

The receptor for TSH (TSHR) has captured our interest and imagination since it was first demonstrated to be a major human antigen in autoimmune thyroid disease. The original realization that the immunoglobulin fraction of sera from patients with Graves' disease incorporated unique human autoantibodies to the TSHR led to the unraveling of the great complexity that the TSHR exhibits in comparison with its orthologous partners; the LH and FSH receptors (1, 2). The TSHR is a member of the class A G-protein coupled receptor (GPCR) family with 7 transmembrane regions. The holoreceptor consists of 764 amino acids divided into a large, highly glycosylated, ectodomain of 415 amino acids incorporating 11 leucine rich repeats and which has been crystallized bound to a stimulating TSHR antibody (3). The ectodomain is linked to a distal signal-specific domain, which is a hinge region of 130 amino acids. The hinge region is attached to a transmembrane domain (TMD) of 349 amino acids typical of the GPCR family incorporating 7 transmembrane helices (TMHs) joined by extracellular (ECL) and intracellular (ICL) loops (Figure 1).
Figure 1.

An homology model of the entire TSH holoreceptor This model highlights the tripartite structure of the TSHR. The ectodomain, shown in gray/black, is made up of 10 leucine-rich repeat domains (LRD) characterized as a “scythe-blade”-shaped structure with loops and β-pleated sheets obtained from the published crystal structure (3) (PDB:3G04). The region connecting the LRD and TMD, known as the “hinge” region, has recently been crystallized for the FSH receptor (46) (PDB:4AY9) and is shown as a looped structure (orange) with a helix conformation close to the carboxyl end of the LRD. The hinge in the TSHR has an additional sequence insert and is larger than in the FSH receptor. Therefore, amino acids 305–381 are missing in the illustrated model (47), and this insert is depicted as a closed dotted loop. The TMD (yellow), with its 7 helices, is depicted as cylindrical structures connected to each other by the specific TSHR intra- and ECLs. The TMD is the region that harbors the allosteric binding pockets for the SMLs. ICL, intracellular loops; PDB, Protein Data Base. ECL, extracellular loops.
The TSHR undergoes a complicated posttranslational modification schedule that has taken years to fully define. It turns out that, in addition to the common posttranslational effects such as palmitoylation, sulfation, glycosylation, phosphorylation, and the expected receptor life cycle involving membrane endocytosis (where it continues to signal) (4) it then undergoes further processing that involves intramolecular cleavage of the ectodomain (5, 6), with likely surface membrane shedding (7), and dimerization and multimerization (8–10).
Because of its primary role in thyroid cell growth and function, as well as disease, the TSHR has been the target of a variety of therapeutic approaches. Although the original clinical use of semipurified porcine TSH was possible for short-term thyroid testing of TSHR function, it proved to have too many immune-related side effects in clinical practice, and the use of TSH was not widely adopted until the introduction of recombinant human TSH for detecting thyroglobulin release from metastatic thyroid cancer and for enhancing radioactive iodine uptake into thyroid glands (11–13). In addition to the high cost of recombinant TSH, which is also a large glycosylated complex protein, there has been difficulty in maintaining a steady supply of high-quality material; therefore, a search for cheaper and more reliable TSHR agonists has been ongoing including the long search for more stably glycosylated superagonist TSH forms (14). But the advent of small molecular ligand (SML) pharmacology has now opened a window onto new therapeutic potential at the GPCR level hot on the trail of the now widely available kinase inhibitors (15). The search for such TSHR-active molecules is further advanced with the paper in this issue by Neumann et al (16) describing a potent SML TSHR antagonist characterized by a group that has been successfully pioneering this avenue after a number of false starts from other investigators.
Receptor-active SMLs may act on targets in a variety of ways. Direct blockade of an active site, for example, an enzyme about to phosphorylate a molecule, is the mechanism employed by the common kinase inhibitors. A second mechanism of action is via allosteric modulation. Allosteric means regulation from a distance and, in this capacity, suggests that a small molecule binding to a receptor is capable of regulating its function through changes in receptor conformation. This action may mimic endogenous ligand interaction as seen at the β-adrenergic receptor, which has been shown on crystallization to result in lengthening of TMH5 by 2 helical turns and a 14-Å outward movement of TMH6 (17). Such allosteric activity may result in positive or negative modulation, and these small molecules are, therefore, referred to as positive allosteric regulators or negative allosteric modulators.
