

Abstract
Autoimmune pancreatitis (AIP) is a form of chronic pancreatitis that is categorized as type 1 or type 2 according to the clinical profile. Type 1 AIP, which predominantly presents in a few Asian countries, is a hyper-IgG4-related disease. We report a case of IgG4-related AIP overlapping with Mikulicz’s disease and lymphadenitis, which is rare and seldom reported in literature. A 63-year male from Northeast China was admitted for abdominal distension lasting for one year. He presented symmetric swelling of the parotid and submandibular glands with slight dysfunction of salivary secretion for 6 mo. He had a 2-year history of bilateral submandibular lymphadenopathy without pain. He underwent surgical excision of the right submandibular lymph node one year prior to admission. He denied any history of alcohol, tobacco, or illicit drug use. Serological examination revealed high fasting blood sugar level (8.8 mmol/L) and high level of IgG4 (15.2 g/L). Anti-SSA or anti-SSB were negative. Computed tomography of the abdomen showed a diffusely enlarged pancreas with loss of lobulation. Immunohistochemical stain for IgG4 demonstrated diffuse infiltration of IgG4-positive plasma cells in labial salivary gland and lymph node biopsy specimens. The patient received a dose of 30 mg/d of prednisone for three weeks. At this three-week follow-up, the patient reported no discomfort and his swollen salivary glands, neck lymph node and pancreas had returned to normal size. The patient received a maintenance dose of 10 mg/d of prednisone for 6 mo, after which his illness had not recurred.
Keywords: IgG4-related disease, Type 1 autoimmune pancreatitis, Mikulicz’s disease, Lymphadenitis
Core tip: We report a rare case of a 63-year-old Northeast Chinese man who suffered from IgG4-related disease (RD) which involved the salivary glands, lymph node and pancreas. The patient responded promptly to prednisone therapy. Further identification and characterization of such cases is required to elucidate the prevalence and clinical features of IgG4-RD in China.
INTRODUCTION
Autoimmune pancreatitis (AIP) is an uncommon form of chronic pancreatitis that was first described in Japan in 1995[1]. Two subtypes of AIP have been so far recognized[2,3]. Type 1 AIP is related to high levels of serum IgG4, dense periductal lymphoplasmacytic infiltration and obliterative venulitis, while type 2 AIP is an IgG4-independent pancreatic disease that is characterized by neutrophilic infiltration into the epithelium of the pancreatic duct[3,4]. Although 20%-40% of AIP cases are type 2 in the United States and Europe, most cases of AIP in Japan and Korea are type 1, and type 2 is quite rare[5]. The prevalence and clinical features of AIP in China has not been fully clarified so far.
Mikulicz’s disease (MD) refers to bilateral and symmetrical swelling of the lacrimal, parotid, and submandibular glands. Based on histological similarities reported by Morgan et al[6] in 1953, MD was considered a subtype of Sjögren’s syndrome (SS). However, several recent reports from Japan have revealed that MD is associated with elevated serum IgG4 levels and prominent infiltration of IgG4-positive plasmacytes[7,8]; these findings are distinct from those of SS and have resulted in the recognition of MD as a singular systemic IgG4-related plasmacytic disease[9].
In this report, we describe a case from Northeast China of IgG4-related autoimmune pancreatitis overlapping with Mikulicz’s disease and lymphadenitis. This rare clinical condition has seldom been reported in literature.
CASE REPORT
A 63-year male from Northeast China was admitted for abdominal distension lasting for one year. He presented symmetric swelling of the parotid, and submandibular glands with slight dysfunction of salivary secretion for 6 mo. He had a 2-year history of bilateral submandibular lymphadenopathy without pain. He underwent surgical excision of the swollen lymph node in the right submandibular region one year prior to hospital admission. The patient denied any history of alcohol, tobacco, or illicit drug use. On admission, his blood pressure was 136/88 mmHg, pulse rate was 72/min, and body temperature was 36.7 °C. On examination, he had bilateral swelling of the parotid and submandibular glands as well as left swelling of the submandibular lymph node. His mouth was dry. Abdominal examination revealed mild epigastric tenderness to deep palpation without rebound.
