

Abstract
The current diversity of life on earth is the product of macroevolutionary processes that have shaped the dynamics of diversification. Although the tempo of diversification has been studied extensively in macroorganisms, much less is known about the rates of diversification in the exceedingly diverse and species-rich microbiota. Decreases in diversification rates over time, a signature of explosive radiations, are commonly observed in plant and animal lineages. However, the few existing analyses of microbial lineages suggest that the tempo of diversification in prokaryotes may be fundamentally different. Here, we use multilocus and genomic sequence data to test hypotheses about the rate of diversification in a well-studied pathogenic bacterial lineage, Borrelia burgdorferi sensu lato (sl). Our analyses support the hypothesis that an explosive radiation of lineages occurred near the origin of the clade, followed by a sharp decay in diversification rates. These results suggest that explosive radiations may be a general feature of evolutionary history across the tree of life.
Keywords: Diversification, microbial macroevolution, phylogeny, radiation
Explosive evolutionary radiations are one of the most conspicuous and intriguing features of evolutionary biology (Simpson 1953; Schluter 2000; Rundell and Price 2009). Explosive radiations have occurred in a wide variety of animal and plant lineages, identified using both fossil and molecular data, and may be a general pattern of the diversity of life (Sanderson and Donoghue 1994; Purvis et al. 1995; Zink and Slowinski 1995; Alroy 1999; Harmon et al. 2003; McPeek 2008; Phillimore and Price 2008; Rabosky and Lovette 2008; Morlon et al. 2010). In comparison with the wealth of evidence supporting rapid radiations of macroorganisms, very little is known about the tempo of microbial diversification (Martin et al. 2004). Given the vast diversity of microbes and their crucial roles in human health and economics, as well as in natural ecosystem processes, it is essential to investigate the pattern of species diversification in the microbial world.
Analyses of molecular phylogenies of extant taxa under birth–death models of cladogenesis are widely used to investigate diversification patterns. In particular, they are used to test the hypothesis that diversification rates have decayed over time, which is commonly interpreted as an ecological niche-filling process associated with radiations (Nee et al. 1994; Pybus and Harvey 2000; Rabosky and Lovette 2008; Glor 2010; Morlon et al. 2010). Time decays in diversification rates are often investigated by measuring the relative position of nodes in a phylogeny using the gamma (γ) statistic (Pybus and Harvey 2000; Harmon et al. 2003; Martin et al. 2004; Phillimore and Price 2008; Brock et al. 2011), or by analyzing the likelihood of the waiting times between branching events in a phylogeny under various birth–death models of cladogenesis (Nee et al. 1992, 1994; Rabosky 2006; Rabosky and Lovette 2008; Morlon et al. 2010; Morlon et al. 2011; Stadler 2011). Such analyses have revealed time decays in diversification rates in many plant and animal clades (Davis et al. 2005; Kozak et al. 2006; McPeek 2008; Phillimore and Price 2008; Schuettpelz and Pryer 2009; Morlon et al. 2010; Valente et al. 2010), and even in the Kingdom Animalia itself (Rokas et al. 2005). By contrast, the sole analysis of the tempo of diversification in prokaryotic phylogenies that we know of did not detect a time decay in diversification rates (Martin et al. 2004), a surprising result given that prokaryote lineages rapidly and adaptively diversify in experimental systems (reviewed in MacLean 2005; Kassen 2009).
There are three main complications associated with phylogeny-based macroevolutionary analyses of microbial lineages. First, the true diversity is rarely well sampled as the majority of bacterial species have yet to be described; second, there is no broadly accepted species concept for microbes; and lastly, the horizontal gene transfer that is common among prokaryotes renders birth–death models of cladogenesis less suited to analyze diversification in microbial clades. The evolutionary clade containing the Lyme disease-causing spirochetes, Borrelia burgdorferi sensu lato (sl), avoids many of these complications and thus provides an excellent opportunity to study rates of diversification in a prokaryotic group.
Borrelia burgdorferi is obligatorily vectored among vertebrate hosts by ticks in the Ixodes ricinus species complex (Barbour and Hayes 1986; Nuttall 1999; van Dam 2002; Piesman and Gern 2004). This host specialization potentially limits species diversity by limiting habitat suitability, and makes scientific sampling easier than for free-living groups. Further, the implications of B. burgdorferi sl for human health has lead to an exhaustive examination of vertebrates and hematophagous arthropods around the world for both human infectious and wildlife-specific Borrelia species through both cultivation-dependent and cultivation-independent methods. These exhaustive field studies to identify B. burgdorferi sl have resulted in a very high coverage of the diversity of this clade, revealing 15 described and two recently proposed phylotypes that are readily distinguishable by DNA sequence divergence and phylogenetic clustering (Gern 2008; Margos et al. 2009; Rudenko et al. 2009, 2011). Genetic diversity within each phylotype is small compared to divergence among phylotypes, suggesting that each lineage is an independently evolving, phylogenetically distinct group comparable to species in eukaryotes (Avise and Wollenberg 1997; Richter et al. 2006; Postic et al. 2007; Margos et al. 2011). Finally, horizontal gene transfer among phylotypes is rare for chromosomal genes (Dykhuizen and Baranton 2001; Qiu et al. 2004) allowing application of birth–death models of cladogenesis to study diversification rates.
The present study provides a comprehensive assessment of the pattern of phylotype diversification in B. burgdorferi sl. We construct phylogenetic hypotheses for B. burgdorferi sl using multiple loci and chromosome sequences. We then analyze variation in the rate of diversification of B. burgdorferi phylotypes over the evolutionary history of the species group using the γ-statistic as well as a recently developed hypothesis-testing framework that accounts for the possibility that some lineages have not been sampled (Morlon et al. 2010). Finally, we discuss our results in light of classical results found for macroorganisms and of specific biological characteristics of B. burgdorferi sl that may influence diversification rates in this group.
