

Abstract
Development of SAR in an N-acyl-N′-arylpiperazine series of negative allosteric modulators of mGlu1 using a functional cell-based assay is described in this Letter. Characterization of selected compounds in protein binding assays was used to aid in selecting VU0469650 for further profiling in ancillary pharmacology assays and pharmacokinetic studies. VU0469650 demonstrated an excellent selectivity profile and good exposure in both plasma and brain samples following intraperitoneal dosing in rats.
Keywords: Glutamate, GPCR, mGlu1, Allosteric modulator, CNS, Piperazine
l-Glutamic acid (glutamate) is the major excitatory neurotransmitter in the mammalian central nervous system (CNS). Glutamate produces its effects through binding to both ionotropic and metabotropic glutamate receptors. The metabotropic glutamate receptors (mGlus) belong to family C of the G protein-coupled receptors (GPCRs). Further classification of the mGlus discovered to date has been according to their structure, preferred signal transduction mechanisms, and pharmacology (Group I: mGlu1 and mGlu5; Group II: mGlu2 and mGlu3; Group III: mGlu4, mGlu6, mGlu7, and mGlu8).1 Many of these receptors have attracted significant attention as promising targets for the treatment of a variety of CNS related disorders. The design of drug-like compounds that selectively target a specific mGlu has been complicated by the fact that the orthosteric binding site of the mGlus is highly conserved. An alternative strategy that has proven successful for overcoming such selectivity hurdles has been the optimization of compounds that modulate the function of the receptor through interaction with an allosteric binding site.2
An area within the mGlu allosteric modulator field that has garnered substantial interest has been the design and application of selective small molecule negative allosteric modulators (NAMs) of mGlu1.3 Several tool compounds have been discovered during recent years further establishing the link between mGlu1 antagonism and the potential treatment of CNS related disorders such as addiction,4 anxiety,5 epilepsy,6 pain,5a,7 and psychotic disorders. 5a,8 Compounds 1–6 (Fig. 1) are representative of the mGlu1 NAM chemotypes that have been discovered to date and examples of molecules with demonstrated efficacy in vivo. For example, JNJ16259685 (1) was efficacious in a rat model of anxiety9 as well as rat10 and squirrel monkey11 models of addiction. A-841720 (2) was established as an analgesic in rat models of neuropathic pain12 and post-operative pain.13 Inhibition of nociceptive pain was observed with 3 in rats14 and with YM298198 (4) in mice.15 Antipsychotic activity as measured by acoustic prepulse inhibition models was noted with 516 and 617 in rats and with 18 and 48 in mice. Also of interest, recent research has identified a potential role for mGlu1 inhibition in the treatment melanoma18 and certain types of breast cancer.19
Figure 1.

mGlu1 NAM compounds with in vivo efficacy.
We have recently become interested in the discovery and development of structurally novel mGlu1 NAMs with properties suitable for evaluation in rodent models of a variety of CNS disorders. A potential starting point for a hit to lead optimization program focused on mGlu1 NAMs was identified through cross screening during the course of our recent work in the mGlu5 NAM field (Fig. 2).20 The adamantyl carboxamides 7 and 8 were mGlu5 NAMs that were also found to behave as mGlu1 NAMs in a functional cell based assay.21 Our mGlu1 assay measures the ability of the compound to block the mobilization of calcium by an EC80 concentration of glutamate in cells expressing human mGlu1.
Figure 2.

mGlu1 NAM starting points for hit to lead optimization.
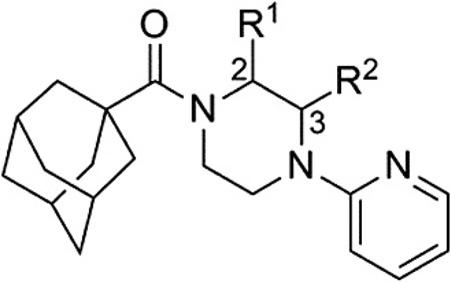
One attractive feature of this scaffold was the fact that robust chemistry would allow to us to rapidly vary three distinct portions of the chemotype and evaluate considerable synthetic diversity (Scheme 1). For instance, commercially available and enantiopure piperazines 9 (R1 = CH3) were useful for evaluating substitution of the piperazine core. For the purpose of evaluating SAR about the aryl portion of the chemotype (R2), substituted piperazines 9 were reacted with adamantoyl chloride to afford amide intermediates 10. Removal of the carbamate protecting group under acidic conditions gave amine intermediates 11. Target compounds were accessible from amines 11 through nucleophilic aromatic substitution reactions with aryl fluorides or Buchwald–Hartwig22 amination reactions with suitable aryl halides.23 For evaluation of SAR around the amide portion of the scaffold, a simple reorganization of the synthetic transformations was required. Reaction of amines 9 with either 2-fluoropyridine or 2-bromopyridine under appropriate conditions provided intermediate 12. Following cleavage of the protecting group, amines 13 were readily converted to the target amide compounds using well established methods.24
Scheme 1.