The laboratory test data on admission revealed an elevated neutrophil ratio of 76%, and an elevated fasting blood sugar level of 8.8 mmol/L. Serum amylase was 42 U/L and serum lipase was 65 U/L, both within normal limits. Serological testing for autoimmune function displayed high levels of IgG4 (15.2 g/L) and IgG (18.5 g/L), and negative values of anti-SSA and anti-SSB. A computed tomography (CT) scan of the abdomen revealed diffuse enlargement of the pancreas and loss of normal pancreatic lobulation, consistent with autoimmune pancreatitis (Figure 1A).
Figure 1.
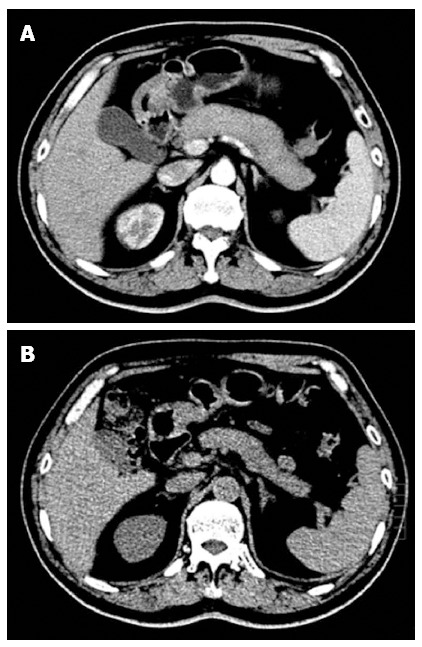
Typical imaging features of type 1 autoimmune pancreatitis. Computed tomography (CT) scan showing diffuse swelling of the pancreas with loss of lobulation (A), and a dramatic decrease in swelling of the pancreas after 3 wk of steroid treatment (B).
The patient underwent a minor labial salivary gland biopsy for a possible diagnosis of MD. Labial gland specimens stained with hematoxylin and eosin revealed significant infiltration of lymphocytoplasma cells in the patient, but no infiltration of these cells in a healthy individual (Figure 2A and B). Immunohistochemical staining showed numerous IgG4-positive plasmacytes in the labial gland of the patient, with a ratio of IgG4/IgG-positive plasmacytes of more than 50% (Figure 2C and D).
Figure 2.
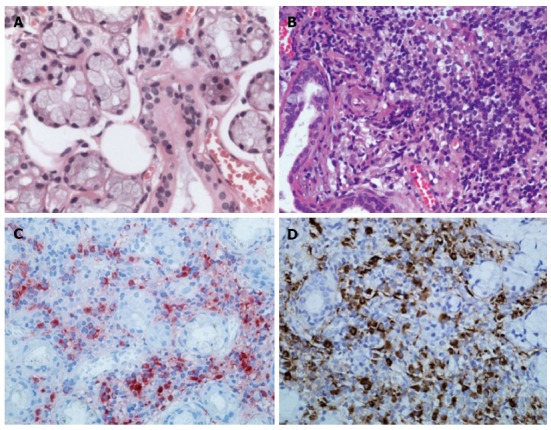
Histological findings of labial salivary gland specimens. A: Hematoxylin and eosin stain showing normal labial gland; B: Diffuse infiltration of lymphoplasma cells from the patient; C, D: Immunohistochemical staining for IgG4 (C) or IgG (D) in plasma cells from the patient, consistent with Mikulicz’s disease. Original magnification, × 400.
Since both autoimmune pancreatitis and MD meet the criteria for IgG4-related disease, we investigated the IgG4 status of the patient’s swollen lymph nodes. Lymph node specimens collected from the patient by excision of the right submandibular lymph node one year prior to admission ago were examined for IgG4 and IgG using immunohistochemistry. As shown in Figure 3, there were diffuse infiltrations of IgG4-positive plasma cells in the patient’s lymph node. The ratio of IgG4/IgG-positive cells was greater than 40% thus meeting the diagnostic criteria for IgG4-related lymphadenitis.