Materials and Methods
LOCUS SEQUENCE DATA
The NCBI nucleotide database was searched in November 2010 for B. burgdorferi sl sequences that meet three criteria. First, only DNA sequence data from chromosomal loci were used as plasmid genes are more prone to horizontal gene transfer (Qiu et al. 2004; Margos et al. 2009). Second, we used only locus datasets for which meaningful comparisons could be made across at least 11 B. burgdorferi sl taxa. Lastly, locus datasets with homologs in at least two taxa in the relapsing fever Borrelia group (B. hermsii, B. duttonii, B. recurrentis, or B. turracatae) were desired, to root the trees. Nucleotide sequences from seven loci—fla (flagellin, 122 sequences), hbb (histone-like protein, 77 sequences), rpoB (RNA polymerase, 66 sequences), RRS (16S rDNA, 256 sequences), recA (recombinase A, 108 sequences), 5S–23S internal transcribed rDNA sequence (ITS, 798 sequences), and groEL (chaperone protein, 116 sequences)—fit these criteria. Sequences for each locus in the NCBI nucleotide database were downloaded and aligned using ClustalX (Higgins and Sharp 1988; Larkin et al. 2007) and Muscle (Edgar 2004). This resulted in a total of 1543 sequences. Data across all 15 recognized B. burgdorferi sl phylotypes were available for only three loci (fla, groEL, and RRS). We thus constructed two phylogenies: a phylogeny comprising all phylotypes based on these three loci, and a phylogeny using all seven loci comprising 11 phylotypes (hereafter referred to as “three-locus” and “seven-locus” trees, respectively).
CHROMOSOME SEQUENCE DATA
Primary shotgun sequences from 15 Borrelia genome projects, 11 of which are from the B. burgdorferi sl group, were acquired from NCBI. These 11 genome projects covered six of the recognized phylotypes: three B. burgdorferi sensu stricto (ss) genome projects (B31, CA-11.2, ZS7), three B. garinii (Far04, PBi, PBr), two B. afzelii (ACA-1, PKo), one B. sp SV1 (SV1), one B. valaisiana (VS116), and one B. spielmanii (A14S). The chromosome sequences were aligned to the annotated chromosome of the B. burgdorferi sensu stricto (ss) type-strain (B31) using Blast (Altschul et al. 1990) and Muscle (Edgar 2004). All ambiguous nucleotides were excluded from analyses. The data represent orthologous genes shared across all sampled organisms. The final dataset comprised 551,152 nucleotides (58% of the approimately 0.95 Mb B31 reference chromosome). Of the 551,152 chromosomal nucleotides, 95,018 were polymorphic (14.7%) of which 41,042 (7.4%) were parsimony informative. Sites with evidence of multiple mutations—those with three or more divergent bases—made up only 7021 of the polymorphic sites suggesting that mutation saturation is unlikely. The tree was rooted using 118,709 nucleotides from B. hermsii, B. duttonii, B. reccurentis, and B. turicatae genomic sequences, hereafter referred to as the “chromosome” tree.
DELIMITING PHYLOTYPES
Although there is no universally accepted species definition for prokaryotes, there are well-recognized phylotypes in B. burgdorferi sl (Richter et al. 2006; Margos et al. 2009; Rudenko et al. 2009, 2011). To test that the genetic diversity of each of the seven loci clusters into independently evolving lineages that correspond to the recognized phylotypes, we first built single-locus ultrametric trees using MrBayes and penalized likelihood (PL), as described below. We then used the Generalized Mixed Yule Coalescent model, implemented in the GMYC package in R (splits http://r-forge.rproject.org/projects/splits/), which tests the hypothesis that the data represent several independently evolving populations against the null hypothesis that it represents a single population (in practice, that the tree conforms to a neutral coalescent, Pons et al. 2006; Barraclough et al. 2009). The GMYC model estimates a threshold that defines clusters of sequences corresponding to independently evolving populations. The clusters formed with this approach matched the phylotypes given by GenBank for each locus, which were used to infer the multilocus trees described below.
PHYLOGENETIC RECONSTRUCTION
The phylogenetic history of B. burgdorferi sl was inferred by Bayesian inference in MrBayes 3.0 version 4 (Huelsenbeck and Ronquist 2001). Two types of phylogenetic trees were constructed, corresponding to the multilocus and partial-chromosome sequence data (the three- and seven-locus trees, and the chromosome tree, respectively). The multiloci trees were obtained by concatenating the individual gene sequences. In practice, for a given phylotype, a concatenated sequence was obtained by randomly sampling one sequence per locus in the cluster of sequences corresponding to that phylotype. We checked that the resulting trees were robust to the set of sequences sampled, and used one set for further analyses. This procedure resulted in an average of approximately five concatenated sequences per phylotype. To circumvent any potential complications that could arise through the concatenation of loci with different phylogenetic signals, we also analyzed the diversification rates in each of the seven individual locus trees.
For both the multilocus and chromosomal sequence data, we used the HKY85 model of molecular evolution with invariant sites and γ distributed rates, as selected by the Akaike Information Criterion by MrModeltest version 2.2 (Nylander 2004). This highly parameterized model of molecular evolution is necessary to avoid biasing branch length estimates that could result in biases in diversification rate estimates (Revell et al. 2005). Bayesian reconstructions using a Markov Chain Monte Carlo (MCMC) algorithm with four chains were run for 15,000,000 generations each and sampled every 250 generations. The first five million generations were discarded as burn-in, and convergence of the model was detected by assessing stationarity of logL values, plotting the posterior probabilities of nodes as a function of the number of generations, and by examining standard deviations of split frequencies for independent runs (Huelsenbeck et al. 2001; Rambaut and Drummond 2007). Three Bayesian runs using different seeds were implemented to verify convergence to the same posterior distribution of trees. To overcome uncertainty in the branching times and branching order, all further analyses were conducted on the 100 trees with the greatest posterior probability support from the Bayesian phylogenetic reconstruction (Huelsenbeck and Ronquist 2001). Ultrametric trees were obtained by applying PL to each phylogram using the software package r8s (Sanderson 2003). In the resulting trees, the nodes corresponded to divergences among phylotypes as well as divergences within phylotypes. Because we were interested in the diversification patterns of independently evolving lineages comparable to macroeukaryote species, that is, the diversification patterns of phylotypes, we had to clump within-phylotype sequences into a single lineage. We thus ran the GMYC model on the ultrametric trees containing all sequences, and checked that they formed clusters consistent with the accepted phylotypes. One branch from each phylotype was then retained for subsequent analyses.
