Kordower et al., 2008; Li et al., 2008). The clear implication is that the normal synuclein expressed by these cells begins to misfold and aggregate after exposure to the abundant misfolded α-synuclein of the host. This has indicated limits to the therapeutic potential of grafts, but also suggested a key feature of prions, the ability of misfolded protein to template conversion of the normal species to an abnormal conformation. Like the form of α-synuclein associated with membranes, the normal cellular form of the prion protein PrP(c) indeed appears predominantly helical whereas the pathogenic form Prp(Sc), like the α-synuclein in Lewy pathology, is mostly β-sheet (Colby and Prusiner, 2011). In the absence of spread between organisms, PD clearly differs from typical prion disorders such as Jakob-Creutzfeldt disease, scrapie and bovine spongiform encephalopathy, but may use a similar mechanism to amplify the pathogenic species at the level of the protein, without a need for nucleic acid (Prusiner, 2001). Second, the apparent inability of oligodendrocytes to make α-synuclein under either normal or pathologic circumstances (Miller et al., 2005; Spillantini et al., 1998a; Tu et al., 1998) requires a mechanism for transfer from the site of production, presumably in neurons, to the GCIs of MSA.
It was not initially clear how a cytosolic protein like synuclein might spread between cells—PrP is a lipid-anchored protein facing the cell exterior. However, it was recognized even before recent interest in the prion hypothesis for PD that small amounts of α-synuclein can undergo secretion through a vesicular mechanism (Lee et al., 2005). More recently, it has become apparent that synuclein release can involve exosomes, the luminal membranes of multivesicular bodies (mvbs) normally targeted for degradation by the lysosome (Emmanouilidou et al., 2010). This is particularly plausible because mvbs form through the invagination of endosomal membranes, and would thus be expected to trap cytosolic proteins such as synuclein. Of course, this would also imply the regulated release of other cytosolic proteins, and the full extent of this mechanism for release remains unclear. It is also possible that oligomeric forms of synuclein, perhaps enriched on the pathway to degradation by the lysosome, become particularly susceptible to release. In addition, this release appears capable of calcium-dependent regulation (Lee et al., 2005; Paillusson et al., 2013), providing an activity-dependent mechanism for propagation that may be relevant for spread along synaptically connected pathways. However, it remains unclear how much of the synuclein released occurs through exosomes as opposed to other, less well-characterized pathways.
Several reports have also documented the uptake of fibrillar synuclein by cells and its ability to produce aggregates composed primarily of the endogenous, host cell protein. Initially, propagation involved either cell extracts including proteins other than α-synuclein, or required transduction of preformed recombinant fibrils into cells over-expressing synuclein (Desplats et al., 2009; Luk et al., 2009). It was shown subsequently, however, that fibrils of recombinant synuclein can enter neurons directly by endocytosis and seed the formation of aggregates resembling Lewy pathology in cells that express only endogenous levels of synuclein (Volpicelli-Daley et al., 2011). The mechanism of uptake remains poorly understood, but glia can also take synuclein derived from neurons, suggesting a mechanism for the formation of GCIs in MSA (Lee et al., 2010a), although it remains unclear how the process could propagate in the absence of any endogenous glial α-synuclein.
Synuclein also appears capable of spread between cells in vivo. Similar to the human transplants described above, cells transplanted into a transgenic animal model can acquire misfolded synuclein from the adjacent tissue and form aggregates (Desplats et al., 2009). Direct injection of fibrillar recombinant synuclein into transgenic mice over-expressing the PD-associated A53T mutant also promotes aggregate formation and disease, with knockouts protected against any pathologic changes (Luk et al., 2012b). However, these transgenic animals would develop degeneration even without injection. More recently, it has been possible to inject fibrils of recombinant mouse α-synuclein into the striatum, resulting in synuclein aggregates in the substantia nigra, dopamine cell loss and parkinsonian deficits (Luk et al., 2012a), and this model of propagation has come the closest yet to demonstrating propagation of the misfolded protein under relatively normal circumstances in vivo. Nonetheless, it still involves injection of extremely large amounts of fibrillar synuclein, and the involvement of dopamine neurons requires only uptake of the fibrils in the striatum, not actually propagation between neurons. Deposits were described in other brain regions such as the cortex and thalamus (Luk et al., 2012a), but at least some of these also project directly to the dorsal striatum, and do not require spread between neurons. Regardless, a prion-like mechanism of transmission suggests that improved clearance of synuclein with circulating antibodies has considerable therapeutic potential (Bae et al., 2012).
Although the data are thus far consistent with a prion-like mechanism for the transmission of misfolded synuclein between cells, there are several important differences between PD and known prion disorders. First, although the incubation time can be long, most prion disorders result in a subacute clinical decline over weeks to months, and PD progresses over decades, particularly since certain features can long precede the onset of typical parkinsonian symptoms. Second, it is important to note that although Lewy pathology was recognized in a few cells of some human transplants, many of the grafts and indeed most of the transplanted cells even in affected grafts appeared entirely normal (Mendez et al., 2008). The process thus does not seem very efficient. Third, the misfolded state in typical prion disorders is quite stable, and indeed, heritable—different strains of the same misfolded protein reproducibly produce distinct forms of degeneration. However, very recent work has suggested that the conformation of misfolded synuclein can change over time, and indeed promote the aggregation of an entirely distinct protein (tau) (Fig. 3) (Guo et al., 2013). Considering the importance of tau for neurodegenerative disease as a whole and PD in particular (Simon-Sanchez et al., 2009), this work expands the relevance of synuclein aggregation but suggests important differences from typical prion disorders.
Figure 3.

Synuclein exhibits prion strain-like properties. Fibrils of recombinant synuclein were taken up by primary neurons and the derived fibrils used for repetitive seeding of additional primary cultures. Early passages yield fibrils capable of forming only inclusions of synuclein (strain A). Fibrils derived from subsequent passages produced robust tau pathology with less deposition of synuclein (strain B). Both strains may derive from the same initial fibrillization reaction (gray arrows), but it seems more likely that strain A converts into strain B. (Adapted from Guo et al., 2013)
Fourth and perhaps most important, sporadic prion disorders presumably involve a very rare misfolding event which then propagates through the prion mechanism. Consistent with this, over-expression of wild type PrP does not by itself usually suffice to produce prion disease. In the case of human PD, however, over-expression of wild type α-synuclein due to gene triplication produces more severe disease than the point mutations, even though several of these apparently increase the propensity to aggregate. For PD, the amount of protein expressed thus appears particularly important, suggesting differences from the prion disorders. PD may simply reflect an increase in monomeric, rather than misfolded or oligomeric, synuclein. In addition, the particular sensitivity to expression may reflect the enhancement of a less rare misfolding event by increased protein. Alternatively, wild type synuclein may misfold at such a high rate that its concentration is more important than any small difference in aggregation tendency. Interestingly, the recent over-expression of wild type bank vole PrP in mice has been found to produce degeneration and prions, but only one variant does this and bank vole PrP appears unusually susceptible to prion formation (Watts et al., 2012). Rather than a rare misfolding event that requires propagation to cause disease, the misfolding of α-synuclein (and possibly bank vole PrP) might therefore originate at multiple sites, with less requirement for transmission between cells.