Figure 3.
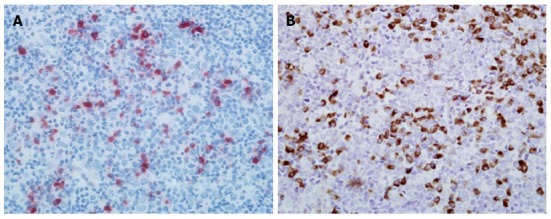
Histological findings of submandibular lymph node specimen. Immunohistochemical staining showing IgG4-positive plasma cells (A) and IgG-positive plasma cells (B) in lymph node sections of the patient. Original magnification, × 400.
On the 8th d after admission, the patient was diagnosed with IgG4-related systemic disease. He received 30 mg/d of prednisone for three days without any side effects, and was then discharged with the same steroid dose for the following 3 wk. At three-week follow-up the patient exhibited no signs of either a dry mouth or abdominal distension. His swollen glands including parotid and submandibular glands as well as left submandibular lymph node were no longer palpable. The enlarged pancreas had returned to its normal size (Figure 1B) and elevated morning glucose levels were within the normal range. The patient then received a long-term maintenance dose of 10 mg/d of prednisone after steroid tapering. At six-month follow-up, his illness had not recurred.
DISCUSSION
Yoshida et al[1] first proposed the concept of AIP in 1995 based on observations of patients who had hyper-γ globulinemia, various autoantibodies, lymphocytic infiltration into pancreatic tissue, and good steroid responsiveness. In 2002 Hamano et al[10] reported high serum IgG4 concentrations in Japanese AIP patients and abundant IgG4-producing plasma cell infiltration in pancreatic tissue. Some cases of AIP in Europe or America appear to represent an “idiopathic duct-centric chronic pancreatitis”, which are caused by neutrophilic granulocyte infiltration and are not related to IgG4[11]. In 2009, Sugumar et al[12] suggested that IgG4-related AIP should be named as type 1 and neutrophilic granulocyte lesions of AIP as type 2[12]. Over the last decade, international consensus diagnostic criteria for AIP were established to be applicable worldwide and to distinguish between the two types of AIP. The diagnosis of AIP can be usually made based on the presence of at least one of the five cardinal features (i.e., imaging, serology, other organ involvement, histology, and response to steroid therapy)[3]. However, sufficient biopsy specimens from the pancreas are difficult to obtain using standard procedures, except laparotomy. The clinical diagnostic criteria to establish type 1 AIP from Japan were the presence of pancreatic swelling together with high serum levels of IgG4 and/or prominent IgG4-producing plasma cell infiltration in pancreas tissue[11]. By contrast, in the case reported here, the patient presented a diffuse swelling of the pancreas with loss of lobulation, high serum IgG4 concentration, abundant IgG4-positive plasma cell infiltration into labial or lymph node tissues, and good steroid responsiveness, thus fully meeting multiple type 1 AIP diagnostic criteria[3,11]. Additionally, in this case report, three-week steroid treatment caused dramatic improvements in either exocrine insufficiency (abdominal distention) or endocrine insufficiency (elevated fasting blood sugar level) as well as dramatic reduction of the enlarged pancreas, suggesting that both exocrine and endocrine insufficiency might be reversible in IgG4-related type 1 AIP.