The sensitivity of our results to the method of phylogenetic reconstruction was assessed by constructing ultrametric trees with two additional methods. The Bayesian Estimation of Species Trees (BEST) leverages multilocus data while accounting for the fact that gene trees are embedded within a common phylogeny (Edwards et al. 2007; Liu and Pearl 2007; Liu 2008). The BEST analyses using all sequences from the seven loci were initiated from random starting trees and sampled every 1000 generations over a 100 million generation run. The posterior distribution of species trees (post burn-in) was summarized as the 50% majority-rule consensus tree to obtain posterior probability values for species relationships. We also constructed phylogenetic trees using maximum-likelihood methods assuming a molecular clock (MLMC) in PAUP* version 4.0b10 (Swofford 2003) for both the partial-chromosome and concatenated multilocus datasets. MODELTEST version 3.06 (Posada and Crandall 1998) was used to choose the best evolutionary model (HKY85+I+Γ). Maximum-likelihood analyses were subjected to 1000 bootstrap replicates.
DETECTING TIME VARIATION IN DIVERSIFICATION RATES
Both the γ-statistic (Pybus and Harvey 2000) and the coalescent approach of Morlon et al. (2010, codes available at http://www.cmap.polytechnique.fr/~morlon/resources.html) were employed to test for temporal variation in diversification rates. Both methods are robust to absolute age estimates and require only relative divergence times. These approaches also allow us to statistically account for phylotypes that are not included in the dataset. Accounting for unsampled phylotypes is essential as missing phylotypes can cause the spurious detection of declines in diversification rates (Pybus and Harvey 2000). Completing the analyses of γ with another method is also essential, as a negative γ does not necessarily indicate an early burst of diversification (Fordyce 2010). Other methods that allow for periods of diversity decline (Morlon et al. 2011) and rate heterogeneity across lineages (Alfaro et al. 2009; Morlon et al. 2011) are also applicable, but not required to address our central question of whether diversification rates have varied over time.
To assess the robustness of our results to incomplete phylotype sampling, we analyzed all datasets using both the γ and the coalescent with the assumption that the true number of existing phylotypes (N) is 15 (the current number of recognized phylotypes), 17 (the current number of recognized phylotypes plus the two newly proposed phylotypes), 20, 30, 50, 100, 150, 200, and 250. An upper limit of 250 phylotypes, more than 15-fold the total number of currently described B. burgdorferi sl phylotypes, seems a reasonable upper bound on the true number of phylotypes given the extensive effort that has been made to sample this clade.
We used the Monte Carlo Constant Rates (MCCR) γ approach detailed in Pybus and Harvey (2000) to test for a decline in diversification rates while accounting for potentially missing taxa. This approach assumes that missing taxa are randomly distributed in the tree. Violations of this assumption could lead to spurious detection of rate declines (Cusimano and Renner 2010), extensions of the approach exist that can account for overdispersed or underdispersed sampling (Brock et al. 2011). However, we chose not to employ these methods here as we had no a priori expectations of the form of the sampling. For each phylogeny, we calculated the γ-value of the 100 Bayesian phylogenies with best posterior support. We then compared these values to the critical MCCR corrected γ-value indicating a significant decay in diversification rate (P < 0.05). The critical MCCR corrected γ-value was estimated for each phylogeny and each assumed number of total phylotypes (N).
The coalescent approach employed yields an approximate likelihood expression for internode distances in a phylogeny under a variety of diversification scenarios (Fig. 1 and Table 1 from Morlon et al. 2010). The coalescent framework was introduced in population genetics to study the genealogy of samples, and is thus particularly well suited to the study of phylogenies that may not be completely sampled. We compared the fit of four diversification models to the empirical data: the Yule model (constant diversification rate, no extinction), the YuleVAR model (varying diversification rate, no extinction), the Birth–Death model (constant diversification and extinction rates), and the BirthVAR-Death model (varying diversification rate, constant extinction rate). When rates varied over time, we used an exponential form of rate variation with time, t. Time is measured from the present to the past, such that t = 0 denotes the present.
Figure 1.

Bayesian phylogenetic reconstruction (left) and ultrametric phylogenies (right) of all available B. burgdorferi sensu lato (sl) phylotypes, based on a total of 1543 gene sequences. The triangles represent the maximum genetic distance among sequences within phylotypes in the Bayesian phylogenies. The majority of among-phylotype diversification events occur near the root of the phylogenies suggesting a rapid burst of diversification early in the evolutionary history of the group. Phylogenies were constructed using: (A) the three loci for which data were available from 15 phylotypes, (B) seven loci for which data were available from 11 phylotypes, and (C) the partial chromosome sequence data (chromosome). Nodes marked with asterisks are supported by posterior probabilities of more than 0.8 (*), 0.9 (**), or 0.95 (***).
To test the hypothesis that diversification rates have varied over time, we used likelihood ratio tests for the two pairs of models with constant versus varying rate (i.e., Yule vs. YuleVAR, and Birth–Death vs. BirthVAR-Death). The possibility that phylotypes exist but are not represented in our datasets was accounted for by computing the sampling fraction f (Morlon et al. 2010) for each reconstructed phylogeny and each hypothesized number of phylotypes (N) and fitting the various diversification models with the corresponding sampling fraction. We tested the robustness of the results to topological and branch-length uncertainty by running the likelihood ratio tests on the 100 Bayesian phylogenies with best posterior support.
Results
The Bayesian inference of the evolutionary relationships among the three-locus tree revealed 15 phylotypes separated by long branches with relatively little variation among sequences within each phylotype (Fig. 1A). This visual inspection was supported by results from the Generalized Mixed Yule Coalescent (GMYC) model, which identified a threshold at 1.4% sequence divergence resulting in 15 independently evolving clusters. Hence, the number of phylotypes delimited with our phylogenetic reconstruction matched the number of described B. burgdoferi sl phylotypes. Similarly, the number of phylotypes delimited by the GMYC model for the seven-loci tree (11 phylotypes with less than 1.3% sequence divergence, Fig. 1B) and the partial chromosome tree (six phylotypes with less than 1.9% sequence divergence, Fig. 1C) matched the number of described B. burgdoferi sl phylotypes expected given the sequences used to reconstruct the phylogenies. Importantly, the GYMC models clustered sequences by the currently accepted genospecies names in all cases. These results indicate that each phylotype can be treated as an independently evolving lineage comparable to species in sexual eukaryotes (Avise and Wollenberg 1997; Richter et al. 2006; Postic et al. 2007).
