
FIGURE 6.
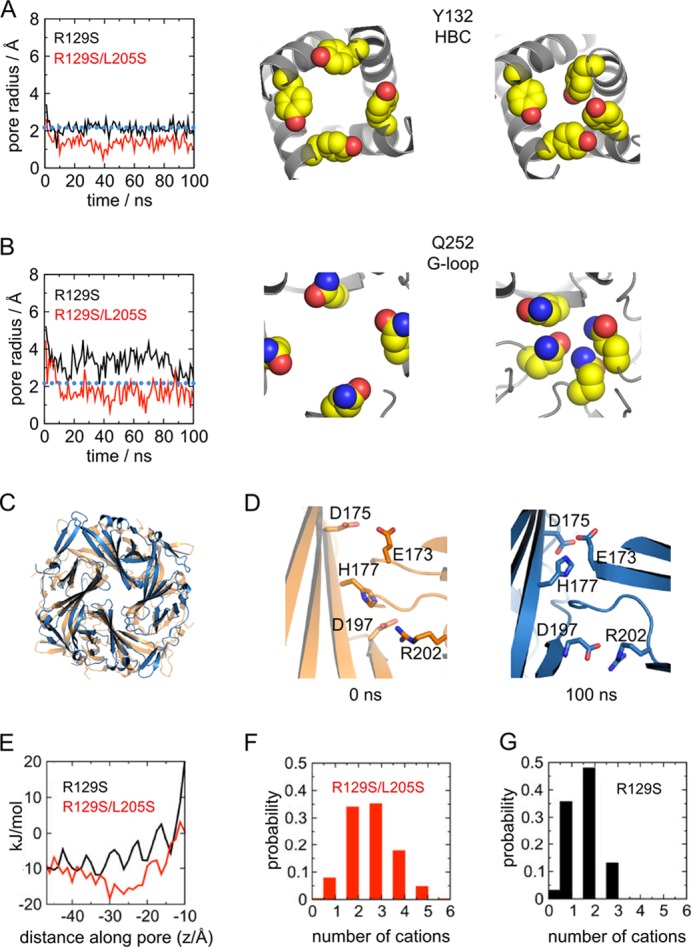
Effect of S205L on stability of the open state conformation. A, molecular dynamics simulations of the single-mutant (R129S) and double-mutant (R129S/L205S) structures, i.e. where residues in the mutant structure were converted back to wild-type serine. Left, the pore radius, calculated by HOLE (40) along the pore axis, is shown for the HBC at Tyr132. The radius of a hydrated K+ ion is indicated by the dotted blue line. For times t = 0 ns and t = 100 ns, snapshots of the respective locations obtained in the R129S/L205S simulation are shown (right). B, as shown for A, but the radius was calculated at the position of the G-loop (Gln252). Although the G-loop closes only in the R129S/L205S simulation, the side chains of the HBC close the pore in both simulations. C, overlay of the CTD of the S129R/S205L mutant structure with a snapshot at t = 100 ns of the R129S/L205S simulation. No significant rotation of the CTD is observed. D, comparison of the intersubunit interface in the R129S/L205S simulation. At 0 ns, the side chains of Glu173 and Asp175 point toward the pore, and His177 points down, interacting with Asp197 and Arg202. At 100 ns, His177 has adopted the closed state conformation, and the side chains of Glu173 and Asp175 turn into the outward-facing position, also adopting the closed state orientation. E, APBS profiles of S129R and S129R/S205L. The Poisson-Boltzmann potential energy of a K+ ion, calculated along the pore axis of the CTD, is markedly lower in S129R/S205L due to reorientation of Glu173 and Asp175, rendering passage of K+ through the CTD pore more energetically favorable. F, number of cations (Na+) in the cytoplasmic pore (−16 Å ≤ z ≤ −37 Å) during the R129S/L205S simulation. G, number of cations (Na+) in the cytoplasmic pore during the R129S simulation.