Reagents and conditions: (a) 1-adamantoyl chloride, DIEA, CH2Cl2; (b) HCl, MeOH, dioxanes; (c) R2F, NMP, μ wave, 250 °C, 10 min; (d) R2X (X = Cl, Br, or I), Pd2(dba)3 or Pd(OAc)2, Xantphos, NaOtBu or Cs2CO3, dioxanes, μ wave, 120 °C, 10 min or 100 °C, 18 h; (e) R3COCl, DIEA, CH2Cl2; (f) R3CO2H, HATU, DIEA, CH2Cl2.
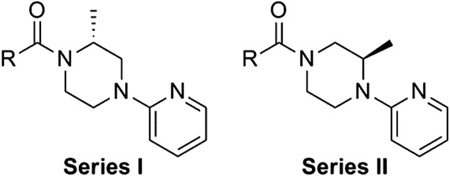
Evaluation of methyl substituted piperazines was one of our first areas of investigation (Table 1). New compounds were prepared with the 2-pyridyl group found in hit 7 (14–17). Evaluation of these enantiopure analogs yielded some interesting results. A preference with regard to the mGlu1 NAM activity was observed for one enantiomer versus the other in the case of both the 2-methyl and 3-methyl analogs. With the 2-methyl analogs a 15- fold preference was observed for the R-enantiomer (15 vs 14), while with the 3-methyl analogs a sixfold preference was observed for the R-enantiomer (17 vs 16). Furthermore, both 15 and 17 were approximately fourfold more potent than the unsubstituted comparator 7. Moreover, an equally substantial discovery was noted upon testing of 15 and 17 in our functional mGlu5 assay.25 Both compounds were only weak antagonists in this assay (IC50 >10 ¼M***). In short order, we had moved from a hit compound with little to no group I mGlu selectivity to two compounds with more than 60-fold selectivity for mGlu1 versus mGlu5.
Table 1.
Chiral piperazine analogs
 | |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | mGlu1 pIC50a (±SEM) | mGlu1 IC50 (nM) | % Glu maxa,b (±SEM) |
| 7 | H | H | 6.20 ± 0.21 | 630 | 1.1 ± 0.8 |
| 14 | (S)-CH3 | H | 5.62 ± 0.18 | 2420 | −1.4 ± 2.5 |
| 15 | (R)-CH3 | H | 6.79 ± 0.15 | 161 | 2.5 ± 0.5 |
| 16 | H | (S)-CH3 | 6.01 ± 0.13 | 988 | 1.8 ± 1.0 |
| 17 | H | (R)-CH3 | 6.78 ± 0.14 | 166 | 2.0 ± 0.4 |
Calcium mobilization mGlu1 assay; values are average of n≥3.
Amplitude of response in the presence of 30 µM test compound as a percentage of maximal response (100 µM glutamate); average of n≥3.
We next chose to evaluate SAR around the amide portion of the chemotype in the context of both the (R)-2-methyl piperazine (series I) and the (R)-3-methyl piperazine (series II) cores (Table 2). A number of analogs (18–35) were prepared in an attempt to identify replacements for the 1-adamantyl group. This was not generally successful; however, there were some notable exceptions. In the context of the (R)-2-methyl piperazine but not the (R)-3-methyl piperazine, the cyclooctyl (26) and the trans-(tert-butyl)cyclohexyl (30) groups demonstrated weak to moderate NAM activity against mGlu1. Additionally, the bicyclo[3.3.1]nonan-3-yl (32) group was essentially equipotent to 15, though this was again not effective in the case of the (R)-3-methyl piperazine (33 vs 17). It has been reported that the cubyl group can function as an effective and less lipophilic isosteric replacement for the adamantyl group.26 Unfortunately, such a modification was not effective in our chemotype (34 and 35).
Table 2.
Amide SAR
 | |||||
|---|---|---|---|---|---|
| Compd | Series | R | mGlu1 pIC50a (±SEM) | mGlu1 IC50 (nM) | % Glu maxa,b (±SEM) |
| 15 | I | 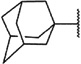 |
6.79 ± 0.15 | 161 | 2.5 ± 0.5 |
| 17 | II | 6.78 ± 0.14 | 166 | 2.0 ± 0.4 | |
| 18 | I | <4.5 | >30,000 | — | |
| 19 | II | <4.5 | >30,000 | — | |
| 20 | I | <4.5 | >30,000 | — | |
| 21 | II | <4.5 | >30,000 | — | |
| 22 | I | <5.0c | >10,000 | 51.8 ± 3.3 | |
| 23 | II | <4.5 | >30,000 | — | |
| 24 | I | 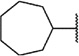 |
<5.0c | >10,000 | 29.9 ± 2.1 |
| 25 | II | <5.0c | >10,000 | 62.6 ± 3.3 | |
| 26 | I | 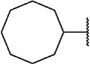 |
5.18 ± 0.06 | 6630 | 0.0 ± 8.2 |
| 27 | II | <5.0c | >10,000 | 52.9 ± 3.3 | |
| 28 | I | <5.0c | >10,000 | 32.1 ± 3.1 | |
| 29 | II | <4.5 | >30,000 | — | |
| 30 | I | 5.44 ± 0.08 | 3610 | 0.3 ± 0.4 | |
| 31 | II | <5.0c | >10,000 | 44.1 ± 3.7 | |
| 32 | I | 6.78 ± 0.11 | 167 | 1.3 ± 0.2 | |
| 33 | II | <5.0c | >10,000 | 12.2 ± 10.6 | |
| 34 | I | <5.0c | >10,000 | 62.7 ± 9.6 | |
| 35 | II | <4.5 | >30,000 | — | |
Calcium mobilization mGlu1 assay; values are average of n≥3.