The Bayesian trees reconstructed from the three-locus dataset, the seven-locus dataset, and the partial-chromosome dataset featured similar topologies and shapes, with many nodes toward the root and long branches leading to each phylotypic cluster of sequences (Fig. 1). It is unlikely that the long terminal branches observed in the ultrametric trees (Fig. 1, right column) resulted from high among-lineage variation in molecular evolutionary rates, because the nonclocklike trees (Fig. 1, left column) also featured long terminal branches and likelihood ratio tests did not detect deviations from clocklike evolution (P > 0.1 for all comparisons). Further, the phylogenetic trees inferred by maximum likelihood with a forced molecular clock (Fig. S1) and the BEST tree (Fig. S2) displayed internal nodes near the root and long branches leading to each phylotype. The evolutionary relationships differed slightly among the phylogenies in some cases (ex B. valaisiana, Fig. 1) and statistical support for several internal nodes was not significant. However, the consistent shape across trees indicates phylotype diversification occurred early and rapidly in the history of the clade, potentially explaining the uncertainty in phylogenetic resolution and incongruence between trees.
Macroevolutionary analyses of the phylogenies support the hypothesis that the present-day diversity of B. burgdorferi sl results from rapid-early diversification (Figs. 2 and 3). The γ-values calculated from the three- and seven-locus phylogenies were less than the smallest γ-value of the phylogenies simulated under constant rate (hence, the null hypothesis of constant diversification rates was rejected, P < 0.001), even when assuming a true diversity of as many as 250 B. burgdorferi sl phylotypes (Fig. 2A, B). The MCCR analyses of the partial chromosome phylogeny also significantly supported a time-decaying diversification rate (P < 0.05) unless the currently described phylotypes represent less than 10% of the true number of extant phylotypes (Fig. 2C). These results strongly suggest that diversification occurred rapidly in the early history of the clade and has since slowed.
Figure 2.
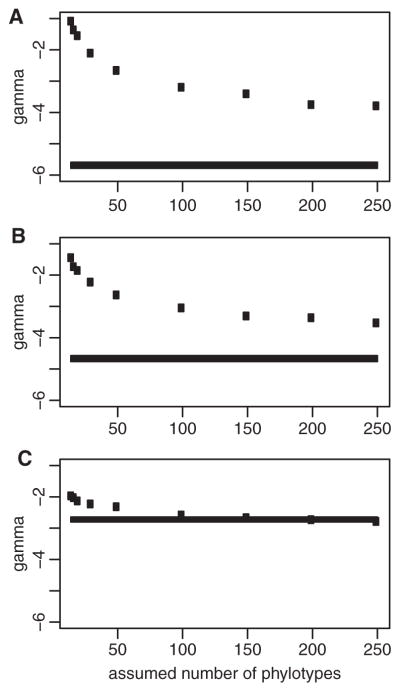
Monte Carlo Constant Rates (MCCR) tests reject the hypothesis that diversification rates have been constant over time even if considerable species diversity in the B. burgdorferi sl clade has yet to be discovered. Black lines represent the γ-values of the 100 phylogenies with maximal Bayesian support. Black dots represent the critical γ-values of the constant-rates test corresponding to each assumed number of phylotypes. γ-values below the dots indicate significant rate-decay (P < 0.05). The MCCR analyses of the three-locus (A) and seven-locus (B) phylogenies support a time-decaying diversification rate, even if the currently described phylotypes represent less than 6% of the true number of extant phylotypes. Similarly, the hypothesis of constant diversification rate can be rejected for the chromosome phylogeny even if the actual number of existing phylotypes exceeds 100 (C).
Figure 3.

Likelihood ratio tests indicate that models with varying diversification rates are significantly more likely than models with constant rates regardless of the assumed number of species in the B. burgdorferi sl clade. The likelihood ratios ( ) between models with constant versus varying diversification rates are plotted as a function of the assumed number of extant phylotypes. The dashed lines indicate the chi-square values corresponding to P < 0.001, 0.01, and 0.05 (from top to bottom). The pure-birth model with decaying diversification rate is more likely given the data than a pure-birth model with constant rates (left column), and the birth–death model with decaying diversification rate is more likely than the constant-rate birth–death model (right column). Models with time-varying rates were consistently supported even when we assumed that as many as 250 B. burgdorferi sl phylotypes exist at present.
The time-decaying diversification rate hypothesis was further supported by the coalescent-based analyses. B. burgdorferi sl phylogenies were consistent with models that included time-varying rates. Both the pure-birth and the birth-death models with varying diversification rates were significantly more likely than models with constant diversification rates (Fig. 3). Both models posit that diversification rates have decayed sharply since the diversification events that happened early in the history of the clade. The strong support we found for time-decaying rates is all the more striking given that there are few B. burgdorferi sl phylotypes and thus a priori little statistical power to select complex (time-varying) models over simpler (time-constant) models. Our conclusions were robust to potential biases introduced by missing phylotypes as the models with time-varying rates were consistently supported even when we assumed that as many as 250 B. burgdorferi sl phylotypes exist today (Fig. 3). The conclusions were also supported by our analyses of the individual-locus trees (Fig. S3), suggesting that our results are not an artifact of concatenating loci with different phylogenetic signal.
Discussion
Although explosive radiations followed by rapid decreases in diversification rates is a common pattern for plant and animal lineages, little is known about the macroevolutionary patterns of prokaryotes, which constitute most of the diversity of life on earth. Contrary to a previous result suggesting that the tempo of diversification in microorganisms is fundamentally different from that of macroorganisms (Martin et al. 2004), our study suggests that the variation in the rates of cladogenesis over time in bacteria can be comparable to those reported in classic examples of radiations (Harmon et al. 2003; Phillimore and Price 2008; Rabosky and Lovette 2008; Morlon et al. 2010). The rapid diversification of B. burgdorferi phylotypes early in the history of the clade followed by a decrease in diversification rates observed in this study closely resembles the patterns described in many plant and animal lineages.