The most common site of GPCR-active negative allosteric modulators and positive allosteric regulators has turned out not to be the receptor ectodomain but rather the TMD. This is almost certainly because of the complexity of binding by the large ligands that bind to such receptors. TSH itself, for example, using homology modeling, is considered to have more than 52 binding sites (33 residues from TSHα and 19 residues from TSHβ) each contributing a binding affinity to the hormone receptor-binding interaction (18). Clearly, because stimulation or blockade of all such binding sites could not be achieved with one small molecule, effective small molecules are more likely to activate or derail the signal transduction pathways rather than influencing the hormone-receptor interface. Hence, the advent of data-rich high-throughput functional screening methods has resulted in the identification of a large number of TMD-active compounds capable of influencing receptor function. In the case of the TSHR, the first compounds were found to be nonspecific, influencing the FSH, LH and TSH receptors with both agonist and antagonist activities (19–21). Neumann et al (21, 22) began to obtain specificity at the TSHR for a stimulating SML by chemically modifying an LH receptor-active agonist small molecule and showed its potency both in vitro and in vivo, and we have developed a structurally distinct and highly specific stimulating SML via high-throughput screening (23). However, the clinical need has also been for a more potent TSHR antagonist that could block the TSHR antibodies of hyperthyroid Graves' disease allowing us to dispense with the side effects of the common antithyroid drugs (methimazole and propylthiouracil), which deter many physicians from their long-term use (24). Neuman et al's first antagonist in 2008 was active in micromolar concentrations after which Van Koppen at al (25) reported a TSHR antagonist active in nanomolar concentrations in 2012, but both small molecules had questionable specificity. The new report from Neumann et al (16) in this issue of Endocrinology again used a chemical modification approach to increase the selectivity and potency of their SML and demonstrated its effectiveness in vivo. Their SML ANTAG3 inhibited the action not only of TSH but of a potent stimulating TSHR antibody (M22), suggesting its usefulness in the treatment of Graves' disease. However, the interpretation of the in vivo data is not as easy as it seems. Neumann et al first gave normal mice TRH to increase T4 levels and found this to be approximately 50% inhibited by a large 2-mg dose of ANTAG3 given by ip injection. In a subsequent study they used the M22-stimulating TSHR antibody (Ab) to release T4 in mice with their endogenous TSH suppressed by T3 and again found approximately 50% inhibition. The lack of dose-response studies in the report suggests that a high concentration of antagonist must have been needed to achieve only 50% inhibition, certainly insufficient inhibition for clinical application. No doubt these modest effects will be improved with further increases in SML potency.
So how can a small molecule activate or inhibit TSHR signaling via allosteric changes? The recent advances in our knowledge of GPCR structure subsequent to successful crystallography has allowed molecular modeling approaches to be applied to SML pharmacology. This has provided insight into critical aspects of binding of both endogenous ligands and SMLs in order to understand the receptor activation process (26). The information gained from these models has provided structural explanations for the role of specific sequences and residues within the TMDs of these receptors. From the studies of patients with activating and inactivating TSHR mutations (27, 28) it is now known that the TSHR TMD is rich with such mutations that are peppered within the different helices as well as the intracellular and extracellular loops. Studies (29, 30) using model-driven site-directed mutagenesis have indicated that these signaling-sensitive amino acids often correspond with SML-binding sites and can be referred to as TMD “hot spots.” Using site-directed mutagenesis and functional characterization, these allosteric binding regions for SMLs, both agonists and antagonists, distributed mostly in TMH 1–7 and ECL2 form 2 main structural clusters (29). Cluster I is made up of residues in TMHs 3, 4, 5, and ECL2, and Cluster II consists of residues in TMHs 1, 2, and 7. These allosteric binding sites are independent of TSH binding and, therefore, have evolved much greater diversity than orthosteric sites and thus offer greater specificity. This has been of great advantage in not only redesigning molecules by chemical modifications in order to improve their potency and specificity, but also provided more discriminative pharmacophores as shown by Neumann et al (21), who evolved their first TSHR antagonist from an agonist. Therefore, the “hot spots,” both activating and inactivating mutations, within the TSHR TMD offer the possibility of finding newer more powerful agonists and antagonists using molecular modeling and high-throughput screening.
The data presented by Neumann et al (16) in this issue is strong proof of principle that SMLs directed to the allosteric binding pockets have the ability to tone down the signaling ability of the TSHR. One possible mechanism by which the inhibition of G protein signaling can be carried out by such an antagonist is by stabilization of the “ionic lock,” a polar interaction between an arginine (R) located at the bottom of TMH3, which is part of a conserved, recurrent D/E-R-Y/W motif (aspartic acid/glutamate-arginine-tyrosine/tryptophan) and the partly conserved glutamate (E)/aspartic acid (D) at the bottom of TMH6 (Figure 2). This locking would prevent the transmission of structural change when a stimulating TSHR antibody or TSH ligand itself binds to the receptor ectodomain by locking-in the basal conformation of the TSHR, which is constrained by the polar contacts between TMHs 2, 3, 6, and 7. Thus, the mechanistic ability to inhibit signaling can be considered analogous to a variable steering lock in a car, which will prevent the outward movement of helix 6, as earlier proposed, and thus prevent G protein coupling and activation by binding to the ICLs of the TSHR. On the other hand, molecules that are agonists to the TSHR may activate the receptor by destabilizing the ionic lock (17, 23, 26) and polar interaction between the helices. The next phase must be development of the molecular evidence in support of such models.