Amplitude of response in the presence of 30 µM test compound as a percentage of maximal response (100 µM glutamate); average of n≥3.
CRC does not plateau.
Our attention next moved from the amide to the aryl portion of the template (Table 3). Using an approach similar to that previously described, we again observed only weak antagonists (37, 41, 43, 45) and a compound (39) with no activity at the top concentration tested (30 µM) in the context of the (R)-3-methyl piperazine. Fortunately, the same was not observed with the (R)-2- methyl piperazine analogs. Though 4-pyridyl analog 38 demonstrated only weak mGlu1 NAM activity, 3-pyridyl analog 36 exhibited good potency. Good to moderate potency was also noted with pyrimidines 40 and 44 and pyrazine 42. Finally, the 2-cyanophenyl analog 46 had potency within twofold of that observed with 15 and represented an improvement over the unsubstituted comparator piperazine 8 (Fig. 2).
Table 3.
First generation aryl SAR
 | |||||
|---|---|---|---|---|---|
| Compd | Series | R | mGlu1 pIC50a (±SEM) | mGlu1 IC50 (nM) | % Glu maxa,b (±SEM) |
| 15 | I | 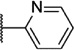 |
6.79 ± 0.15 | 161 | 2.5 ± 0.5 |
| 17 | II | 6.78 ± 0.14 | 166 | 2.0 ± 0.4 | |
| 36 | I | 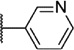 |
6.22 ± 0.06 | 596 | 1.8 ± 0.2 |
| 37 | II | <5.0c | >10,000 | 32.3 ± 5.2 | |
| 38 | I | <5.0c | >10,000 | 49.2 ± 4.5 | |
| 39 | II | <4.5 | >30,000 | — | |
| 40 | I | 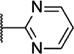 |
6.15 ± 0.07 | 714 | 0.9 ± 0.4 |
| 41 | II | <5.0 | >10,000 | 17.0 ± 5.7 | |
| 42 | I | 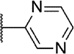 |
5.93 ± 0.08 | 1190 | −0.3 ± 0.8 |
| 43 | II | <5.0 | >10,000 | 34.0 ± 5.1 | |
| 44 | I | 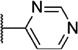 |
5.88 ± 0.07 | 1330 | −1.0 ± 2.2 |
| 45 | II | <5.0 | >10,000 | 34.7 ± 7.2 | |
| 46 | I | 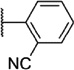 |
6.58 ± 0.06 | 264 | 2.1 ± 0.3 |
| 47 | II | 5.61 ± 0.09 | 2460 | 5.3 ± 1.7 | |
Calcium mobilization mGlu1 assay; values are average of n≥3.
Amplitude of response in the presence of 30 µM test compound as a percentage of maximal response (100 µM glutamate); average of n≥3.
CRC does not plateau.
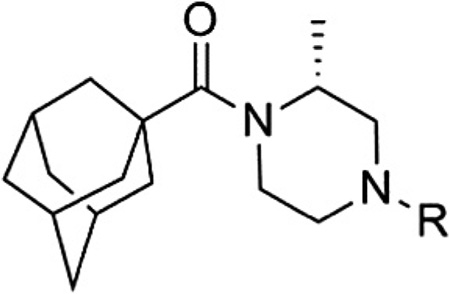
Having seen little progress in identifying improved analogs with the (R)-3-methyl piperazine core, we chose to focus our remaining efforts on attempting to improve upon the results observed with (R)-2-methyl piperazine analogs 15 and 46 (Table 4). Knowing that very subtle structural modifications can dramatically affect activity with allosteric modulators, we prepared fluorinated analogs of 15 (48–50). Though all three compounds exhibited good potency, 6-fluoropyridine 48 was the most potent and only slightly less potent than 15.We also prepared heteroaryl analogs (51–54) of 46 as a means to reduce lipophilicity. Most of these derivatives demonstrated good (51 and 53) to moderate (52) potency; however, analog 54 represented a nearly threefold improvement relative to 46 and the most potent compound from within the chemotype identified to date.
Table 4.