Our results conflict with previous findings that suggest that the diversification rates of microorganisms are constant over time (Martin et al. 2004). This discrepancy may stem from either methodological artifacts or differing biological characteristics of the clades analyzed. Martin et al. (2004) pointed out that the broad phylogenetic clade they investigated was substantially un-dersampled and exhibited significant substitution rate variation among lineages. Undersampling and substitution rate variation are thought to bias γ negatively rather than positively (Revell et al. 2005), which would lead to a false detection of rate declines rather than the opposite. These two factors nevertheless create biases in the distribution of node heights, which weakens the general confidence one can have in the inferred patterns of lineage diversification. By contrast, the focus on a narrow clade in the present study resulted in no departure from clocklike assumptions, allowing a more accurate assessment of node heights and thus the relative timing of diversification. Further, it is likely that the B. burgdorferi sl clade has been thoroughly sampled due to its role in human Lyme disease and because of its obligatory association with arthropod vectors and vertebrate hosts that are easy to capture and investigate for infection (Barbour and Hayes 1986; Nuttall 1999; van Dam 2002; Piesman and Gern 2004). Although novel phylotypes will doubtless be described in the future, it is unlikely that the 15 phylotypes analyzed here represent a small enough fraction of the total diversity to lose the statistical support we found for the decay in diversification rate over time (Figs. 2 and 3).
One difference between our analyses and that of Martin et al. (2004) is that we grouped individual sequences into phylotypes before analyzing diversification patterns. Our resulting phylogenies therefore represent divergences among phylotypes. By contrast, Martin et al. (2004) analyzed the lineage-through-time plot of a phylogeny without grouping sequences; their phylogeny represents divergences both within and between phylotypes. Hence, a potential slowdown in the diversification rate of phylotypes in Martin’s study could have been masked by recent divergences of individuals within phylotypes. Because our goal was to analyze the diversification patterns of independently evolving bacterial lineages—comparable to species in macroorganisms—we defined the independently evolving lineages (the phylotypes) prior to analyzing their phylogenetic relationships and diversification dynamics. The 15 phylotypes we defined, based on molecular data from multiple loci, conform to the 15 B. burgdorferi sl phylotypes recognized in the literature.
The patterns of diversification observed in the B. burgdorferi clade may indicate that time-decaying diversification rates are a common pattern in the microbial world, as they are in macroeukaryote clades. However, the rapid-early diversification observed here may result from specific biological features of B. burgdorferi that differ from free-living microbes like those previously analyzed (Martin et al. 2004). B. burgdorferi are obligatorily associated with vertebrates and hematophagous arthropods and thus may be subject to similar ecological processes that structure the macroevolutionary patterns of macroeukaryotes. For example, the geographical processes that influence diversification in macroeukaryotes by creating ecological opportunity and reducing gene flow among populations (Day 2000) may be less important for many free-living prokaryotes that are more readily transported over large distances. The constant association of B. burgdorferi with either ticks or vertebrate hosts (Barbour and Hayes 1986; Nuttall 1999; van Dam 2002; Piesman and Gern 2004) may limit gene flow between isolated populations, rendering macroevolutionary patterns more similar to those of macroorganisms. Hence, time variation in diversification rates may be rare among free-living bacteria as previously suggested (Martin et al. 2004) but common in bacteria that are obligatorily associated with macroorganisms. This hypothesis could be tested using a broader array of well-sampled free-living and nonfree-living bacterial phylogenies.
The fast-early diversification followed by a strong decay in diversification rates observed in the B. burgdorferi phylogenies is consistent with the hypothesis that ecological opportunity promoted diversification (Schluter 2000; Yoder et al. 2010), that is, that diversification is constrained by ecological factors (Rabosky and Lovette 2008; Rabosky 2009; Morlon et al. 2010; Rabosky and Glor 2010). An intriguing hypothesis is that diversification of B. burgdorferi phylotypes may have resulted from adaptive specialization to ecological niches, such as specific vertebrate species or tick vectors (Kurtenbach et al. 2002, 2006; Xu et al. 2003). Indeed, each phylotype infects only a subset of vertebrate species collectively infected by all B. burgdorferi phylotypes (Kurtenbach et al. 2002, 2006); for example, human infectiousness is restricted primarily to three of the 15 phylotypes (Baranton et al. 1992). Although current data suggest that B. burgdorferi phylotypes partition potential niches only into broad categories such as association with either birds, mammals, or both (Kurtenbach et al. 2002), additional studies on host species specificity or organ tropism may reveal further niche partitioning. Recent evidence suggests that lineages within two of the human infectious B. burgdorferi phylotypes specialize on specific host species and maybe organs within hosts (Kurtenbach et al. 2002; Hanincova et al. 2003; Brisson and Dykhuizen 2004; Hanincova et al. 2006; Brisson et al. 2008; Brisson et al. 2011). Finer scale investigation into host species use by B. burgdorferi phylotypes that are not medically relevant may also reveal greater specialization than is currently acknowledged.
Evidence of rapid diversification in bacteria gathered from experimental microcosm studies (reviewed in MacLean 2005; Kassen 2009) is consistent with the analyses supporting time-decaying diversification rates in B. burgdorferi sl. A wide variety of bacterial lineages readily and repeatedly diversify to adapt to a myriad of structured environmental and biotic niches, and to specialize on alternative resources during experimental evolution studies (Hardin 1960; Friesen et al. 2004; Kassen and Rainey 2004; Barrett et al. 2005; Barrett and Bell 2006; Brockhurst et al. 2006; Habets et al. 2006; Jasmin and Kassen 2007). These experiments suggest that diversification rates should be rapid initially when ecological opportunities are abundant and decrease as opportunities become less abundant (MacLean 2005; Kassen 2009). Additionally, nonadaptive radiations, potentially caused by vicariance processes, could also lead to a time decline in diversification rates as space is occupied (Kozak et al. 2006). Therefore, the signature of decaying diversification rates is likely to be common and readily observable in many prokaryotic phylogenies.
There are many challenges associated with the study of macroevolutionary patterns in microorganisms. Although many microbial groups must be analyzed to establish that the rapid-early diversification hypothesis is a common macroevolutionary feature across the entire tree of life, the analyses presented here suggest that rapid-early diversification can occur in bacterial lineages.