Toxicity: Membranes
How does synuclein cause toxicity? The analysis of synuclein in multiple systems has suggested a role for its interaction with membranes. As noted above, synuclein oligomers can permeabilize membranes in vitro (Rochet et al., 2004; Tsigelny et al., 2007; Volles et al., 2001), but the relevance of this observation for cells has remained unclear. To study its toxicity in vivo, over-expression of human α-synuclein in yeast has provided an experimentally tractable system (Outeiro and Lindquist, 2003). Although yeast do not contain any sequences resembling synuclein, over-expression of the human protein appears to interfere with transport through the early secretory pathway, and genes that modify the toxicity of synuclein in yeast also tend to involve lipid metabolism and membrane trafficking (Willingham et al., 2003). The small GTPase rab1 that operates early in the secretory pathway rescues synuclein toxicity, both in yeast and in mammalian cells over-expressing a PD-associated mutant (Cooper et al., 2006; Gitler et al., 2008), This might be considered a nonspecific effect, but additional work has suggested an interaction of synuclein with rabs (Chen et al., 2013; Dalfo et al., 2004; Lee et al., 2011; Rendon et al., 2013). In the absence of a clear rab-related defect in synuclein knockout mice, the physiological significance remains unclear, but it may have a role in degeneration.
In yeast, over-expressed α-synuclein localizes to punctate structures. EM has shown that these accumulations are in fact clusters of vesicles rather than proteinaceous deposits, and synuclein appears to act by inhibiting membrane fusion (Gitler et al., 2008; Soper et al., 2008), similar to what has been reported in chromaffin cells (Larsen et al., 2006) (see Role in Neurotransmitter Release above). Human synuclein can also produce lipid droplets in yeast (Outeiro and Lindquist, 2003). Regardless of mechanism, a mutational analysis of synuclein has also shown that toxicity in yeast correlates with the strength of membrane interactions rather than the tendency to aggregate (Volles and Lansbury, 2007). However, the behavior of synuclein in mammalian cells differs in many respects from that observed in yeast, with less obvious membrane association and toxicity, particularly with the wild type protein. In addition, human synuclein cannot form lipid droplets in mammalian cells, but does coat and stabilize the fat droplets formed by feeding with oleic acid (Cole et al., 2002). Perhaps most dramatically, the γ-synuclein knockout shows resistance to obesity and increased lipolysis in white adipose tissue, apparently through increased access of lipolytic enzymes to fat droplets (Millership et al., 2012). The effect of this knockout on brain phospholipids is modest (Guschina et al., 2011), but the effect on adipose tissue strongly supports a role for the other isoforms as well in membrane access and modification.
In recent years, structural studies in vitro have suggested that when synuclein binds to membranes, it can remodel them (Bodner et al., 2009; Diao et al., 2013). The analysis of mitochondrial morphology has now corroborated this possibility in cells. Implicated in the pathogenesis of Parkinson’s disease by the MPTP model and the role in mitochondrial autophagy of recessive PD genes parkin and PINK1 (Narendra et al., 2012), mitochondria have been found to interact with synuclein in several ways. In addition to protection against MPTP toxicity by inactivation of the α-synuclein gene (Dauer et al., 2002; Fornai et al., 2005; Fountaine et al., 2008; Klivenyi et al., 2006), both over-expression and loss of synuclein have been reported to impair mitochondrial function (Ellis et al., 2005; Martin et al., 2006; Smith et al., 2005a; Stichel et al., 2007). Synuclein also binds to mitochondria (Cole et al., 2008; Devi et al., 2008; Li et al., 2007; Liu et al., 2009; Nakamura et al., 2008). More recently, however, over-expression of α-synuclein in a variety of cell types including neurons has been found to fragment mitochondria (Kamp et al.; Nakamura et al., 2011). The change in mitochondrial morphology appears specific, with minimal morphological change in other organelles (such as the Golgi complex), and precedes any deterioration in mitochondrial function (Nakamura et al., 2011).
How does synuclein over-expression fragment mitochondria? Using a direct assay, synuclein does not prevent mitochondrial fusion, but rather promotes fission, and acts independently of the major fission protein Drp1 (Nakamura et al., 2011). The fragmentation also requires access of synuclein to mitochondria, suggesting a direct effect of synuclein on mitochondrial membranes. Indeed, recombinant synuclein can fragment artificial membranes containing the mitochondrial lipid cardiolipin, but not membranes without an acidic phospholipid headgroup (Nakamura et al., 2011). Consistent with sequence conservation of the N-terminal membrane-binding domain, β- and γ-synuclein can also affect mitochondrial morphology (Nakamura et al., 2011).
The Langen group has independently provided compelling evidence that synuclein can remodel membranes (Jao et al., 2008; Varkey et al., 2010). Dramatically, high concentrations of recombinant synuclein can turn an opaque solution of artificial membranes clear (Fig. 4A).
Figure 4.
α-Synuclein tubulates membranes in vitro. A, Recombinant α-synuclein (600 μM) clarifies an opaque solution containing phosphatidylglycerol with palmitoyl and oleoyl side chains (60 μM). (Varkey et al., 2010) (Republished with the permission of JBC). B, 20 μM recombinant α-synuclein was added to nonextruded, ~0.4 μm membranes containing a 1:1 mixture of dioleoyl phosphatidyl choline and dioleoyl phosphatidic acid (400 μM), and examined by negative staining electron microscopy (T.L., R.H.E., unpublished observations). Size bar indicates 100 nm.
At intermediate concentrations, synuclein produces massive tubulation, very similar to Bin-amphysin-Rvs (BAR) domain proteins that bend membranes (McMahon and Boucrot, 2011; Mim and Unger, 2012) (Fig. 4B). However, synuclein seems to act through a distinct mechanism that may involve electrostatic interaction with the phospholipid headgroups to bind membranes but requires insertion into the bilayer for membrane bending (Jensen et al., 2011). In particular, synuclein appears to insert in an extended helical conformation parallel to the long axis of the tubule (Mizuno et al., 2012). The preference of synuclein for highly curved membranes and the ability to tubulate membranes may indeed derive from the same ability to recognize defects in the lipid bilayer (Ouberai et al., 2013; Pranke et al., 2011), similar to the amphipathic lipid packing sensor motifs desribed in ArfGAP1 (Antonny, 2011). Synuclein can also cluster artificial membranes, and consistent with previous work, the A30P mutation disrupts membrane tubulation (Diao et al., 2013; Nakamura et al., 2011; Zigoneanu et al., 2012).
The analysis of knockout mice further supports a role for synuclein in membrane bending. A proteomic analysis of the triple knockouts shows reciprocal changes in BAR domain proteins, in particular endophilin (Westphal and Chandra, 2013). This work also demonstrates the effect of synuclein on membrane curvature in vitro. In contrast to another study suggesting that a multimeric form of synuclein was responsible for tubulation (Nakamura et al., 2011), however, this report indicated a requirement for the monomeric protein (Westphal and Chandra, 2013).
Despite these observations, synuclein normally resides at presynaptic boutons, and most mitochondria localize to the cell body and dendrites. How then can synuclein influence mitochondrial behavior in neurons? We hypothesize that synuclein localizes to mitochondria only when up-regulated. The high presynaptic concentration of synaptic vesicles with high curvature presumably accounts for the normal localization of synuclein to this site. If expression is increased, however, synuclein may then also associate with other membranes such as the mitochondrial inner membrane, which has high curvature at particular sites and is exceptionally rich in the acidic phospholipid cardiolipin. Indeed, the level of expression correlates with mitochondrial fragmentation (Nakamura et al., 2011).
Conclusions
Recent work now indicates the potential for propagation of misfolded synuclein between cells, through a prion-like mechanism. However, the events that trigger misfolding of synuclein in the first place remain poorly understood. A simple increase in the amount of α-synuclein appears sufficient, but its interaction with membranes probably has a crucial role. Lower levels of synuclein contribute to its physiological role at the nerve terminal, influencing the amount of SNARE complex either directly, as a chaperone, or indirectly through other effects on the synaptic vesicle cycle. When up-regulated through physiological or pathological mechanisms, synuclein may target other membranes such as mitochondria, and this presumably accounts for the toxicity observed in human PD. The interaction with membranes thus appears central to both the normal function of synuclein and its role in degeneration. Determining how this interaction influences the conformation of synuclein will help to understand the misfolding that occurs in PD. Similarly, understanding how the interaction affects membrane behavior will illuminate the normal function of the protein, provide a biological context to understand its regulation, and indicate mechanisms responsible for toxicity. However, all of these questions await better methods to understand the behavior and activity of synuclein within the cell.
Acknowledgments
This work was supported by fellowships from NIH (to T.L.), the Giannini Foundation (to J.B.) and a grant from NIH (NS062715) to R.H.E.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Ahlskog JE. Beating a dead horse: dopamine and Parkinson disease. Neurology. 2007;69:1701–1711. doi: 10.1212/01.wnl.0000296942.14309.4a. [DOI] [PubMed] [Google Scholar]
- Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, Kim YK, Kim SE, Kim JM, Kim HJ, et al. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology. 2008;70:43–49. doi: 10.1212/01.wnl.0000271080.53272.c7. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A, Durr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, Morrison KE, Renton A, Sussmuth SD, Landwehrmeyer BG, et al. Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS One. 2009;4:e7114. doi: 10.1371/journal.pone.0007114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, Kaji H, Shinoda T, Hisanaga S, Ueda K. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem. 2002;277:2112–2117. doi: 10.1074/jbc.M102981200. [DOI] [PubMed] [Google Scholar]
- Alonso-Canovas A, Katschnig P, Tucci A, Carecchio M, Wood NW, Edwards M, Martinez Castrillo JC, Burke D, Heales S, Bhatia KP. Atypical parkinsonism with apraxia and supranuclear gaze abnormalities in type 1 Gaucher disease. Expanding the spectrum: case report and literature review. Mov Dis. 2010;25:1506–1509. doi: 10.1002/mds.23109. [DOI] [PubMed] [Google Scholar]
- Annerino DM, Arshad S, Taylor GM, Adler CH, Beach TG, Greene JG. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol. 2012;124:665–680. doi: 10.1007/s00401-012-1040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B. Mechanisms of membrane curvature sensing. Ann Rev Biochem. 2011;80:101–123. doi: 10.1146/annurev-biochem-052809-155121. [DOI] [PubMed] [Google Scholar]
- Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, Threlfell S, Kooner G, Deacon RM, Bannerman DM, et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci. 2011;31:7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, et al. Alpha-synuclein p. H50Q, a novel pathogenic mutation for Parkinson’s disease. Movement Dis. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- Assayag K, Yakunin E, Loeb V, Selkoe DJ, Sharon R. Polyunsaturated fatty acids induce alpha-synuclein-related pathogenic changes in neuronal cells. Am J Path. 2007;171:2000–2011. doi: 10.2353/ajpath.2007.070373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Gen. 2009;18:872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, Fox M, Goldstein JM, Soriano F, Seubert P, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegen Dis. 2008;5:55–59. doi: 10.1159/000112832. [DOI] [PubMed] [Google Scholar]
- Barcelo-Coblijn G, Golovko MY, Weinhofer I, Berger J, Murphy EJ. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J Neurochem. 2007;101:132–141. doi: 10.1111/j.1471-4159.2006.04348.x. [DOI] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Gedalya T, Loeb V, Israeli E, Altschuler Y, Selkoe DJ, Sharon R. Alpha-synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic. 2009;10:218–234. doi: 10.1111/j.1600-0854.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of alpha-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- Bertoncini CW, Fernandez CO, Griesinger C, Jovin TM, Zweckstetter M. Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. J Biol Chem. 2005;280:30649–30652. doi: 10.1074/jbc.C500288200. [DOI] [PubMed] [Google Scholar]
- Bisaglia M, Tosatto L, Munari F, Tessari I, de Laureto PP, Mammi S, Bubacco L. Dopamine quinones interact with alpha-synuclein to form unstructured adducts. Bioc Biop Res Comm. 2010;394:424–428. doi: 10.1016/j.bbrc.2010.03.044. [DOI] [PubMed] [Google Scholar]
- Bloch A, Probst A, Bissig H, Adams H, Tolnay M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol and Appl Neurobiol. 2006;32:284–295. doi: 10.1111/j.1365-2990.2006.00727.x. [DOI] [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, Terada M, Hu J, Bushong EA, Hwang M, Masliah E, George JM, Ellisman MH. Mapping the subcellular distribution of alpha-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson’s disease pathogenesis. J Neurosci. 2013;33:2605–2615. doi: 10.1523/JNEUROSCI.2898-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodner CR, Dobson CM, Bax A. Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J Mol Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher JM, Hartman KL, Ladror DT, Qi Z, Woods WS, George JM, Rienstra CM. Membrane-induced folding of the cAMP-regulated phosphoprotein endosulfine-alpha. Biochemistry. 2008;47:12357–12364. doi: 10.1021/bi801450t. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Brenz Verca MS, Bahi A, Boyer F, Wagner GC, Dreyer JL. Distribution of alpha- and gamma-synucleins in the adult rat brain and their modification by high-dose cocaine treatment. Eur J Neurosci. 2003;18:1923–1938. doi: 10.1046/j.1460-9568.2003.02913.x. [DOI] [PubMed] [Google Scholar]
- Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson’s disease. Bioc Biop Acta. 2012;1822:261–285. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Vivona S, Diao J, Sharma M, Brunger AT, Sudhof TC. Properties of native brain alpha-synuclein. Nature. 2013;498:E4–6. doi: 10.1038/nature12125. discussion E6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DJ, Morgan JR. Synuclein accumulation is associated with cell-specific neuronal death after spinal cord injury. J Comp Neurol. 2012;520:1751–1771. doi: 10.1002/cne.23011. [DOI] [PubMed] [Google Scholar]