Figure 2.
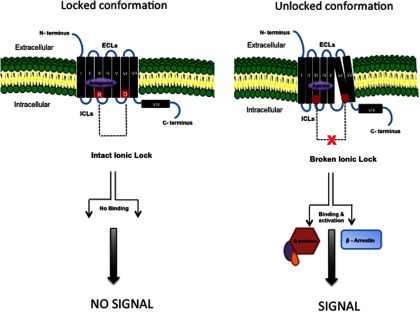
Interaction of SMLs with the TSHR Similar to most GPCRs, the basal state of TSHR activity is hypothesized to be constrained by polar interactions between different TMHs forming an “ionic lock” between an arginine (R) at the base of transmembrane TMH3 and partly conserved residues such as glutamate (E) or aspartic acid (D) at the base of TM6 as shown in red (left panel). When a SML antagonist binds to the allosteric pocket (purple) on the TMD, the ionic lock remains intact even if TSH ligand binds to the ectodomain because there can be no outward movement of TMH6. Hence, the basal conformation of the receptor is locked and stabilized. In contrast, when a SML agonist binds to the allosteric pocket (right panel), there is an outward movement of TMH6 as a result of destabilization of the ionic lock and loss of constrained polar interactions. This movement causes conformational changes in the intracellular loops, which, in turn, allow binding of G proteins and β-arrestin activation leading to signal transduction. ICL, intracellular loops; ECL, extracellular loops.
Lastly, we need to consider that the TSHR is expressed in more places than the thyroid gland and can be expressed even in embryonic stem cells (31). TSHRs are widely expressed in fibroblasts, adipocytes, bone cells, and a variety of additional cell types (32, 33) but have attracted a lot of attention in the retro-orbit (34–36). This is because the Graves' Triad Syndrome consists of thyroid disease with a dermopathy referred to as “pretibial myxedema” and an orbitopathy referred to as “Graves' Ophthalmopathy.” There is considerable evidence that retro-orbital expression of the TSHR in the fibroblasts and adipocytes behind the eye may contribute to this difficult-to-treat orbitopathy, and serum TSHR-Ab levels tend to correlate with eye disease (37–39). It has been hypothesized that blockade of the receptor may be a useful mode of therapy (40, 41). However, it is not clear that merely inhibiting TSHR signaling, rather than removing the TSHR antigen from immune presentation, would help such a disease. One way such blockade could help is by reducing TSHR-Ab-induced cytokine release from retro-orbital fibroblasts. Such cytokines contribute to glycosaminoglycan generation and disrupt the osmotic pressure behind the eyes, causing muscle fiber damage and swelling (41, 42). On the other hand, we also need to consider the potential side effects of widespread TSHR inhibition at the adipocyte (43) and especially on osteoblasts and osteoclasts where TSH action is osteoprotective and where long-term TSHR blockade may induce bone loss (44, 45). Clearly, we have much to learn about the pharmacology and physiology of TSHR-active SMLs, and the journey is only beginning.
Acknowledgments
This work was supported, in part, by National Institutes of Health Grants DK069713 and DK052464 and the Veterans Affairs Merit Award program.
Disclosure Summary: The authors have nothing to disclose.
For article see page 310
- Ab
- antibody
- ECL
- extracellular loop
- GPCR
- G-protein coupled receptor
- SML
- small molecular ligand
- TMD
- transmembrane domain
- TMH
- transmembrane helix
- TSHR
- TSH receptor.