Supplementary Material
Acknowledgments
We thank P. Sniegowski and D. Dykhuizen for helpful advice and comments on the manuscript. We also thank P. Turner, M. Pennell, and two anonymous reviewers for comments that improved the manuscript. This work was supported by grants from the NIAID (AI076342) to DB. HM acknowledges support from the Centre National de la Recherche Scientifique and the Agence Nationale de la Recherche (ECOEVOBIO—CHEX2011). JBP acknowledges support from the James S. McDonnell Foundation, the Alfred P. Sloan Foundation, the David and Lucille Packard Foundation, the Burroughs Wellcome Fund, and the Defense Advanced Research Projects Agency (HR0011–05-1–0057).
Footnotes
The following supporting information is available for this article:
Figure S1. Phylogenetic trees inferred by maximum likelihood with a forced molecular clock for: (A) the three-locus tree (multilocus3), (B) the seven-locus tree (multilocus7), and (C) the chromosome tree (chromosome).
Figure S2. Phylogenetic tree inferred by Bayesian Estimation of Species Trees (BEST).
Figure S3. For most of the seven individual locus phylogenies trees, likelihood ratio tests indicate that the pure-birth model with decaying diversification rate is significantly more likely than a pure-birth model with constant rate regardless of the assumed number of species in the B. burgdorferi sl clade.
Supporting Information may be found in the online version of this article.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
LITERATURE CITED
- Alfaro ME, Santini F, Brock C, Alamillo H, Dornburg A, Rabosky DL, Carnevale G, Harmon LJ. Nine exceptional radiations plus high turnover explain species diversity in jawed vertebrates. Proc Natl Acad Sci USA. 2009;106:13410–13414. doi: 10.1073/pnas.0811087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alroy J. The fossil record of North American mammals: evidence for a Paleocene evolutionary radiation. Syst Biol. 1999;48:107–118. doi: 10.1080/106351599260472. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Avise JC, Wollenberg K. Phylogenetics and the origin of species. Proc Natl Acad Sci USA. 1997;94:7748–7755. doi: 10.1073/pnas.94.15.7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranton G, Postic D, Saint Girons I, Boerlin P, Piffaretti JC, Assous M, Grimont PA. Delineation of Borrelia burgdorferi sensu stricto, Borrelia garinii sp. nov., and group VS461 associated with Lyme borreliosis. Int J Syst Bacteriol. 1992;42:378–383. doi: 10.1099/00207713-42-3-378. [DOI] [PubMed] [Google Scholar]
- Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev. 1986;50:381–400. doi: 10.1128/mr.50.4.381-400.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraclough TG, Hughes M, Ashford-Hodges N, Fujisawa T. Inferring evolutionarily significant units of bacterial diversity from broad environmental surveys of single-locus data. Biol Lett. 2009;5:425–428. doi: 10.1098/rsbl.2009.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RD, Bell G. The dynamics of diversification in evolving Pseudomonas populations. Evolution. 2006;60:484–490. [PubMed] [Google Scholar]
- Barrett RD, MacLean RC, Bell G. Experimental evolution of Pseudomonas fluorescens in simple and complex environments. Am Nat. 2005;166:470–480. doi: 10.1086/444440. [DOI] [PubMed] [Google Scholar]
- Brisson D, Dykhuizen DE. ospC diversity in Borrelia burgdorferi: different hosts are different niches. Genetics. 2004;168:713–722. doi: 10.1534/genetics.104.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisson D, Dykhuizen DE, Ostfeld RS. Conspicuous impacts of inconspicuous hosts on the Lyme disease epidemic. Proc R Soc Lond B. 2008;275:227–235. doi: 10.1098/rspb.2007.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisson D, Baxamusa N, Schwartz I, Wormser GP. Biodiversity of Borrelia burgdorferi strains in tissues of Lyme disease patients. PLoS One. 2011;6:e22926. doi: 10.1371/journal.pone.0022926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock CD, Harmon LJ, Alfaro ME. Testing for temporal variation in diversification rates when sampling is incomplete and nonrandom. Syst Biol. 2011;60:410–419. doi: 10.1093/sysbio/syr007. [DOI] [PubMed] [Google Scholar]
- Brockhurst MA, Hochberg ME, Bell T, Buckling A. Character displacement promotes cooperation in bacterial biofilms. Curr Biol. 2006;16:2030–2034. doi: 10.1016/j.cub.2006.08.068. [DOI] [PubMed] [Google Scholar]
- Cusimano N, Renner SS. Slowdowns in diversification rates from real phylogenies may not be real. Syst Biol. 2010;59:458–464. doi: 10.1093/sysbio/syq032. [DOI] [PubMed] [Google Scholar]
- Davis CC, Webb CO, Wurdack KJ, Jaramillo CA, Donoghue MJ. Explosive radiation of Malpighiales supports a mid-cretaceous origin of modern tropical rain forests. Am Nat. 2005;165:E36–E65. doi: 10.1086/428296. [DOI] [PubMed] [Google Scholar]
- Day T. Competition and the effect of spatial resource heterogeneity on evolutionary diversification. Am Nat. 2000;155:790–803. doi: 10.1086/303356. [DOI] [PubMed] [Google Scholar]
- Dykhuizen DE, Baranton G. The implications of a low rate of horizontal transfer in Borrelia. Trends Microbiol. 2001;9:344–350. doi: 10.1016/s0966-842x(01)02066-2. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SV, Liu L, Pearl DK. High-resolution species trees without concatenation. Proc Natl Acad Sci USA. 2007;104:5936–5941. doi: 10.1073/pnas.0607004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce JA. Interpreting the gamma statistic in phylogenetic diversification rate studies: a rate decrease does not necessarily indicate an early burst. PloS One. 2010;5:e11781. doi: 10.1371/journal.pone.0011781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen ML, Saxer G, Travisano M, Doebeli M. Experimental evidence for sympatric ecological diversification due to frequency-dependent competition in Escherichia coli. Evolution. 2004;58:245–260. [PubMed] [Google Scholar]