- Bussell R, Jr, Ramlall TF, Eliezer D. Helix periodicity, topology, and dynamics of membrane-associated alpha-synuclein. Protein Sci. 2005;14:862–872. doi: 10.1110/ps.041255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ. Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal. 2010;22:821–827. doi: 10.1016/j.cellsig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caumont AS, Galas MC, Vitale N, Aunis D, Bader MF. Regulated exocytosis in chromaffin cells. Translocation of ARF6 stimulates a plasma membrane-associated phospholipase D. J Biol Chem. 1998;273:1373–1379. doi: 10.1074/jbc.273.3.1373. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Chen L, Jin J, Davis J, Zhou Y, Wang Y, Liu J, Lockhart PJ, Zhang J. Oligomeric alpha-synuclein inhibits tubulin polymerization. Bioc Biop Res Comm. 2007;356:548–553. doi: 10.1016/j.bbrc.2007.02.163. [DOI] [PubMed] [Google Scholar]
- Chen RH, Wislet-Gendebien S, Samuel F, Visanji NP, Zhang G, Marsilio D, Langman T, Fraser PE, Tandon A. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J Biol Chem. 2013;288:7438–7449. doi: 10.1074/jbc.M112.439497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet. 2001;10:3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- Cockcroft S, Way G, O’Luanaigh N, Pardo R, Sarri E, Fensome A. Signalling role for ARF and phospholipase D in mast cell exocytosis stimulated by crosslinking of the high affinity FcepsilonR1 receptor. Mol Immunol. 2002;38:1277–1282. doi: 10.1016/s0161-5890(02)00075-5. [DOI] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harbor Persp Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008;314:2076–2089. doi: 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J Biol Chem. 2002;277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- Collaboration MSAR. Mutations in COQ2 in familial and sporadic multiple-system atrophy. New Engl J Med. 2013;369:233–244. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997;7:191–201. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, Rienstra CM. Structured regions of alpha-synuclein fibrils include the early-onset Parkinson’s disease mutation sites. J Mol Biol. 2011;411:881–895. doi: 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM. Structural intermediates during alpha-synuclein fibrillogenesis on phospholipid vesicles. J Am Chem Soc. 2012;134:5090–5099. doi: 10.1021/ja209019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dalfo E, Barrachina M, Rosa JL, Ambrosio S, Ferrer I. Abnormal alpha-synuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol Dis. 2004;16:92–97. doi: 10.1016/j.nbd.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt DC, Rhoades E. alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry. 2013;52:2385–2387. doi: 10.1021/bi4002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Burre J, Vivona S, Cipriano DJ, Sharma M, Kyoung M, Sudhof TC, Brunger AT. Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife. 2013;2:e00592. doi: 10.7554/eLife.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA, Frigerio R, Burnett M, Parisi JE, Klos KJ, Ahlskog JE. Neuropathology of non-motor features of Parkinson disease. Parkinson & Rel Dis. 2009;15(Suppl 3):S1–5. doi: 10.1016/S1353-8020(09)70769-2. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y. Evidence in favor of Braak staging of Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S78–82. doi: 10.1002/mds.22637. [DOI] [PubMed] [Google Scholar]
- Ding ZT, Wang Y, Jiang YP, Hashizume Y, Yoshida M, Mimuro M, Inagaki T, Iwase T. Characteristics of alpha-synucleinopathy in centenarians. Acta Neuropathol. 2006;111:450–458. doi: 10.1007/s00401-005-0015-y. [DOI] [PubMed] [Google Scholar]
- Drolet RE, Behrouz B, Lookingland KJ, Goudreau JL. Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicol. 2004;25:761–769. doi: 10.1016/j.neuro.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, Nussbaum RL. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol. 2005;25:10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- Fan Y, Limprasert P, Murray IV, Smith AC, Lee VM, Trojanowski JQ, Sopher BL, La Spada AR. Beta-synuclein modulates alpha-synuclein neurotoxicity by reducing alpha-synuclein protein expression. Hum Mol Genet. 2006;15:3002–3011. doi: 10.1093/hmg/ddl242. [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Williams LN, Algom AA, Kachergus J, Hulihan MM, Ross OA, Rajput A, Papapetropoulos S, Mash DC, Dickson DW. Glucosidase-beta variations and Lewy body disorders. Parkinsonism Relat Disord. 2009;15:414–416. doi: 10.1016/j.parkreldis.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Wolfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Munoz M, Rosenmund C, Montesinos ML, Sanes JR, Schneggenburger R, et al. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. 2004;42:237–251. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet MF. Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur J Neurosci. 2008;28:247–256. doi: 10.1111/j.1460-9568.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountaine TM, Venda LL, Warrick N, Christian HC, Brundin P, Channon KM, Wade-Martins R. The effect of alpha-synuclein knockdown on MPP+ toxicity in models of human neurons. Eur J Neurosci. 2008;28:2459–2473. doi: 10.1111/j.1460-9568.2008.06527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Sugama S, Sekiyama K, Sekigawa A, Tsukui T, Nakai M, Waragai M, Takenouchi T, Takamatsu Y, Wei J, et al. A beta-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat Comm. 2010;1:110. doi: 10.1038/ncomms1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Giasson B, Hurtig HI, Lee VM, Trojanowski JQ. Neurodegeneration with brain iron accumulation, type 1 is characterized by alpha-, beta-, and gamma-synuclein neuropathology. Am J Pathol. 2000;157:361–368. doi: 10.1016/s0002-9440(10)64548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, Schuck TM, Lee VM, Trojanowski JQ. Differential expression and distribution of alpha-, beta-, and gamma-synuclein in the developing human substantia nigra. Exp Neurol. 2001;168:347–355. doi: 10.1006/exnr.2000.7615. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000a;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VM. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson’s disease. J Neurosci Res. 2000b;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- Gillette JS, Bloomquist JR. Differential up-regulation of striatal dopamine transporter and alpha-synuclein by the pyrethroid insecticide permethrin. Toxicol Appl Pharmacol. 2003;192:287–293. doi: 10.1016/s0041-008x(03)00326-0. [DOI] [PubMed] [Google Scholar]
- Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, et al. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–429. doi: 10.1016/s1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies. Phil Trans Royal Soc Series B, Biol Sci. 1999;354:1101–1118. doi: 10.1098/rstb.1999.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry. 2003;42:8465–8471. doi: 10.1021/bi0341152. [DOI] [PubMed] [Google Scholar]
- Gohil K, Schock BC, Chakraborty AA, Terasawa Y, Raber J, Farese RV, Jr, Packer L, Cross CE, Traber MG. Gene expression profile of oxidant stress and neurodegeneration in transgenic mice deficient in alpha-tocopherol transfer protein. Free Rad Biol Med. 2003;35:1343–1354. doi: 10.1016/s0891-5849(03)00509-4. [DOI] [PubMed] [Google Scholar]
