
Figure 7.
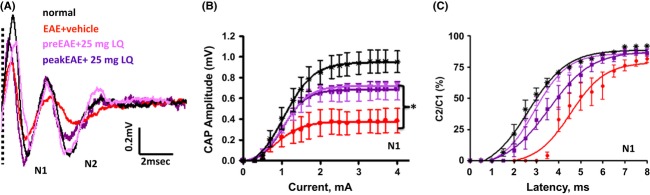
Therapeutic treatment with 25 mg/kg laquinimod (LQ) after onset of clinical EAE attenuates callosal conduction deficits and demyelination. (A) Compound action potential (CAP) responses were recorded from slices with midline-crossing segments of the CC overlying the middorsal hippocampus. Typical CC CAP from normal (black), vehicle-treated EAE (red), pre-EAE+25 mg/kg LQ (magenta), and peak EAE+25 mg/kg LQ (purple) brain slices on day 34 are shown. There is a decrease in N1 and N2 CAP amplitude in the vehicle-treated EAE group, as well as latency shift in the N2 peak. Both pre-EAE and peak EAE LQ treatment induced an increase in N1 and N2 CAP amplitude compared to vehicle alone, and normalization of N2 peak latency. (Dashed vertical line represents CAP beyond the stimulus artifact.) (B) Quantification of CAP amplitudes in the CC of vehicle-treated EAE mice showed a significant reduction in N1 amplitude. LQ pre- and post-treatment showed a significant improvement in CAP response. (C) Average C2/C1 ratio analysis of LQ-treated EAE callosal axons show a significant increase (a leftward shift in the curve) in refractoriness of N1 compared to vehicle treatment. The interpulse interval (IPI) values (mean ± SD) of N1 component are normal = 2.6 ± 0.1 msec, EAE+vehicle = 4.5 ± 0.2 msec, pre-EAE+LQ = 2.7 ± 0.1 msec, and peak EAE+LQ = 3.5 ± 0.1 msec. Similar trends were seen with the N2 IPI (data not shown). Statistically significant compared with normal controls at 2–4 mA stimulus strength (*P < 0.05; ** P < 0.001; ANOVAs; Bonferroni's multiple comparison post-test; n = 6 mice/group).