Second generation aryl SAR
 | |||||
|---|---|---|---|---|---|
| Compd | R | mGlu1 pIC50a (±SEM) | mGlu1 IC50 (nM) | % Glu maxa,b (±SEM) | |
| 15 | 6.79 ± 0.15 | 161 | 2.5 ± 0.5 | ||
| 48 | 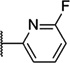 |
6.59 ± 0.11 | 255 | 2.0 ± 0.4 | |
| 49 | 6.38 ± 0.07 | 414 | 1.3 ± 0.9 | ||
| 50 | 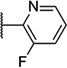 |
6.15 ± 0.05 | 712 | 2.4 ± 0.7 | |
| 46 | 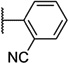 |
6.58 ± 0.06 | 264 | 2.1 ± 0.3 | |
| 51 | 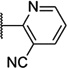 |
6.46 ± 0.14 | 344 | 3.3 ± 1.0 | |
| 52 | 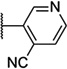 |
5.86 ± 0.04 | 1400 | −0.8 ± 3.3 | |
| 53 | 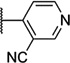 |
6.40 ± 0.06 | 398 | 0.3 ± 2.2 | |
| 54 | 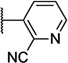 |
7.01 ± 0.03 | 99 | 2.8 ± 0.3 | |
Calcium mobilization mGlu1 assay; values are average of n≥3.
Amplitude of response in the presence of 30 µM test compound as a percentage of maximal response (100 µM glutamate); average of n≥3.
Calculation of the ligand-lipophilicity efficiency (LLE) values for our most potent analogs identified analog 54 as the compound with the best balance of potency and lipophilicity, two factors linked to drug-likeness (Table 5).27 We also decided to examine these same compounds for their propensity to non-specifically bind to rat plasma proteins.28 Binding to proteins can limit the amount of drug available to interact with the target, and thus impact efficacy in an in vivo setting. Since we are primarily interested in CNS applications of an mGlu1 NAM, we also chose to further profile interesting compounds by measuring their binding to rat brain homogenates. Not surprisingly, the measured fraction unbound tracked well with the calculated logP values. In the instances where brain homogenate binding was measured, there was reduced fraction unbound in brain homogenates relative to plasma. Our experience is that such a relationship between the unbound fraction in brain homogenates versus plasma is often but not always the case. Gratifyingly, the most potent analog 54 also demonstrated an increased fraction unbound compared to most of the other compounds tested.
Table 5.
Protein binding results
| Compd | cLogPa | mGlu1 IC50 (nM) | LLEb | Rat PPBc (Fu) | Rat BHBc (Fu) |
|---|---|---|---|---|---|
| 15 | 4.49 | 161 | 2.30 | 0.016 | 0.011 |
| 17 | 4.49 | 166 | 2.29 | 0.017 | 0.008 |
| 46 | 4.76 | 264 | 1.82 | 0.003 | — |
| 48 | 4.74 | 255 | 1.85 | 0.006 | — |
| 49 | 4.65 | 414 | 1.73 | 0.012 | — |
| 51 | 4.22 | 344 | 2.24 | 0.016 | — |
| 53 | 3.51 | 398 | 2.89 | 0.081 | — |
| 54 | 3.60 | 99 | 3.41 | 0.071 | 0.016 |
Calculated using ADRIANA. Code (www.molecular-networks.com).
LLE (ligand-lipophilicity efficiency) = pIC50—cLogP.
Fu = fraction unbound.
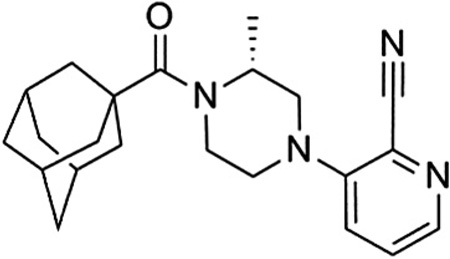
Compound 54 (VU0469650) was also examined in cell based functional assays for its selectivity against the other members of the mGlu family and was determined to be inactive against mGlu2–8 at 10 µM.29 Additionally, we tested the compound in a functional cell based assay against rat mGlu1 and found little difference across species.30 With over 100-fold selectivity against the other mGlus, we also wanted to assess the selectivity of the compound against a wider variety of targets. As such, we submitted the compound to a commercially available radioligand binding assay panel of 68 clinically relevant GPCRs, ion channels, kinases, and transporters,31 and no significant responses were found at a concentration of 10 µM.32
Given the results summarized herein, we chose to evaluate VU0469650 in a rat pharmacokinetic study (Table 6).33 In order to understand the rate of metabolism in vivo, a study was conducted using intravenous (IV) dosing. VU0469650 exhibited moderate to high clearance, a moderate volume of distribution, and a half-life of approximately 1 hour. We reasoned that such a clearance profile was supportive of further studies and proceeded to evaluate the compound in a plasma-brain level experiment using intraperitoneal (IP) dosing. Given the moderate to high clearance observed in the IV experiment, the IP dosing route was chosen in an attempt to maximize exposure. The 30 min post-dose time point was selected as it would be a relevant time point for many behavioral experiments of interest. We were pleased to see that the CNS penetration for VU0469650 was excellent and that the absolute levels in the brain were quite good. Taking into account the aforementioned brain homogenate binding data, we estimate the unbound fraction in the brain to be 32 nM. If that is indeed the case, a threefold increase in dose (30 mg/kg) should provide unbound drug levels near the functional IC50 for mGlu1.