The first case of MD was reported by Mikulicz-Radecki in 1888, which was described as bilateral symmetrical enlargement of the salivary and lacrimal glands, and lymphocytic infiltration into lacrimal and salivary gland tissues[9]. Since 1953, when Morgan et al[6] found similarities in histology between MD and SS, MD was considered as a subtype of SS. However, this concept has been modified over the decade in light of compelling Japanese studies showing that MD is associated with elevated serum IgG4 levels and prominent infiltration of IgG4-positive plasmacytes into lacrimal and salivary glands[7,8]. Thus, a modern clinical concept of MD is that it is distinct from SS and is instead part of the spectrum of IgG4-RD[9]. Diagnostic criteria for IgG4-related MD were approved by the Japanese Sjögren’s Syndrome Society in 2008. According to these critera, IgG4-related MD is defined in the presence of persistent (≥ 3 mo), symmetrical swelling of the lacrimal, parotid and submandibular glands involving at least two pairs, together with either high serum levels of IgG4 (≥ 1.35 g/L) and/or marked IgG4-positive plasmacyte infiltration (≥ 50% IgG4-positive/IgG-positive cells in five high power fields) into lacrimal and salivary gland tissues[9]. In this study, the patient presented six-month symmetric swelling of the parotid and submandibular glands, elevated serum IgG4 levels, and prominent IgG4-positive plasmacyte infiltration into the labial gland tissue, which fully met the diagnostic criteria for IgG4-related MD[9]. Furthermore, both the swollen salivary glands and the dry mouth of the patient promptly recovered in response to prednisone therapy, indicating that IgG4-related MD is a reversible disorder which differs from SS.
Recently, IgG4-RD has been defined as a novel clinical entity with multi-organ involvement and associated abundant infiltration of IgG4-positive cells[11,13]. In this case, the clinical and histopathological features of the patient met diagnostic criteria for IgG4-related type 1 AIP and MD respectively. Additionally, the patient presented abundant infiltration of IgG4-positive plasma cells in lymph node tissue (> 40% IgG4/IgG-positive cells) and good steroid responsiveness, which fully met the diagnostic criteria for IgG4-related lymphadenitis[13].
In summary, we report a rare case of a 63-year-old Northeast Chinese man who suffered from IgG4-RD which involved the salivary glands, lymph node and pancreas. The patient responded promptly to prednisone therapy. Further identification and characterization of such cases is required to elucidate the prevalence and clinical features of IgG4-RD in China and their relationship to similar cases in Japan.
COMMENTS
Case characteristics
The patient presented symmetric swelling of the parotid and submandibular glands as well as a diffusely enlarged pancreas with loss of lobulation.
Clinical diagnosis
The authors report a rare case of a 63-year-old Northeast Chinese man who suffered from IgG4-related disease which involved the salivary glands, lymph node and pancreas.
Differential diagnosis
Immunohistochemical staining for IgG4 and IgG is the major method for differential diagnosis between IgG4-related disease and other diseases.
Laboratory diagnosis
Serological testing for autoimmune function displayed high levels of serum IgG4 and IgG, and negative values of anti-SSA and anti-SSB in the patient.
Imaging diagnosis
A computed tomography scan of the abdomen revealed diffuse enlargement of the pancreas and loss of normal pancreatic lobulation.
Pathological diagnosis
Immunohistochemical staining showed numerous IgG4-positive plasmacytes in labial gland and lymph node of the patient, with a ratio of IgG4/IgG-positive plasmacytes of more than 40% in both tissues.
Treatment
The patient received a dose of 30 mg/d of prednisone for three week, and a long-term maintenance dose of 10 mg/d of prednisone.
Term explanation
Mikulicz’s disease (MD) refers to bilateral and symmetrical swelling of the lacrimal, parotid, and submandibular glands. MD, as a singular systemic IgG4-related plasmacytic disease, was considered a subtype of Sjögren’s syndrome. IgG4-related disease (IgG4-RD) has been defined as a novel clinical entity with multi-organ involvement and associated abundant infiltration of IgG4-positive plasmacytes.
Experiences and lessons
The authors report a rare case of a 63-year-old Northeast Chinese man who suffered from IgG4-RD which involved the salivary glands, lymph node and pancreas. This rare clinical condition has seldom been reported in literature.
Peer review
Type 1 autoimmune pancreatitis is related to high levels of serum IgG4, dense periductal lymphoplasmacytic infiltration and obliterative venulitis. In this manuscript, the authors reported a interesting case of IgG4-related autoimmune pancreatitis overlapping with Mikulicz’s disease and lymphadenitis. This case is very rare and seldom reported in literature.