References
- 1. Adams DD, Purves HD. Abnormal responses in the assay of thyrotropin. Proceedings of the University of Otago Medical School. 1956;34:11–12 [Google Scholar]
- 2. Smith BR, Hall R. Thyroid-stimulating immunoglobulins in Graves' disease. Lancet. 1974;2(7878):427–429 [DOI] [PubMed] [Google Scholar]
- 3. Sanders J, Chirgadze DY, Sanders P, et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid. 2007;17(5):395–410 [DOI] [PubMed] [Google Scholar]
- 4. Calebiro D, Nikolaev VO, Persani L, Lohse MJ. Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci. 2010;31(5):221–228 [DOI] [PubMed] [Google Scholar]
- 5. Tanaka K, Chazenbalk GD, McLachlan SM, Rapoport B. Subunit structure of thyrotropin receptors expressed on the cell surface. J Biol Chem. 1999;274(48):33979–33984 [DOI] [PubMed] [Google Scholar]
- 6. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19(6):673–716 [DOI] [PubMed] [Google Scholar]
- 7. Tanaka K, Chazenbalk GD, McLachlan SM, Rapoport B. The shed thyrotropin receptor is primarily a carboxyl terminal truncated form of the A subunit, not the entire A subunit. Mol Cell Endocrinol. 1999;150(1–2):113–119 [DOI] [PubMed] [Google Scholar]
- 8. Latif R, Graves P, Davies TF. Oligomerization of the human thyrotropin receptor: fluorescent protein-tagged hTSHR reveals post-translational complexes. J Biol Chem. 2001;276(48):45217–45224 [DOI] [PubMed] [Google Scholar]
- 9. Latif R, Michalek K, Davies TF. Subunit interactions influence TSHR multimerization. Mol Endocrinol. 2010;24(10):2009–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Urizar E, Montanelli L, Loy T, et al. Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 2005;24(11):1954–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huber GK, Fong P, Concepcion ES, Davies TF. Recombinant human thyroid-stimulating hormone: initial bioactivity assessment using human fetal thyroid cells. J Clin Endocrinol Metab. 1991;72:1328–1331 [DOI] [PubMed] [Google Scholar]
- 12. Ladenson PW, Braverman LE, Mazzaferri EL, et al. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma [see comments]. N Engl J Med. 1997;337(13):888–896 [DOI] [PubMed] [Google Scholar]
- 13. Mazzaferri EL, Kloos RT. Is diagnostic iodine-131 scanning with recombinant human TSH useful in the follow-up of differentiated thyroid cancer after thyroid ablation? J Clin Endocrinol Metab. 2002;87(4):1490–1498 [DOI] [PubMed] [Google Scholar]
- 14. Mueller S, Kleinau G, Szkudlinski MW, Jaeschke H, Krause G, Paschke R. The superagonistic activity of bovine thyroid-stimulating hormone (TSH) and the human TR1401 TSH analog is determined by specific amino acids in the hinge region of the human TSH receptor. J Biol Chem. 2009;284(24):16317–16324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39 [DOI] [PubMed] [Google Scholar]
- 16. Neumann S, Nir EA, Eliseeva E, et al. A selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice. Endocrinology. 2013;155:310–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benovic JL. G-protein-coupled receptors signal victory. Cell. 2012;151(6):1148–1150 [DOI] [PubMed] [Google Scholar]
- 18. Núñez Miguel R, Sanders J, Chirgadze DY, Furmaniak J, Rees Smith B. Thyroid stimulating autoantibody M22 mimics TSH binding to the TSH receptor leucine rich domain: a comparative structural study of protein-protein interactions. J Mol Endocrinol. 2009;42(5):381–395 [DOI] [PubMed] [Google Scholar]
- 19. Jäschke H, Neumann S, Moore S, et al. A low molecular weight agonist signals by binding to the transmembrane domain of thyroid-stimulating hormone receptor (TSHR) and luteinizing hormone/chorionic gonadotropin receptor (LHCGR). J Biol Chem. 2006;281(15):9841–9844 [DOI] [PubMed] [Google Scholar]
- 20. van Koppen CJ, Zaman GJ, Timmers CM, et al. A signaling-selective, nanomolar potent allosteric low molecular weight agonist for the human luteinizing hormone receptor. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(5):503–514 [DOI] [PubMed] [Google Scholar]
- 21. Neumann S, Kleinau G, Costanzi S, et al. A low-molecular-weight antagonist for the human thyrotropin receptor with therapeutic potential for hyperthyroidism. Endocrinology. 2008;149(12):5945–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neumann S, Gershengorn MC. Small molecule TSHR agonists and antagonists. Ann Endocrinol (Paris). 2011;72(2):74–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Latif R, Morshed SA, Xu T, et al. Pathways to thyroid stimulation: identification of new, potent and selective small molecule agonists to the TSH receptor. Paper presented at the Annual Meeting of The Endocrine Society; June 2013; San Francisco, CA Abstract 28–1 [Google Scholar]