Table 6.
Rat PK results for 54 (VU0469650)
 | |||
|---|---|---|---|
| IV PKa,c | IP PKb,c | ||
| Dose | 0.2 mg/kg | Dose | 10 mg/kg |
| Half-life | 55 min | Time point | 30 min |
| MRT | 43 min | Plasma concd | 509 ng/mL |
| CL | 52 mL/min/kg | Brain concd | 727 ng/mL |
| VSS | 2.2 L/kg | B/P ratio | 1.4 |
Formulation = 10% EtOH, 40% PEG400, 50% DMSO.
Formulation = 10% Tween80 in water.
Male Sprague–Dawley rats (n = 2).
In conclusion, we have identified a new mGlu1 NAM tool compound from within a series of N-acyl-N′-arylpiperazines using a hit to lead optimization plan initiated from a cross screening hit. VU0469650 can be prepared using a simple three step procedure and readily available starting materials. The compound is also potent and selective with excellent CNS penetration in rats. Future plans include evaluation of the compound in rodent behavioral assays associated with CNS disorders linked to mGlu1 signaling. Results and observations from such studies will be the subject of future communications.
Acknowledgments
We thank Seaside Therapeutics (VUMC36176) for their support of our programs in the development of mGlu1 NAMs. We also thank Tammy S. Santomango for technical contributions with the protein binding assays.
References and notes
- 1.(a) Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]; (b) Conn PJ, Pin J-P. Annu. Rev. Pharmacol. Toxicol. 1997;37:205. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.(a) Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J. Med. Chem. 2012;55:1445. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Conn PJ, Christopolous A, Lindsley CW. Nat. Rev. Drug Disc. 2009;8:41. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bridges TM, Lindsley CW. ACS Chem. Biol. 2008;3:530. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]; (d) Ritzén A, Mathiesen JM, Thomsen C. Basic Clin. Pharmacol. Toxicol. 2005;97:202. doi: 10.1111/j.1742-7843.2005.pto_156.x. [DOI] [PubMed] [Google Scholar]; (e) Kew JNC. Pharmacol. Ther. 2004;104:233. doi: 10.1016/j.pharmthera.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 3.(a) Owen DR. ACS Chem. Neurosci. 2011;2:394. doi: 10.1021/cn2000124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lesage A, Steckler T. Eur. J. Pharmacol. 2010;639:2. doi: 10.1016/j.ejphar.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kotlinska JH, Bochenski M, Danysz W. Eur. J. Pharmacol. 2011;670:154. doi: 10.1016/j.ejphar.2011.09.025. [DOI] [PubMed] [Google Scholar]; (b) Dravolina OA, Zakharova ES, Shekunova EV, Zvartau EE, Danysz W, Bespalov AY. Neuropharmacology. 2007;52:263. doi: 10.1016/j.neuropharm.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 5.(a) Satow A, Maehara S, Ise S, Hikichi H, Fukushima M, Suzuki G, Kimura T, Tanaka T, Ito S, Kawamoto H, Ohta H. J. Pharmacol. Exp. Ther. 2008;326:577. doi: 10.1124/jpet.108.138107. [DOI] [PubMed] [Google Scholar]; (b) Rorick-Kehn LM, Hart JC, McKinzie DL. Psychopharmacology. 2005;183:226. doi: 10.1007/s00213-005-0169-2. [DOI] [PubMed] [Google Scholar]
- 6.(a) Shannon HA, Peters SC, Kingston AE. Neuropharmacology. 2005;49:188. doi: 10.1016/j.neuropharm.2005.05.010. [DOI] [PubMed] [Google Scholar]; (b) Barton ME, Peters SC, Shannon HE. Epilepsy Res. 2003;56:17. doi: 10.1016/j.eplepsyres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bennett CE, Burnett DA, Greenlee WJ, Knutson CE, Korakas P, Li C, Tulshian D, Wu W-L, Bertorelli R, Fredduzzi S, Grilli M, Lozza G, Reggiani A, Veltri A. Bioorg. Med. Chem. Lett. 2012;22:1575. doi: 10.1016/j.bmcl.2011.12.131. [DOI] [PubMed] [Google Scholar]; (b) Sasikumar TK, Qiang L, Burnett DA, Greenlee WJ, Li C, Heimark L, Pramanik B, Grilli M, Bertorelli R, Lozza G, Reggiani A. Bioorg. Med. Chem. Lett. 2009;19:3199. doi: 10.1016/j.bmcl.2009.04.104. [DOI] [PubMed] [Google Scholar]; (c) Wu W-L, Burnett DA, Domalski M, Greenlee WJ, Li C, Bertorelli R, Fredduzzi S, Lozza G, Veltri A, Reggiani A. J. Med. Chem. 2007;50:5550. doi: 10.1021/jm070590c. [DOI] [PubMed] [Google Scholar]; (d) Varty GB, Grilli M, Forlani A, Fredduzzi S, Grzelak ME, Guthrie DH, Hodgson RA, Lu SX, Nicolussi E, Pond AJ, Parker EM, Hunter JC, Higgins GA, Reggiani A, Bertorelli R. Psychopharmacology. 2005;179:207. doi: 10.1007/s00213-005-2143-4. [DOI] [PubMed] [Google Scholar]