- Gern L. Borrelia burgdorferi sensu lato, the agent of Lyme borreliosis: life in the wilds. Parasite. 2008;15:244–247. doi: 10.1051/parasite/2008153244. [DOI] [PubMed] [Google Scholar]
- Glor RE. Phylogenetic insights on adaptive radiation. Annu Rev Ecol Evol Syst. 2010;41:251–270. [Google Scholar]
- Habets MG, Rozen DE, Hoekstra RF, de Visser JA. The effect of population structure on the adaptive radiation of microbial populations evolving in spatially structured environments. Ecol Lett. 2006;9:1041–1048. doi: 10.1111/j.1461-0248.2006.00955.x. [DOI] [PubMed] [Google Scholar]
- Hanincova K, Taragelova V, Koci J, Schafer SM, Hails R, Ullmann AJ, Piesman J, Labuda M, Kurtenbach K. Association of Borrelia garinii and B. valaisiana with songbirds in Slovakia. Appl Environ Microbiol. 2003;69:2825–2830. doi: 10.1128/AEM.69.5.2825-2830.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanincova K, Kurtenbach K, Diuk-Wasser M, Brei B, Fish D. Epidemic spread of Lyme borreliosis, northeastern United States. Emerg Infect Dis. 2006;12:604–611. doi: 10.3201/eid1204.051016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin G. The competitive exclusion principle. Science. 1960;131:1292–1297. doi: 10.1126/science.131.3409.1292. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, Schulte JA, Larson A, Losos JB. Tempo and mode of evolutionary radiation in iguanian lizards. Science. 2003;301:961–964. doi: 10.1126/science.1084786. [DOI] [PubMed] [Google Scholar]
- Higgins DG, Sharp PM. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene. 1988;73:237–244. doi: 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Jasmin JN, Kassen R. On the experimental evolution of specialization and diversity in heterogeneous environments. Ecol Lett. 2007;10:272–281. doi: 10.1111/j.1461-0248.2007.01021.x. [DOI] [PubMed] [Google Scholar]
- Kassen R. Toward a general theory of adaptive radiation: insights from microbial experimental evolution. Ann N Y Acad Sci. 2009;1168:3–22. doi: 10.1111/j.1749-6632.2009.04574.x. [DOI] [PubMed] [Google Scholar]
- Kassen R, Rainey PB. The ecology and genetics of microbial diversity. Annu Rev Microbiol. 2004;58:207–231. doi: 10.1146/annurev.micro.58.030603.123654. [DOI] [PubMed] [Google Scholar]
- Kozak KH, Weisrock DW, Larson A. Rapid lineage accumulation in a non-adaptive radiation: phylogenetic analysis of diversification rates in eastern North American woodland salamanders (Plethodontidae: Plethodon) Proc R Soc Lond B. 2006;273:539–546. doi: 10.1098/rspb.2005.3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach K, De Michelis S, Etti S, Schafer SM, Sewell HS, Brade V, Kraiczy P. Host association of Borrelia burgdorferi sensu lato–the key role of host complement. Trends Microbiol. 2002;10:74–79. doi: 10.1016/s0966-842x(01)02298-3. [DOI] [PubMed] [Google Scholar]
- Kurtenbach K, Hanincova K, Tsao JI, Margos G, Fish D, Ogden NH. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nat Rev Microbiol. 2006;4:660–669. doi: 10.1038/nrmicro1475. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Liu L. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics. 2008;24:2542–2543. doi: 10.1093/bioinformatics/btn484. [DOI] [PubMed] [Google Scholar]
- Liu L, Pearl DK. Species trees from gene trees: reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst Biol. 2007;56:504–514. doi: 10.1080/10635150701429982. [DOI] [PubMed] [Google Scholar]
- MacLean RC. Adaptive radiation in microbial microcosms. J Evol Biol. 2005;18:1376–1386. doi: 10.1111/j.1420-9101.2005.00931.x. [DOI] [PubMed] [Google Scholar]
- Margos G, Vollmer SA, Cornet M, Garnier M, Fingerle V, Wilske B, Bormane A, Vitorino L, Collares-Pereira M, Drancourt M, et al. A new Borrelia species defined by multilocus sequence analysis of housekeeping genes. Appl Environ Microbiol. 2009;75:5410–5416. doi: 10.1128/AEM.00116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margos G, Vollmer SA, Ogden NH, Fish D. Population genetics, taxonomy, phylogeny and evolution of Borrelia burgdorferi sensu lato. Infect Genet Evol. 2011;11:1545–1563. doi: 10.1016/j.meegid.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AP, Costello EK, Meyer AF, Nemergut DR, Schmidt SK. The rate and pattern of cladogenesis in microbes. Evolution. 2004;58:946–955. doi: 10.1111/j.0014-3820.2004.tb00429.x. [DOI] [PubMed] [Google Scholar]
- McPeek MA. The ecological dynamics of clade diversification and community assembly. Am Nat. 2008;172:E270–E284. doi: 10.1086/593137. [DOI] [PubMed] [Google Scholar]
- Morlon H, Potts MD, Plotkin JB. Inferring the dynamics of diversification: a coalescent approach. PLoS Biol. 2010;8:e1000493. doi: 10.1371/journal.pbio.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlon H, Parsons TL, Plotkin JB. Reconciling molecular phylogenies with the fossil record. Proc Natl Acad Sci USA. 2011;108:16327–16332. doi: 10.1073/pnas.1102543108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nee S, Mooers AO, Harvey PH. Tempo and mode of evolution revealed from molecular phylogenies. Proc Natl Acad Sci USA. 1992;89:8322–8326. doi: 10.1073/pnas.89.17.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nee S, Holmes EC, May RM, Harvey PH. Extinction rates can be estimated from molecular phylogenies. Phil Trans R Soc Lond B. 1994;344:77–82. doi: 10.1098/rstb.1994.0054. [DOI] [PubMed] [Google Scholar]
- Nuttall PA. Pathogen-tick-host interactions: Borrelia burgdorferi and TBE virus. Zentralbl Bakteriol. 1999;289:492–505. doi: 10.1016/s0934-8840(99)80002-4. [DOI] [PubMed] [Google Scholar]
- Nylander JAA. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University; 2004. [Google Scholar]
- Phillimore AB, Price TD. Density-dependent cladogenesis in birds. PLoS Biol. 2008;6:e71. doi: 10.1371/journal.pbio.0060071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piesman J, Gern L. Lyme borreliosis in Europe and North America. Parasitology. 2004;129(Suppl):S191–S220. doi: 10.1017/s0031182003004694. [DOI] [PubMed] [Google Scholar]
- Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol. 2006;55:595–609. doi: 10.1080/10635150600852011. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Postic D, Garnier M, Baranton G. Multilocus sequence analysis of atypical Borrelia burgdorferi sensu lato isolates–description of Borrelia californiensis sp. nov., and genomospecies 1 and 2. Int J Med Microbiol. 2007;297:263–271. doi: 10.1016/j.ijmm.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Purvis A, Nee S, Harvey PH. Macroevolutionary inferences from primate phylogeny. Proc R Soc Lond B. 1995;260:329–333. doi: 10.1098/rspb.1995.0100. [DOI] [PubMed] [Google Scholar]
- Pybus OG, Harvey PH. Testing macro-evolutionary models using incomplete molecular phylogenies. Proc R Soc Lond B. 2000;267:2267–2272. doi: 10.1098/rspb.2000.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WG, Schutzer SE, Bruno JF, Attie O, Xu Y, Dunn JJ, Fraser CM, Casjens SR, Luft BJ. Genetic exchange and plasmid transfers in Borrelia burgdorferi sensu stricto revealed by three-way genome comparisons and multilocus sequence typing. Proc Natl Acad Sci USA. 2004;101:14150–14155. doi: 10.1073/pnas.0402745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky DL. Likelihood methods for detecting temporal shifts in diversification rates. Evolution. 2006;60:1152–1164. [PubMed] [Google Scholar]
- Rabosky DL. Ecological limits and diversification rate: alternative paradigms to explain the variation in species richness among clades and regions. Ecol Lett. 2009;12:735–743. doi: 10.1111/j.1461-0248.2009.01333.x. [DOI] [PubMed] [Google Scholar]
- Rabosky DL, Glor RE. Equilibrium speciation dynamics in a model adaptive radiation of island lizards. Proc Natl Acad Sci USA. 2010;107:22178–22183. doi: 10.1073/pnas.1007606107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky DL, I, Lovette J. Explosive evolutionary radiations: decreasing speciation or increasing extinction through time? Evolution. 2008;62:1866–1875. doi: 10.1111/j.1558-5646.2008.00409.x. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ. [Accessed September 2009];Tracer. 2007 1.4 Available at: beast.bio.ac.uk/Tracer. [Google Scholar]
- Revell LJ, Harmon LJ, Glor RE. Underparameterized model of sequence evolution leads to bias in the estimation of diversification rates from molecular phylogenies. Syst Biol. 2005;54:973–983. doi: 10.1080/10635150500354647. [DOI] [PubMed] [Google Scholar]
- Richter D, Postic D, Sertour N, Livey I, Matuschka FR, Baranton G. Delineation of Borrelia burgdorferi sensu lato species by multi-locus sequence analysis and confirmation of the delineation of Borrelia spielmanii sp. nov. Int J Syst Evol Microbiol. 2006;56:873–881. doi: 10.1099/ijs.0.64050-0. [DOI] [PubMed] [Google Scholar]
- Rokas A, Krüger D, Carroll SB. Animal evolution and the molecular signature of radiations compressed in time. Science. 2005;310:1933–1938. doi: 10.1126/science.1116759. [DOI] [PubMed] [Google Scholar]
- Rudenko N, Golovchenko M, Grubhoffer L, Oliver JH., Jr Borrelia carolinensis sp. nov., a new (14th) member of the Borrelia burgdorferi sensu lato complex from the southeastern region of the United States. J Clin Microbiol. 2009;47:134–141. doi: 10.1128/JCM.01183-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko N, Golovchenko M, Grubhoffer L, Oliver JH., Jr Borrelia carolinensis sp. nov., a new species of Borrelia burgdorferi sensu lato isolated from rodents and tick from the southeastern United States. Int J Syst Evol Microbiol. 2011;61:381–383. doi: 10.1099/ijs.0.021436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundell RJ, Price TD. Adaptive radiation, nonadaptive radiation, ecological speciation and nonecological speciation. Trends Ecol Evol. 2009;24:394–399. doi: 10.1016/j.tree.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Donoghue MJ. Shifts in diversification rate with the origin of angiosperms. Science. 1994;264:1590–1593. doi: 10.1126/science.264.5165.1590. [DOI] [PubMed] [Google Scholar]
- Schluter D. The ecology of adaptive radiation. Oxford Univ. Press; New York: 2000. [Google Scholar]
- Schuettpelz E, Pryer KM. Evidence for a Cenozoic radiation of ferns in an angiosperm-dominated canopy. Proc Natl Acad Sci USA. 2009;106:11200–11205. doi: 10.1073/pnas.0811136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson GG. The major features of evolution. Columbia Univ. Press; New York: 1953. [Google Scholar]
- Stadler T. Mammalian phylogeny reveals recent diversification rate shifts. Proc Natl Acad Sci USA. 2011;108:6187–6192. doi: 10.1073/pnas.1016876108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods) Sinauer Associates; Sunderland, MA: 2003. [Google Scholar]
- Valente LM, Savolainen V, Vargas P. Unparalleled rates of species diversification in Europe. Proc R Soc Sci Lond B. 2010;277:1489–1496. doi: 10.1098/rspb.2009.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam AP. Diversity of Ixodes-borne Borrelia species–clinical, pathogenetic, and diagnostic implications and impact on vaccine development. Vector Borne Zoonotic Dis. 2002;2:249–254. doi: 10.1089/153036602321653833. [DOI] [PubMed] [Google Scholar]
- Xu G, Fang QQ, Keirans JE, Durden LA. Molecular phylogenetic analyses indicate that the Ixodes ricinus complex is a paraphyletic group. J Parasitol. 2003;89:452–457. doi: 10.1645/0022-3395(2003)089[0452:MPAITT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Yoder JB, Clancey E, Des Roches S, Eastman JM, Gentry L, Godsoe W, Hagey TJ, Jochimsen D, Oswald BP, Robertson J, et al. Ecological opportunity and the origin of adaptive radiations. J Evol Biol. 2010;23:1581–1596. doi: 10.1111/j.1420-9101.2010.02029.x. [DOI] [PubMed] [Google Scholar]
- Zink RM, Slowinski JB. Evidence from molecular systematics for decreased avian diversification in the pleistocene Epoch. Proc Natl Acad Sci USA. 1995;92:5832–5835. doi: 10.1073/pnas.92.13.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.