- Golbe LI, Di Iorio G, Sanges G, Lazzarini AM, La Sala S, Bonavita V, Duvoisin RC. Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol. 1996;40:767–775. doi: 10.1002/ana.410400513. [DOI] [PubMed] [Google Scholar]
- Golovko MY, Faergeman NJ, Cole NB, Castagnet PI, Nussbaum RL, Murphy EJ. Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of alpha-synuclein palmitate binding. Biochemistry. 2005;44:8251–8259. doi: 10.1021/bi0502137. [DOI] [PubMed] [Google Scholar]
- Golovko MY, Rosenberger TA, Faergeman NJ, Feddersen S, Cole NB, Pribill I, Berger J, Nussbaum RL, Murphy EJ. Acyl-CoA synthetase activity links wild-type but not mutant alpha-synuclein to brain arachidonate metabolism. Biochemistry. 2006;45:6956–6966. doi: 10.1021/bi0600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovko MY, Rosenberger TA, Feddersen S, Faergeman NJ, Murphy EJ. Alpha-synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J Neurochem. 2007;101:201–211. doi: 10.1111/j.1471-4159.2006.04357.x. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S, Sondel J, Kotilinek L, Day J, Schwarzschild MA, et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24:245–258. doi: 10.1016/s0197-4580(02)00091-x. [DOI] [PubMed] [Google Scholar]
- Goncalves S, Outeiro TF. Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol Neurobiol. 2013;47:1081–1092. doi: 10.1007/s12035-013-8406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, et al. Distinct alpha-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschina I, Millership S, O’Donnell V, Ninkina N, Harwood J, Buchman V. Lipid classes and fatty acid patterns are altered in the brain of gamma-synuclein null mutant mice. Lipids. 2011;46:121–130. doi: 10.1007/s11745-010-3486-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- Herrera FE, Chesi A, Paleologou KE, Schmid A, Munoz A, Vendruscolo M, Gustincich S, Lashuel HA, Carloni P. Inhibition of alpha-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS One. 2008;3:e3394. doi: 10.1371/journal.pone.0003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry. 2004;43:16233–16242. doi: 10.1021/bi048453u. [DOI] [PubMed] [Google Scholar]
- Hua H, Xu L, Wang J, Jing J, Luo T, Jiang Y. Up-regulation of gamma-synuclein contributes to cancer cell survival under endoplasmic reticulum stress. J Pathol. 2009;217:507–515. doi: 10.1002/path.2465. [DOI] [PubMed] [Google Scholar]
- Hughes WE, Elgundi Z, Huang P, Frohman MA, Biden TJ. Phospholipase D1 regulates secretagogue-stimulated insulin release in pancreatic beta-cells. J Biol Chem. 2004;279:27534–27541. doi: 10.1074/jbc.M403012200. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Vitale N, Chasserot-Golaz S, Dupont JL, Du G, Frohman MA, Bader MF, Poulain B. A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci U S A. 2001;98:15300–15305. doi: 10.1073/pnas.261358698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Inaba S, Li C, Shi YE, Song DQ, Jiang JD, Liu J. Synuclein gamma inhibits the mitotic checkpoint function and promotes chromosomal instability of breast cancer cells. Breast Cancer Res Treat. 2005;94:25–35. doi: 10.1007/s10549-005-6938-0. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Iwanaga K, Wakabayashi K, Yoshimoto M, Tomita I, Satoh H, Takashima H, Satoh A, Seto M, Tsujihata M, Takahashi H. Lewy body-type degeneration in cardiac plexus in Parkinson’s and incidental Lewy body diseases. Neurology. 1999;52:1269–1271. doi: 10.1212/wnl.52.6.1269. [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci U S A. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Synuclein deposition and non-motor symptoms in Parkinson disease. J Neurol Sci. 2011;310:107–111. doi: 10.1016/j.jns.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Lantos PL. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update. Acta Neuropathol. 2010;119:657–667. doi: 10.1007/s00401-010-0672-3. [DOI] [PubMed] [Google Scholar]
- Jenco JM, Rawlingson A, Daniels B, Morris AJ. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998;37:4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Bhatia VK, Jao CC, Rasmussen JE, Pedersen SL, Jensen KJ, Langen R, Stamou D. Membrane Curvature Sensing by Amphipathic Helices: A SINGLE LIPOSOME STUDY USING alpha-SYNUCLEIN AND ANNEXIN B12. J Biol Chem. 2011;286:42603–42614. doi: 10.1074/jbc.M111.271130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- Ji H, Liu YE, Jia T, Wang M, Liu J, Xiao G, Joseph BK, Rosen C, Shi YE. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 1997;57:759–764. [PubMed] [Google Scholar]
- Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J Mol Biol. 2002;315:799–807. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. alpha-Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodilov NG, Neystat M, Oo TF, Lo SE, Larsen KE, Sulzer D, Burke RE. Increased expression of rat synuclein in the substantia nigra pars compacta identified by mRNA differential display in a model of developmental target injury. J Neurochem. 1999;73:2586–2599. doi: 10.1046/j.1471-4159.1999.0732586.x. [DOI] [PubMed] [Google Scholar]
- Kim M, Jung W, Lee IH, Bhak G, Paik SR, Hahn JS. Impairment of microtubule system increases alpha-synuclein aggregation and toxicity. Bioc Biop Res Comm. 2008;365:628–635. doi: 10.1016/j.bbrc.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, Ferrante RJ, Kowall NW, Abeliovich A, Beal MF. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis. 2006;21:541–548. doi: 10.1016/j.nbd.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Gen. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Krueger MJ, Singer TP, Casida JE, Ramsay RR. Evidence that the blockade of mitochondrial respiration by the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) involves binding at the same site as the respiratory inhibitor, rotenone. Biochem Biophys Res Comm. 1990;169:123–128. doi: 10.1016/0006-291x(90)91442-u. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Gen. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kubo SI, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y, Edwards RH, Fortin DL. A combinatorial code for the interaction of alpha -synuclein with membranes. J Biol Chem. 2005;280:31164–31672. doi: 10.1074/jbc.M504894200. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM, Bruneau BG, Giasson BI, Smeyne RJ, Gershon MD, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Gen. 2010;19:1633–1650. doi: 10.1093/hmg/ddq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002a;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Kang SJ, Lee K, Im H. Human alpha-synuclein modulates vesicle trafficking through its interaction with prenylated Rab acceptor protein 1. Bioc Biop Res Comm. 2011;412:526–531. doi: 10.1016/j.bbrc.2011.07.028. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Lee S, Lee SJ. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur J Neurosci. 2006;24:3153–3162. doi: 10.1111/j.1460-9568.2006.05210.x. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24:1888–1896. doi: 10.1523/JNEUROSCI.3809-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010a;285:9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002b;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010b;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leftin A, Job C, Beyer K, Brown MF. Solid-State C NMR Reveals Annealing of Raft-Like Membranes Containing Cholesterol by the Intrinsically Disordered Protein alpha-Synuclein. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.04.002. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013 doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB, Kukull WA, Lopez O, Galasko D, Masliah E, et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol. 2008;18:220–224. doi: 10.1111/j.1750-3639.2007.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry. 2001;40:11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Li W, Lesuisse C, Xu Y, Troncoso JC, Price DL, Lee MK. Stabilization of alpha-synuclein protein with aging and familial parkinson’s disease-linked A53T mutation. J Neurosci. 2004;24:7400–7409. doi: 10.1523/JNEUROSCI.1370-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R, et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:5500–5505. doi: 10.1073/pnas.0401081101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, Yang H, Ueda K, Chan P, Yu S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosc Lett. 2009;454:187–192. doi: 10.1016/j.neulet.2009.02.056. [DOI] [PubMed] [Google Scholar]
- Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, Brecha N, Edwards RH. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992a;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- Liu Y, Roghani A, Edwards RH. Gene transfer of a reserpine-sensitive mechanism of resistance to N-methyl-4-phenylpyridinium. Proc Natl Acad Sci U S A. 1992b;89:9074–9078. doi: 10.1073/pnas.89.19.9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012a;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012b;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285:13621–13629. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci. 2003;23:3095–3099. doi: 10.1523/JNEUROSCI.23-08-03095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Bog AB, Schule B, Langston JW. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicol. 2009;30:1127–1132. doi: 10.1016/j.neuro.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. 2009;68:816–822. doi: 10.1097/NEN.0b013e3181ac10a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–343. doi: 10.1016/0169-328x(91)90043-w. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marui W, Iseki E, Nakai T, Miura S, Kato M, Ueda K, Kosaka K. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195:153–159. doi: 10.1016/s0022-510x(02)00006-0. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, et al. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Hyman BT. Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience. 2001;104:901–912. doi: 10.1016/s0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Ribich S, Hyman BT. Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J Neural Trans Supp. 2000:53–63. doi: 10.1007/978-3-7091-6284-2_5. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- McNulty BC, Young GB, Pielak GJ. Macromolecular crowding in the Escherichia coli periplasm maintains alpha-synuclein disorder. J Mol Biol. 2006;355:893–897. doi: 10.1016/j.jmb.2005.11.033. [DOI] [PubMed] [Google Scholar]
- Mendez I, Vinuela A, Astradsson A, Mukhida K, Hallett P, Robertson H, Tierney T, Holness R, Dagher A, Trojanowski JQ, et al. Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat Med. 2008;14:507–509. doi: 10.1038/nm1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton ER, Rhoades E. Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Trans. 2005;112:1613–1624. doi: 10.1007/s00702-005-0378-1. [DOI] [PubMed] [Google Scholar]
- Millership S, Ninkina N, Guschina IA, Norton J, Brambilla R, Oort PJ, Adams SH, Dennis RJ, Voshol PJ, Rochford JJ, et al. Increased lipolysis and altered lipid homeostasis protect gamma-synuclein-null mutant mice from diet-induced obesity. Proc Natl Acad Sci U S A. 2012;109:20943–20948. doi: 10.1073/pnas.1210022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mim C, Unger VM. Membrane curvature and its generation by BAR proteins. Trends Bioc Sci. 2012;37:526–533. doi: 10.1016/j.tibs.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng N, Langen R, Steven AC. Remodeling of lipid vesicles into cylindrical micelles by alpha-synuclein in an extended alpha-helical conformation. J Biol Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K. Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res. 2002;941:118–126. doi: 10.1016/s0006-8993(02)02643-4. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VM. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, et al. Direct membrane association drives mitochondrial fission by the Parkinson Disease-associated protein {alpha}-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J Neurosci. 2008;28:12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harbor Perspect Biol. 2012;4:1–19. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. Q J Med. 1996;89:691–694. doi: 10.1093/qjmed/89.9.691. [DOI] [PubMed] [Google Scholar]
- Ninkina N, Peters O, Millership S, Salem H, van der Putten H, Buchman VL. Gamma-synucleinopathy: neurodegeneration associated with overexpression of the mouse protein. Hum Mol Genet. 2009;18:1779–1794. doi: 10.1093/hmg/ddp090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina N, Peters OM, Connor-Robson N, Lytkina O, Sharfeddin E, Buchman VL. Contrasting effects of alpha-synuclein and gamma-synuclein on the phenotype of cysteine string protein alpha (CSPalpha) null mutant mice suggest distinct function of these proteins in neuronal synapses. J Biol Chem. 2012;287:44471–44477. doi: 10.1074/jbc.M112.422402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishie M, Mori F, Fujiwara H, Hasegawa M, Yoshimoto M, Iwatsubo T, Takahashi H, Wakabayashi K. Accumulation of phosphorylated alpha-synuclein in the brain and peripheral ganglia of patients with multiple system atrophy. Acta Neuropathol. 2004a;107:292–298. doi: 10.1007/s00401-003-0811-1. [DOI] [PubMed] [Google Scholar]
- Nishie M, Mori F, Yoshimoto M, Takahashi H, Wakabayashi K. A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol Appl Neurobiol. 2004b;30:546–554. doi: 10.1111/j.1365-2990.2004.00564.x. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Wider C, Vilarino-Guell C, Soto-Ortolaza AI, Lincoln SJ, Kachergus JM, Jasinska-Myga B, Ross OA, Rajput A, Robinson CA, et al. Association of alpha-, beta-, and gamma-Synuclein with diffuse lewy body disease. Arch Neurol. 2010;67:970–975. doi: 10.1001/archneurol.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorian AR, Rha J, Annerino DM, Bernhard D, Taylor GM, Greene JG. Alpha-synuclein transgenic mice display age-related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol Dis. 2012;48:9–19. doi: 10.1016/j.nbd.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem. 2005;280:21212–21219. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- Ohtake H, Limprasert P, Fan Y, Onodera O, Kakita A, Takahashi H, Bonner LT, Tsuang DW, Murray IV, Lee VM, et al. Beta-synuclein gene alterations in dementia with Lewy bodies. Neurology. 2004;63:805–811. doi: 10.1212/01.wnl.0000139870.14385.3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Prusiner SB. Is Parkinson’s disease a prion disorder? Proc Natl Acad Sci USA. 2009;106:12571–12572. doi: 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain. 2008;131:642–650. doi: 10.1093/brain/awm302. [DOI] [PubMed] [Google Scholar]
- Ouberai MM, Wang J, Swann MJ, Galvagnion C, Guilliams T, Dobson CM, Welland ME. alpha-Synuclein Senses Lipid Packing Defects and Induces Lateral Expansion of Lipids Leading to Membrane Remodeling. J Biol Chem. 2013;288:20883–20895. doi: 10.1074/jbc.M113.478297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson’s disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast Cells Provide Insight into Alpha-Synuclein Biology and Pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Healy DG, Abou-Sleiman PM, Ahmadi KR, Quinn N, Lees AJ, Shaw K, Wullner U, Berciano J, Moller JC, et al. The alpha-synuclein gene in multiple system atrophy. J Neurol Neurosurg Psych. 2006;77:464–467. doi: 10.1136/jnnp.2005.073528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik SR, Shin HJ, Lee JH, Chang CS, Kim J. Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem J. 1999;340(Pt 3):821–828. [PMC free article] [PubMed] [Google Scholar]