- 8.Hikichi H, Nishino M, Fukushima M, Satow A, Maehara S, Kawamoto H, Ohta H. Eur. J. Pharmacol. 2010;639:99. doi: 10.1016/j.ejphar.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 9.Steckler T, Lavreysen H, Oliveira AM, Aerts N, Van Craenendonck H, Prickaerts J, Megens A, Lesage ASJ. Psychopharmacology. 2005;179:198. doi: 10.1007/s00213-004-2056-7. [DOI] [PubMed] [Google Scholar]
- 10.Xie X, Ramirez DR, Lasseter HC, Fuchs RA. Psychopharmacology. 2010;208:1. doi: 10.1007/s00213-009-1700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Achat-Mendes C, Platt DM, Spealman RD. J. Pharmacol. Exp. Ther. 2013;343:214. doi: 10.1124/jpet.112.196295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng GZ, Bhatia P, Kolasa T, Patel M, El Kouhen OF, Chang R, Uchic ME, Miller L, Baker S, Lehto SG, Honore P, Wetter JM, Marsh KC, Moreland RB, Brioni JD, Stewart AO. Bioorg. Med. Chem. Lett. 2006;16:4936. doi: 10.1016/j.bmcl.2006.06.053. [DOI] [PubMed] [Google Scholar]
- 13.Zhu CZ, Baker S, El-Kouhen O, Lehto S, Hollingsworth PR, Gauvin DM, Hernandez G, Zheng GZ, Chang R, Moreland RB, Stewart AO, Brioni JD, Honore P. Eur. J. Pharmacol. 2008;580:314. doi: 10.1016/j.ejphar.2007.09.047. [DOI] [PubMed] [Google Scholar]
- 14.Mantell SJ, Gibson KR, Osborne SA, Maw GN, Rees H, Dodd PG, Greener B, Harbottle GW, Million WA, Poinsard C, England S, Carnell P, Betts AM, Monhemius R, Prime RL. Bioorg. Med. Chem. Lett. 2009;19:2190. doi: 10.1016/j.bmcl.2009.02.106. [DOI] [PubMed] [Google Scholar]
- 15.Kohara A, Toya T, Tamura S, Watabiki T, Nagakura Y, Shitaka Y, Hayashibe S, Kawabata S, Okada M. J. Pharmacol. Exp. Ther. 2005;315:163. doi: 10.1124/jpet.105.087171. [DOI] [PubMed] [Google Scholar]
- 16.Satoh A, Nagatomi Y, Hirata Y, Ito S, Suzuki G, Kimura T, Maehara S, Hikichi H, Satow A, Hata M, Ohta H, Kawamoto H. Bioorg. Med. Chem. Lett. 2009;19:5464. doi: 10.1016/j.bmcl.2009.07.097. [DOI] [PubMed] [Google Scholar]
- 17.Ito S, Hirata Y, Nagatomi Y, Satoh A, Suzuki G, Kimura T, Satow A, Maehara S, Hikichi H, Hata M, Ohta H, Kawamoto H. Bioorg. Med. Chem. Lett. 2009;19:5310. doi: 10.1016/j.bmcl.2009.07.145. [DOI] [PubMed] [Google Scholar]
- 18.(a) Martino JJ, Wall BA, Mastrantoni E, Wilimczyk BJ, La Cava SN, Degenhardt K, White E, Chen S. Oncogene. 2012 doi: 10.1038/onc.2012.471. http://dx.doi.org/10.1038/onc.2012.471. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ohtani Y, Harada T, Funasaka Y, Nakao K, Takahara C, Abdel-Daim M, Sakai N, Saito N, Nishigori C, Aiba A. Oncogene. 2008;27:7162. doi: 10.1038/onc.2008.329. [DOI] [PubMed] [Google Scholar]; (c) Namkoong J, Shin S-S, Lee HJ, Marín YE, Wall BA, Goydos JS, Chen S. Cancer Res. 2007;67:2298. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- 19.Speyer CL, Smith JS, Banda M, DeVries JA, Mekani T, Gorski DH. Breast Cancer Res. Treat. 2012;132:565. doi: 10.1007/s10549-011-1624-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez AL, Williams R, Zhou Y, Lindsley SR, Le U, Grier MD, Weaver CD, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2009;19:3209. doi: 10.1016/j.bmcl.2009.04.