- 24. Latif R, Morshed SA, David M, Davies TF. TSH receptor transmembrane “hotspots” for thyroid stimulation revealed by small molecule agonism. Paper presented at the Annual Meeting of the American Thyroid Association, October 2013; San Juan, Puerto Rico Abstract 90 [Google Scholar]
- 25. van Koppen CJ, de Gooyer ME, Karstens WJ, et al. Mechanism of action of a nanomolar potent, allosteric antagonist of the thyroid-stimulating hormone receptor. Br J Pharmacol. 2012;165(7):2314–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Audet M, Bouvier M. Restructuring G-protein-coupled receptor activation. Cell. 2012;151(1):14–23 [DOI] [PubMed] [Google Scholar]
- 27. Hoyer I, Haas AK, Kreuchwig A, Schülein R, Krause G. Molecular sampling of the allosteric binding pocket of the TSH receptor provides discriminative pharmacophores for antagonists and agonists. Biochem Soc Trans. 2013;41(1):213–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodien P, Ho SC, Vlaeminck V, Vassart G, Costagliola S. Activating mutations of TSH receptor. Ann Endocrinol (Paris). 2003;64(1):12–16 [PubMed] [Google Scholar]
- 29. Haas AK, Kleinau G, Hoyer I, et al. Mutations that silence constitutive signaling activity in the allosteric ligand-binding site of the thyrotropin receptor. Cell Mol Life Sci. 2011;68(1):159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kleinau G, Haas AK, Neumann S, et al. Signaling-sensitive amino acids surround the allosteric ligand binding site of the thyrotropin receptor. FASEB J. 2010;24(7):2347–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davies TF, Ando T, Lin RY, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J Clin Invest. 2005;115(8):1972–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davies T, Marians R, Latif R. The TSH receptor reveals itself. J Clin Invest. 2002;110(2):161–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci USA. 2002;99(24):15776–15781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bahn RS. Thyrotropin receptor expression in orbital adipose/connective tissues from patients with thyroid-associated ophthalmopathy. Thyroid. 2002;12(3):193–195 [DOI] [PubMed] [Google Scholar]
- 35. Bahn RS. TSH receptor expression in orbital tissue and its role in the pathogenesis of Graves' ophthalmopathy. J Endocrinol Invest. 2004;27(3):216–220 [DOI] [PubMed] [Google Scholar]
- 36. Gershengorn MC, Neumann S, Pope A, Geras-Raaka E, Raaka BM, Bahn RS. A drug-like antagonist inhibits TSH receptor-mediated stimulation of cAMP production in Graves' orbital fibroblasts. Thyroid. 2012;22(8):839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khoo TK, Bahn RS. Pathogenesis of Graves' ophthalmopathy: the role of autoantibodies. Thyroid. 2007;17(10):1013–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumar S, Nadeem S, Stan MN, Coenen M, Bahn RS. A stimulatory TSH receptor antibody enhances adipogenesis via phosphoinositide 3-kinase activation in orbital preadipocytes from patients with Graves' ophthalmopathy. J Mol Endocrinol. 2011;46(3):155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar S, Schiefer R, Coenen MJ, Bahn RS. A stimulatory thyrotropin receptor antibody (M22) and thyrotropin increase interleukin-6 expression and secretion in Graves' orbital preadipocyte fibroblasts. Thyroid. 2010;20(1):59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith BR, Furmaniak J, Sanders J. TSH receptor blocking antibodies. Thyroid. 2008;18(11):1239. [DOI] [PubMed] [Google Scholar]
- 41. Bahn RS. Graves' ophthalmopathy. N Engl J Med. 2010;362(8):726–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hansen C, Rouhi R, Förster G, Kahaly GJ. Increased sulfatation of orbital glycosaminoglycans in Graves' ophthalmopathy. J Clin Endocrinol Metab. 1999;84(4):1409–1413 [DOI] [PubMed] [Google Scholar]
- 43. Elgadi A, Zemack H, Marcus C, Norgren S. Tissue-specific knockout of TSHr in white adipose tissue increases adipocyte size and decreases TSH-induced lipolysis. Biochem Biophys Res Commun. 2010;393(3):526–530 [DOI] [PubMed] [Google Scholar]
- 44. Abe E, Marians RC, Yu W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115(2):151–162 [DOI] [PubMed] [Google Scholar]
- 45. Baliram R, Sun L, Cao J, et al. Hyperthyroid-associated osteoporosis is exacerbated by the loss of TSH signaling. J Clin Invest. 2012;122(10):3737–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang X, Liu H, Chen X, et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci USA. 2012;109(31):12491–12496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krause G, Kreuchwig A, Kleinau G. Extended and structurally supported insights into extracellular hormone binding, signal transduction and organization of the thyrotropin receptor. PLoS One. 2012;7(12):e52920. [DOI] [PMC free article] [PubMed] [Google Scholar]