- Paillusson S, Clairembault T, Biraud M, Neunlist M, Derkinderen P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J Neurochem. 2013;125:512–517. doi: 10.1111/jnc.12131. [DOI] [PubMed] [Google Scholar]
- Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. 2010;30:3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Bruening W, Giasson BI, Lee VM, Godwin AK. Gamma-synuclein promotes cancer cell survival and inhibits stress- and chemotherapy drug-induced apoptosis by modulating MAPK pathways. J Biol Chem. 2002;277:35050–35060. doi: 10.1074/jbc.M201650200. [DOI] [PubMed] [Google Scholar]
- Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain. 1994;117(Pt 2):235–243. doi: 10.1093/brain/117.2.235. [DOI] [PubMed] [Google Scholar]
- Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nature Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- Payton JE, Perrin RJ, Woods WS, George JM. Structural determinants of PLD2 inhibition by alpha-synuclein. J Mol Biol. 2004;337:1001–1009. doi: 10.1016/j.jmb.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007;27:3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. 2001;276:41958–41962. doi: 10.1074/jbc.M105022200. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Postuma RB, Aarsland D, Barone P, Burn DJ, Hawkes CH, Oertel W, Ziemssen T. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Movement Dis. 2012;27:617–626. doi: 10.1002/mds.24996. [DOI] [PubMed] [Google Scholar]
- Pranke IM, Morello V, Bigay J, Gibson K, Verbavatz JM, Antonny B, Jackson CL. alpha-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J Cell Biol. 2011;194:89–103. doi: 10.1083/jcb.201011118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Shattuck lecture--neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Chen Q, Vila M, Giasson BI, Djaldatti R, Vukosavic S, Souza JM, Jackson-Lewis V, Lee VM, Ischiropoulos H. Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J Neurochem. 2001;76:637–640. doi: 10.1046/j.1471-4159.2001.00174.x. [DOI] [PubMed] [Google Scholar]
- Quilty MC, Gai WP, Pountney DL, West AK, Vickers JC. Localization of alpha-, beta-, and gamma-synuclein during neuronal development and alterations associated with the neuronal response to axonal trauma. Exp Neurol. 2003;182:195–207. doi: 10.1016/s0014-4886(03)00108-0. [DOI] [PubMed] [Google Scholar]
- Rappley I, Gitler AD, Selvy PE, LaVoie MJ, Levy BD, Brown HA, Lindquist S, Selkoe DJ. Evidence that alpha-synuclein does not inhibit phospholipase D. Biochemistry. 2009a;48:1077–1083. doi: 10.1021/bi801871h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappley I, Myers DS, Milne SB, Ivanova PT, Lavoie MJ, Brown HA, Selkoe DJ. Lipidomic profiling in mouse brain reveals differences between ages and genders, with smaller changes associated with alpha-synuclein genotype. J Neurochem. 2009b;111:15–25. doi: 10.1111/j.1471-4159.2009.06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekas A, Knott RB, Sokolova A, Barnham KJ, Perez KA, Masters CL, Drew SC, Cappai R, Curtain CC, Pham CL. The structure of dopamine induced alpha-synuclein oligomers. Eur Biophys J. 2010;39:1407–1419. doi: 10.1007/s00249-010-0595-x. [DOI] [PubMed] [Google Scholar]
- Rendon WO, Martinez-Alonso E, Tomas M, Martinez-Martinez N, Martinez-Menarguez JA. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem Cell Biol. 2013;139:671–684. doi: 10.1007/s00418-012-1059-4. [DOI] [PubMed] [Google Scholar]
- Ribeiro CS, Carneiro K, Ross CA, Menezes JR, Engelender S. Synphilin-1 is developmentally localized to synaptic terminals, and its association with synaptic vesicles is modulated by alpha-synuclein. J Biol Chem. 2002;277:23927–23933. doi: 10.1074/jbc.M201115200. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Dietrich P, Savalle M, Dauer WT, Stefanis L. Regulation of alpha-synuclein by bFGF in cultured ventral midbrain dopaminergic neurons. J Neurochem. 2003;84:803–813. doi: 10.1046/j.1471-4159.2003.01574.x. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Intl J Biochem Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Stefanis L. Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci. 2002;21:223–238. doi: 10.1006/mcne.2002.1173. [DOI] [PubMed] [Google Scholar]
- Robertson DC, Schmidt O, Ninkina N, Jones PA, Sharkey J, Buchman VL. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J Neurochem. 2004;89:1126–1136. doi: 10.1111/j.1471-4159.2004.02378.x. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Hansen LA, Mallory M, Trojanowski JQ, Galasko D, Masliah E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res. 2001;914:48–56. doi: 10.1016/s0006-8993(01)02772-x. [DOI] [PubMed] [Google Scholar]
- Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, Nishioka K, Fuchs J, Gasser T, Maraganore DM, et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63:743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, Engelender S. alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A. 2011;108:18666–18671. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Kato T, Arawaka S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev Neurosci. 2013:1–9. doi: 10.1515/revneuro-2012-0071. [DOI] [PubMed] [Google Scholar]
- Sato K, Kaji R, Matsumoto S, Nagahiro S, Goto S. Compartmental loss of striatal medium spiny neurons in multiple system atrophy of parkinsonian type. Movement Dis. 2007;22:2365–2370. doi: 10.1002/mds.21732. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Schluter OM, Fornai F, Alessandri MG, Takamori S, Geppert M, Jahn R, Sudhof TC. Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience. 2003;118:985–1002. doi: 10.1016/s0306-4522(03)00036-8. [DOI] [PubMed] [Google Scholar]
- Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segarane B, Li A, Paudel R, Scholz S, Neumann J, Lees A, Revesz T, Hardy J, Mathias CJ, Wood NW, et al. Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology. 2009;72:1185–1186. doi: 10.1212/01.wnl.0000345356.40399.eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Schols L, Nuber S, Petrasch-Parwez E, Gierga K, Wszolek Z, Dickson D, Gai WP, Bornemann A, Riess O, et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol. 2010;67:684–689. doi: 10.1002/ana.21966. [DOI] [PubMed] [Google Scholar]
- Senior SL, Ninkina N, Deacon R, Bannerman D, Buchman VL, Cragg SJ, Wade-Martins R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci. 2008;27:947–957. doi: 10.1111/j.1460-9568.2008.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Movement Dis. 2012;27:716–719. doi: 10.1002/mds.25020. [DOI] [PubMed] [Google Scholar]
- Sharma M, Burre J, Bronk P, Zhang Y, Xu W, Sudhof TC. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012;31:829–841. doi: 10.1038/emboj.2011.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Burre J, Sudhof TC. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron. 2003a;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ. Altered fatty acid composition of dopaminergic neurons expressing alpha -synuclein and human brains with alpha -synucleinopathies. J Biol Chem. 2003b;278:49874–49881. doi: 10.1074/jbc.M309127200. [DOI] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci U S A. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–10699. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvadchak VV, Yushchenko DA, Pievo R, Jovin TM. The mode of alpha-synuclein binding to membranes depends on lipid composition and lipid to protein ratio. FEBS Lett. 2011;585:3513–3519. doi: 10.1016/j.febslet.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. New Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Gen. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005a;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005b;25:5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma H, Yabe I, Takei A, Fujiki N, Yanagihara T, Sasaki H. Heredity in multiple system atrophy. J Neurol Sci. 2006;240:107–110. doi: 10.1016/j.jns.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Soper JH, Roy S, Stieber A, Lee E, Wilson RB, Trojanowski JQ, Burd CG, Lee VM. {alpha}-Synuclein-induced Aggregation of Cytoplasmic Vesicles in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:1093–1103. doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht CG, Tigaret CM, Rast GF, Thalhammer A, Rudhard Y, Schoepfer R. Subcellular localisation of recombinant alpha- and gamma-synuclein. Mol Cell Neurosci. 2005;28:326–334. doi: 10.1016/j.mcn.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998a;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998b;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Kholodilov N, Rideout HJ, Burke RE, Greene LA. Synuclein-1 is selectively up-regulated in response to nerve growth factor treatment in PC12 cells. J Neurochem. 2001;76:1165–1176. doi: 10.1046/j.1471-4159.2001.00114.x. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:3377–3393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- Taschenberger G, Toloe J, Tereshchenko J, Akerboom J, Wales P, Benz R, Becker S, Outeiro T, Looger L, Bahr M, et al. Beta-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann Neurol. 2013 doi: 10.1002/ana.23905. [DOI] [PubMed] [Google Scholar]
- Thomas B, Mandir AS, West N, Liu Y, Andrabi SA, Stirling W, Dawson VL, Dawson TM, Lee MK. Resistance to MPTP-neurotoxicity in alpha-synuclein knockout mice is complemented by human alpha-synuclein and associated with increased beta-synuclein and Akt activation. PLoS One. 2011;6:e16706. doi: 10.1371/journal.pone.0016706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Garcia Reitbock P, Humby T, Lambourne SL, O’Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1–120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A. 2011;108:17004–17009. doi: 10.1073/pnas.1109356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM. Fatal attractions: abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Diff. 1998;5:832–837. doi: 10.1038/sj.cdd.4400432. [DOI] [PubMed] [Google Scholar]
- Tsigelny IF, Bar-On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, Keller SH, Platoshyn O, Yuan JX, Masliah E. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007;274:1862–1877. doi: 10.1111/j.1742-4658.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44:415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–697. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha -synuclein. J Biol Chem. 2004;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- Ulusoy A, Febbraro F, Jensen PH, Kirik D, Romero-Ramos M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur J Neurosci. 2010;32:409–422. doi: 10.1111/j.1460-9568.2010.07284.x. [DOI] [PubMed] [Google Scholar]
- Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT. In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One. 2010;5:e10589. doi: 10.1371/journal.pone.0010589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Gen. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey J, Mizuno N, Hegde BG, Cheng N, Steven AC, Langen R. alpha-synuclein oligomers with broken helical conformation form lipoprotein nanoparticles. J Biol Chem. 2013 doi: 10.1074/jbc.M113.476697. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N, Caumont AS, Chasserot-Golaz S, Du G, Wu S, Sciorra VA, Morris AJ, Frohman MA, Bader MF. Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 2001;20:2424–2434. doi: 10.1093/emboj/20.10.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Relationships between the Sequence of alpha-Synuclein and its Membrane Affinity, Fibrillization Propensity, and Yeast Toxicity. J Mol Biol. 2007;366:1510–1522. doi: 10.1016/j.jmb.2006.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Fukushima T, Koide R, Horikawa Y, Hasegawa M, Watanabe Y, Noda T, Eguchi I, Morita T, Yoshimoto M, et al. Juvenile-onset generalized neuroaxonal dystrophy (Hallervorden-Spatz disease) with diffuse neurofibrillary and lewy body pathology. Acta Neuropathol. 2000;99:331–336. doi: 10.1007/s004010050049. [DOI] [PubMed] [Google Scholar]
- Wakamatsu M, Ishii A, Iwata S, Sakagami J, Ukai Y, Ono M, Kanbe D, Muramatsu S, Kobayashi K, Iwatsubo T, et al. Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiol Aging. 2008;29:574–585. doi: 10.1016/j.neurobiolaging.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Wang L, Fleming SM, Chesselet MF, Tache Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. Neuroreport. 2008;19:873–876. doi: 10.1097/WNR.0b013e3282ffda5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Giles K, Stohr J, Oehler A, Bhardwaj S, Grillo SK, Patel S, DeArmond SJ, Prusiner SB. Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc Natl Acad Sci U S A. 2012;109:3498–3503. doi: 10.1073/pnas.1121556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Westphal CH, Chandra SS. Monomeric synucleins generate membrane curvature. J Biol Chem. 2013;288:1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietek J, Haralampiev I, Amoussouvi A, Herrmann A, Stockl M. Membrane bound alpha-synuclein is fully embedded in the lipid bilayer while segments with higher flexibility remain. FEBS Lett. 2013 doi: 10.1016/j.febslet.2013.06.034. [DOI] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Dev Brain Res. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- Woods WS, Boettcher JM, Zhou DH, Kloepper KD, Hartman KL, Ladror DT, Qi Z, Rienstra CM, George JM. Conformation-specific binding of alpha-synuclein to novel protein partners detected by phage display and NMR spectroscopy. J Biol Chem. 2007;282:34555–34567. doi: 10.1074/jbc.M705283200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Cochran EJ, Murrell JR, Polymeropoulos MH, Shannon KM, Crowther RA, Goedert M, Ghetti B. Abundant neuritic inclusions and microvacuolar changes in a case of diffuse Lewy body disease with the A53T mutation in the alpha-synuclein gene. Acta Neuropathol. 2005;110:298–305. doi: 10.1007/s00401-005-1042-4. [DOI] [PubMed] [Google Scholar]
- Yavich L, Jakala P, Tanila H. Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice. J Neurochem. 2006;99:724–732. doi: 10.1111/j.1471-4159.2006.04098.x. [DOI] [PubMed] [Google Scholar]
- Yavich L, Tanila H, Vepsalainen S, Jakala P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–859. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberle AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, et al. Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem. 2007;282:21746–21757. doi: 10.1074/jbc.M702968200. [DOI] [PubMed] [Google Scholar]
- Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- Zigoneanu IG, Yang YJ, Krois AS, Haque E, Pielak GJ. Interaction of alpha-synuclein with vesicles that mimic mitochondrial membranes. Bioc Biop Acta. 2012;1818:512–519. doi: 10.1016/j.bbamem.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]