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.HEK 293A TREx cells stably expressing human mGlu1 were plated in black-walled, clear-bottomed, poly-d-lysine coated 384-well plates in 20 µL of assay medium (DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate) at a density of 20 K cells/well. The cells were induced with 50 ng/mL tetracycline and grown overnight at 37 °C in the presence of 5% CO2. The next day, medium was exchanged to assay buffer (Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid) using an ELX405 microplate washer, leaving 20 µL/well, followed by addition of 20 µL/well 2× Fluo-4 AM (2.3 µMfinal) indicator dye (prepared as a DMSO stock and mixed in a 1:1 ratio with pluronic acid F-127) in assay buffer, and incubation for 45 m at 37 °C. The dye was then exchanged to assay buffer using an ELX405, leaving 20 µL/well. Ca2+ flux was measured using the Functional Drug Screening System (FDSS7000, Hamamatsu, Japan). Compounds were serially diluted 1:3 into 10 point concentration response curves and transferred to daughter plates using the Echo acoustic plate reformatter. Compounds were diluted into assay buffer to a 2× stock using a Thermo Fisher Combi which was applied to cells at t = 3 s. Cells were incubated with the test compounds for 2.3 min and then stimulated with an EC20 concentration of glutamate; 1.9 min later an EC80 concentration of glutamate was added and readings taken for an additional 1.7 min. Data were collected at 1 Hz. Concentration response curves were generated using a four point logistical equation with XLfit curve fitting software for Excel.
- 22.(a) Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schlummer B, Scholz U. Adv. Synth. Catal. 2004;346:1599. [Google Scholar]
- 23.Synthesis of 54 (VU0469650) is representative of route I. (R)-4-N-Boc-2-methyl piperazine (665 mg, 3.32 mmol), 1-adamantoyl chloride (990 mg, 4.98 mmol), and DIEA (1.15 mL, 6.64 mmol) were dissolved in 2.0 mL of dichloromethane, and the reaction was monitored by LCMS. Upon completion, 5.0 mL of MeOH was added, and the mixture was concentrated in vacuo. The residue was purified by flash chromatography. The purified product was immediately dissolved in 1.0 mL of MeOH and excess 4 N HCl in dioxanes (4.0 mL, 16 mmol). Once the protecting group had cleaved as judged by LCMS, an aqueous solution of saturated sodium bicarbonate was added until the pH was basic. The mixture was extracted with dichloromethane, and the organic layers were concentrated in vacuo to afford 814 mg (94% yield) of adamantan-1-yl((R)-2-methylpiperazin-1-yl)methanone as a white solid. Adamantan-1-yl((R)-2-methylpiperazin-1-yl)methanone (355 mg, 1.35 mmol) and 2-cyano-3-fluoropyridine (1.42 mL, 5.41 mmol) were dissolved in 4.0 mL of NMP in a microwave reaction vial and heated in the microwave for 10 min at 250 °C. The reaction mixture was purified via reverse phase chromatography (0.1% TFA in H2O/MeCN). The fractions containing the product were combined and neutralized by the addition of aqueous saturated sodium bicarbonate. The mixture was extracted with dichloromethane, and the organic layers were combined and concentrated in vacuo. The residue was dissolved in 1.0 mL of MeOH and 4 N HCl in dioxanes (3.0 mL, 12 mmol) and stirred for 2 h. The mixture was concentrated in vacuo to afford 212 mgs (39% yield) of the HCl salt as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (dd, J = 4.3, 1.4 Hz, 1H), 7.67 (dd, J = 8.6, 1.3 Hz, 1H), 7.62 (dd, J = 8.6, 4.4 Hz, 1H), 4.74 (m, 1H), 4.35 (d, J = 13.7 Hz, 1H), 3.44 (t, J = 12.1 Hz, 2H), 3.27 (m, 1H), 2.93 (dd, J = 12.1, 3.5 Hz, 1H), 2.84 (td, J = 12.8, 2.8 Hz, 1H), 2.02–1.83 (m, 9H), 1.75–1.60 (m, 6H), 1.32 (d, J = 6.7 Hz, 3H). HRMS (TOF, ES+) C22H28N4O [M+H]+ calculated 365.2341; found 365.2344. −45.7° (c = 6.0, MeOH).