Footnotes
Supported by National Natural Scientific Foundation, No. 81070370, 81270544 (to Gao RP) and NIH 5R01AA016003 (to Brigstock D)
P- Reviewers: Lampertico P, Steenholdt C S- Editor: Wang JL L- Editor: A E- Editor: Zhang DN
References
- 1.Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561–1568. doi: 10.1007/BF02285209. [DOI] [PubMed] [Google Scholar]
- 2.Deshpande V, Gupta R, Sainani N, Sahani DV, Virk R, Ferrone C, Khosroshahi A, Stone JH, Lauwers GY. Subclassification of autoimmune pancreatitis: a histologic classification with clinical significance. Am J Surg Pathol. 2011;35:26–35. doi: 10.1097/PAS.0b013e3182027717. [DOI] [PubMed] [Google Scholar]
- 3.Kamisawa T, Ryu JK, Kim MH, Okazaki K, Shimosegawa T, Chung JB. Recent advances in the diagnosis and management of autoimmune pancreatitis: similarities and differences in Japan and Korea. Gut Liver. 2013;7:394–400. doi: 10.5009/gnl.2013.7.4.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sah RP, Chari ST, Pannala R, Sugumar A, Clain JE, Levy MJ, Pearson RK, Smyrk TC, Petersen BT, Topazian MD, et al. Differences in clinical profile and relapse rate of type 1 versus type 2 autoimmune pancreatitis. Gastroenterology. 2010;139:140–148; quiz 140-148. doi: 10.1053/j.gastro.2010.03.054. [DOI] [PubMed] [Google Scholar]
- 5.Kamisawa T, Notohara K, Shimosegawa T. Two clinicopathologic subtypes of autoimmune pancreatitis: LPSP and IDCP. Gastroenterology. 2010;139:22–25. doi: 10.1053/j.gastro.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 6.Morgan WS, Castleman B. A clinicopathologic study of Mikulicz’s disease. Am J Pathol. 1953;29:471–503. [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto M, Takahashi H, Sugai S, Imai K. Clinical and pathological characteristics of Mikulicz’s disease (IgG4-related plasmacytic exocrinopathy) Autoimmun Rev. 2005;4:195–200. doi: 10.1016/j.autrev.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Takano K, Yamamoto M, Takahashi H, Shinomura Y, Imai K, Himi T. Clinicopathologic similarities between Mikulicz disease and Küttner tumor. Am J Otolaryngol. 2010;31:429–434. doi: 10.1016/j.amjoto.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Himi T, Takano K, Yamamoto M, Naishiro Y, Takahashi H. A novel concept of Mikulicz’s disease as IgG4-related disease. Auris Nasus Larynx. 2012;39:9–17. doi: 10.1016/j.anl.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, Nakazawa K, Shimojo H, Kiyosawa K. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359:1403–1404. doi: 10.1016/s0140-6736(02)08359-9. [DOI] [PubMed] [Google Scholar]
- 11.Masaki Y, Kurose N, Umehara H. IgG4-related disease: a novel lymphoproliferative disorder discovered and established in Japan in the 21st century. J Clin Exp Hematop. 2011;51:13–20. doi: 10.3960/jslrt.51.13. [DOI] [PubMed] [Google Scholar]
- 12.Sugumar A, Klöppel G, Chari ST. Autoimmune pancreatitis: pathologic subtypes and their implications for its diagnosis. Am J Gastroenterol. 2009;104:2308–210; quiz 2311. doi: 10.1038/ajg.2009.336. [DOI] [PubMed] [Google Scholar]
- 13.Okazaki K, Uchida K, Ikeura T, Takaoka M. Current concept and diagnosis of IgG4-related disease in the hepato-bilio-pancreatic system. J Gastroenterol. 2013;48:303–314. doi: 10.1007/s00535-012-0744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]