- 24.Synthesis of 17 is representative of route II. (R)-4-N-Boc-2-methyl piperazine (490 mg, 2.44 mmol), 2-bromopyridine (257 µL, 2.69 mmol), Xantphos (212 mg, 0.366 mmol), Pd2(dba)3 (112 mg, 0.112 mmol), and NaOtBu (705 mg, 7.34 mmol) were added to 5.0 mL of dioxanes in a microwave reaction vial. The reaction was heated for 10 min in the microwave at 120 °C and monitored by LCMS. Upon completion, the mixture was cooled and filtered through a pad of celite and concentrated in vacuo. The crude material was purified by flash chromatography and immediately dissolved in 1.0 mL of MeOH with stirring. 4 N HCl in dioxanes (4.0 mL, 16 mmol) was added. Once the protecting group had cleaved as judged by LCMS, an aqueous solution of saturated sodium bicarbonate was added until the pH was basic. The mixture was extracted with dichloromethane, and the organic layers were concentrated in vacuo to afford 260 mg (60%) of (R)-2-methyl-1-(pyridin-2-yl)piperazine as a colorless liquid. (R)-2-Methyl-1-(pyridin-2-yl)piperazine (260 mg, 1.47 mmol), 1-adamantoyl chloride (437 mg, 2.20 mmol), and DIEA (508 µL, 2.93 mmol) were dissolved in 1.0 mL of dichloromethane and monitored by LCMS. Upon completion, 5.0 mL of MeOH was added, and the mixture was concentrated in vacuo. The crude product was purified via reverse phase chromatography (0.1% TFA in H2O/MeCN). The fractions containing the product were combined and neutralized by the addition of aqueous saturated sodium bicarbonate. The mixture was extracted with dichloromethane, and the organic layers were combined and concentrated in vacuo to afford 200 mg (40% yield) of the product as a white solid. 1H NMR 1H NMR (400 MHz, DMSOd6) δ 8.12–8.07 (m, 1H), 7.52 (t, J = 8.0 Hz, 1H), 6.79 (Rotamer 1, d, J = 8.6, 0.65 H) 6.73 (Rotamer 2, d, J = 8.6 Hz, 0.35 H), 6.64–6.59 (m, 1H), 4.64 (Rotamer 1, m, 0.65 H), 4.49 (Rotamer 2, m, 0.35 H), 4.4–4.22 (m, 1H), 4.16–3.95 (m, 2H), 3.27 (m, 2.35 H), 2.80–2.69 (m, 0.65H), 2.02–1.85 (m, 9H), 1.75–1.62 (m, 6H), 1.12 (Rotamer 1, d, J = 6.6 Hz, 2H), 0.95 (Rotamer 2, d, J = 6.6 Hz, 1H); HRMS (TOF, ES+) C21H30N3O [M+H]+ calculated 340.2389; found 340.2389. −20.2° (c = 6.0, MeOH).
- 25.HEK293A cells expressing rat mGlu5 were cultured and plated. The cells were loaded with a Ca2+ sensitive fluorescent dye and the plates were washed and placed in the Functional Drug Screening System (Hamamatsu). Test compound was applied to cells 3 s after baseline readings were taken. Cells were incubated with the test compounds for 140 s and then stimulated with an EC20 concentration of glutamate; 60 s later an EC80 concentration of agonist was added and readings taken for an additional 40 s. Allosteric modulation by the compounds was measured by comparing the amplitude of the responses at the time of glutamate addition plus and minus test compound. For a more detailed description of the assay, see Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:4098. doi: 10.1016/j.bmcl.2008.05.091.
- 26.Gunosewoyo H, Guo JL, Bennett MR, Coster MJ, Kassiou M. Bioorg. Med. Chem. Lett. 2008;18:3720. doi: 10.1016/j.bmcl.2008.05.062. [DOI] [PubMed] [Google Scholar]
- 27.Leeson PD, Springthorpe B. Nat. Rev. Drug Disc. 2007;6:881. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 28.Binding to rat plasma proteins and brain homogenates were measured using equilibrium dialysis according to methods similar to those described in Kalvass JC, Maurer TS. Biopharm. Drug Dispos. 2002;23:327. doi: 10.1002/bdd.325.
- 29.mGlu selectivity assays are described in Noetzel MJ, Rook JM, Vinson PN, Cho H, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharmacol. 2012;81:120. doi: 10.1124/mol.111.075184.
- 30.Analogous to the method described in Ref.21 with the exception that HEK 293A TREx cells stably expressing rat mGlu1 were used.
- 31.LeadProfilingScreen®, Eurofins Panlabs, Inc. http://www.eurofinspanlabs.com.
- 32.Significant responses are defined as those that inhibited more than 50% of radioligand binding. In the case of VU0469650, no inhibition greater than 41% (adenosine A1 and sigma σ1) was observed.
- 33.Male Sprague–Dawley rats weighing between 250 and 300 g were purchased from Harlan Laboratories (Indianapolis, IN). VU0469650 was used as its mono-HCl salt for these studies. For the IV study, cannulated animals with catheters surgically implanted in the carotid artery and jugular vein were used. Blood collections via the carotid artery were performed at 0.033, 0.117, 0.25, 0.5, 1, 2, 4, 7, and 24 h after administration via the jugular vein catheter. For the IP study, rats were euthanized and decapitated 30 min after compound administration, and both blood and brain samples were collected. Following protein precipitation, the supernatants of all plasma and brain homogenate samples were analyzed by means of LC–MS/MS. All animal studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee.