

Abstract
The design and application of rational strategies that rely on accurate species identification are pivotal for effective vector control. When morphological identification of the target vector species is impractical, the use of molecular markers is required. Here we describe a non-coding, single-copy nuclear DNA fragment that contains a single-nucleotide polymorphism (SNP) with the potential to distinguish the important domestic Chagas disease vector, Rhodnius prolixus, from members of the four sylvatic Rhodnius robustus cryptic species complex. A total of 96 primer pairs obtained from whole genome shotgun sequencing of the R. prolixus genome (12,626 random reads) were tested on 43 R. prolixus and R. robustus s.l. samples. One of the seven amplicons selected (AmpG) presented a SNP, potentially diagnostic for R. prolixus, on the 280th site. The diagnostic nature of this SNP was then performed on 154 R. prolixus and R. robustus s.l. samples aimed at achieving the widest possible geographic coverage. The results of a 60% majority rule Bayesian consensus tree and a median-joining network constructed based on the genetic variability observed reveal the paraphyletic nature of the R. robustus species complex, with respect to R. prolixus. AmpG region is located in the fourth intron of the Transmembrane protein 165 gene, which seems to be in the R. prolixus X chromosome. Other possible chromosomal locations of the AmpG region in the R. prolixus genome are also presented and discussed.
Keywords: Rhodnius, cryptic species, SNP, nuclear DNA
1. Introduction
Chagas disease is transmitted through Trypanosoma cruzi infected feces of triatomine bugs (Hemiptera: Reduviidae) and is endemic in Latin America and in the Caribbean (Coura, 2007), ranking fourth in epidemiologic importance among all neglected diseases in those regions (Hotez et al., 2008). Although case incidence estimates show a 90% decline (from 500,000 to 50,000 new cases per year), 8 million people are still infected and 14,000 die every year (Senior, 2007). Domestic insect vectors are responsible for most of the disease transmission to humans. Because no vaccine is available, the elimination of household-infesting triatomines is the major goal of many Chagas disease control programs (Abad-Franch et al., 2010).
The design and implementation of any vector control intervention must rely on accurate species identification. However, the identification of Chagas disease vectors based on morphological characters alone can be technically challenging, since some groups of species are cryptic (i.e. isomorphic), and therefore not amenable to be distinguished in such a manner. This taxonomical issue is even more relevant when applied to the transmission of T. cruzi by Rhodnius prolixus and R. robustus s.l. to humans. Although morphologically indistinguishable, they play very different epidemiological roles: the former are efficient domestic vectors, and the latter are not, as all species of the complex are entirely sylvatic (Monteiro et al., 2003).
Rhodnius prolixus is one of the most important Chagas disease vectors in Latin America, particularly in Venezuela, Colombia, and Central America, due to its ability to efficiently colonize human dwellings. Although certain countries such as Guatemala have successfully controlled the transmission mediated by this species (Guhl et al., 2009), Venezuela and Colombia still face serious problems of re-infestation of insecticide-treated houses by sylvatic populations (Fitzpatrick et al., 2008; Lopez et al., 2007). These events are commonplace in endemic regions where vectors are autochthonous, and will demand the development of innovative surveillance/control strategies that contemplate the complexities of sylvatic populations dynamics (Abad-Franch and Monteiro, 2005).
To allow for the accurate taxonomic identification of R. prolixus and the members of the R. robustus cryptic species complex, Pavan and Monteiro (2007) have developed an objective and cost-effective mitochondrial DNA (mtDNA) multiplex PCR assay. However, due to its mitochondrial-based nature, this method could generate misleading results in areas where R. prolixus and members of the R. robustus complex might come into contact and hybridize, causing mitochondrial DNA introgression (Fitzpatrick et al., 2008).
Thus, in order to overcome such limitation, we describe a new non-coding, single-copy nuclear DNA fragment containing a single-nucleotide polymorphism (SNP) with the potential to separate R. prolixus from members of the R. robustus cryptic species complex.
2. Materials and Methods
2.1. Molecular identification of specimens and sampling
Since accurate taxonomic identification of Rhodnius prolixus and the four cryptic species of R. robustus [sensu Monteiro et al. (2003)] based on morphology alone is problematic, all specimens used in this study were identified through molecular taxonomy based on a fragment of the mitochondrial cytochrome b gene (Pavan and Monteiro, 2007). Moreover, we sequenced the ribosomal second internal transcribed spacer (ITS-2) of all specimens analyzed (data not shown), according to Marcilla and collaborators (2001), to identify possible introgression of mitochondrial DNA specimens.
For an exploratory screening aimed at finding unique, non-coding regions that separate these species, we tested 25 R. prolixus from two well established laboratory colonies kept at the Centers for Disease Control and Prevention (CDC) in Atlanta (19 specimens) and at the Medical Entomology Research and Training Unit in Guatemala (six specimens), plus two field-collected R. prolixus from Venezuela, two R. robustus I, four of each R. robustus II and III, and six R. robustus IV [sensu Monteiro et al. (2003)]. This approach was done during the initial phase of R. prolixus genome project, named survey sequencing, aimed to select a non-introgressed R. prolixus colony with low genetic variability. Fifty-two colony specimens, 43 R. prolixus and nine R. robustus II, and 102 field-collected insects, three R. prolixus, two R. robustus I, 74 R. robustus II, 19 R. robustus III, and four R. robustus IV (Figure 1 and Table 1), were assayed to confirm the diagnostic applicability of possible species-specific SNPs.
Figure 1. Geographical distribution of sampled specimens of R. prolixus and R. robustus s.l.
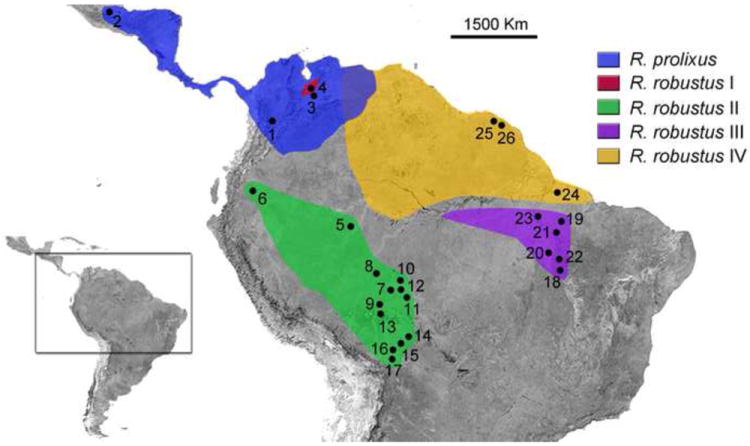
Each location is numbered according to Table 1. The colors indicate the range of R. prolixus and the four members of the R. robustus cryptic species complex (R. robustus I-IV).
Table 1. Information on the samples used in this study.
Field-collected samples are numbered according to Figure 1.
| Species | Collection site/Colony location | Geographical coordinates | NA | NB | Field/Colony | Date of colection | GenBank accession numbers |
|---|---|---|---|---|---|---|---|
| R. prolixus | 1. Colombia / CDC1, Atlanta, USA | - | 19 | 10 | Colony | - | JQ432871 to 432880 |
| 2. Guatemala / MERTU2, Guatemala | - | 6 | 10 | Colony | - | JQ432881 to 432890 | |
| “Unknown” / IOC3, Brazil | - | - | 23 | Colony | - | JQ432844 to 432866 | |
| 3. Barinas, Venezuela | 08° 37’ N 70° 12’ W | 2 | 3 | Field | 2004 | JQ432891 to 432893 | |
| R. robustus I | 4. Trujillo, Venezuela | 09° 22’ N 70° 25’ W | 2 | 2 | Field | 2004 | JQ432894, JQ432895 |
| R. robustus II | 5. Carauarí, Amazonas, Brazil / INPA4, Brazil | - | - | 4 | Colony | - | JQ432867 to 432870 |
| 6. Sucumbios, Ecuador | - | - | 5 | Colony | - | JQ432896 to 432900 | |
| 7. Monte Negro, Rondônia, Brazil | 10° 15’ N 63° 17’ W | 4 | 2 | Field | 2004 | JQ432961, JQ432962 | |
| 8. Porto Velho, Rondônia, Brazil | 08° 44’ S 63° 25’ W | - | 45 | Field | Apr/2009 | JQ432916 to 432960 | |
| 9. Cacoal, Rondônia, Brazil | 11° 26’ S 61° 27’ W | - | 2 | Field | Apr/2009 | JQ432973, JQ432974 | |
| 10. Santo Antônio, Rondônia, Brazil | 12° 31’ S 63° 33’ W | - | 1 | Field | Feb/2010 | JQ432971 | |
| 11. Guajará Mirim, Rondônia, Brazil | 10° 47’ S 65° 20’ W | - | 2 | Field | Jul/2010 | JQ432969, JQ432970 | |
| 12. Ouro Preto do Oeste, Rondônia, Brazil | 10° 43’ S 62° 15’ W | - | 7 | Field | Oct/2009 | JQ432963 to 432968, JQ432972 | |
| 13. San Gabriel, Pando, Bolivia | 10° 16’ S 67° 03’ W | - | 2 | Field | Sep/2007 | JQ432912, JQ432913 | |
| 14. Guarayos, Santa Cruz, Bolivia | 15° 52’ S 63° 20’ W | - | 7 | Field | Mar/2008 | JQ432901 to 432907 | |
| 15. San Ramón, Santa Cruz, Bolivia | 16° 29’ S 62° 29’ W | - | 1 | Field | Mar/2008 | JQ432908 | |
| 16. Montero, Santa Cruz, Bolivia | 17° 17’ S 63° 08’ W | - | 3 | Field | Mar/2008 | JQ432909 to 432911 | |
| 17. El Torno, Santa Cruz, Bolivia | 18° 04’ S 63° 18’ W | - | 2 | Field | Sep/2007 | JQ432914, JQ432915 | |
| R. robustus III | 18. Araguaína, Pará, Brazil | 07° 11’ S 48° 12’ W | - | 2 | Field | Nov/2009 | JQ432975, JQ432976 |
| 19. Ulianópolis, Pará, Brazil | 03° 43’ S 47° 29’ W | - | 7 | Field | Nov/2009 | JQ432977 to 432983 | |
| 20. São Domingos do Araguaia, Pará, Brazil | 05° 30’ S 48° 44’ W | - | 3 | Field | Nov/2009 | JQ432984 to 432986 | |
| 21. Rondon do Pará, Pará, Brazil | 04° 54’ S 48° 20’ W | - | 2 | Field | Nov/2009 | JQ432987, JQ432988 | |
| 22. São Bento do Tocantins, Tocantins, Brazil | 06° 02’ S 47° 55’ W | - | 3 | Field | Nov/2009 | JQ432989 to 432991 | |
| 23. Novo Repartimento, Pará, Brazil | 04° 20’ S 49° 50’ W | 4 | 2 | Field | Aug/1998 | JQ432992, JQ432993 | |
| R. robustus IV | 24. Santa Maria, Pará, Brazil | 01° 20’ S 47° 32’ W | - | 1 | Field | Oct/2009 | JQ432994 |
| 25. Cayenne, French Guiana | 04° 56’ N 52° 20’ W | 5 | 2 | Field | Jan/2003 | JQ432995, JQ432996 | |
| 26. Rémire, French Guiana | 04° 53’ N 52° 16’ W | 1 | 1 | Field | Aug/2001 | JQ432997 | |
| Total | 43 | 154 |
NA - number of individuals tested for seven amplicons (AmpA to AmpG); NB - number of individuals sequenced for AmpG.
Centers for Disease Control and Prevention;
Medical Entomology Research and Training Unit;
Instituto Oswaldo Cruz;
Instituto Nacional de Pesquisa da Amazonia.
2.2. DNA purification and library construction
DNA from the CDC and Guatemala Colony samples were extracted from ovaries or testis using the Promega genomic extraction kit. For all other Rhodnius specimens, one or two legs of a single Rhodnius specimen was placed in a 1.5 mL microtube, dipped into liquid nitrogen until frozen, and ground to a powder with Kimble/Kontes Pellet Pestles® prior to standard Genomic DNA Extraction with a Real Genomics™ kit.
From a small WGS plasmid library, 12,626 random Genome Survey Sequences (GSS) were generated as described by Abubucker et al. (2008), using genomic DNA extracted from a mixed population of Rhodnius prolixus from the Atlanta colony, and running on an ABI 3730 automated sequencer. Ten thousand of the GSS reads were screened for non-coding regions, searching against a transposon database, then against RFAM (Gardner et al., 2011), and finally against a non-redundant protein database (with an e-value cutoff of 10-5). Regions with hits were masked. Vector sequences were removed and sequences with unmasked regions of 400-500 base pairs (bp) were extracted and sent through an in-house primer-calling pipeline. Primers with the following criteria were picked: (1) all bases of each primer in the pair must have a quality score of at least Q25; (2) minimum and maximum acceptable amplicon size was 400 and 500, respectively; and (3) excluded region was 150-bp (i.e. region with repetitive elements in the middle of the amplicon that would compromise primer annealing).
2.3. PCR amplification and DNA sequencing
From the putative unique, non-coding regions, 96 primer pairs were designed for the PCR-amplification of regions of approximately 400-500-bp in length (Table A.1), during the survey sequencing approach.
PCR amplifications of the nuclear gene fragments were performed in a Veriti® 96 Well Thermal Cycler (Applied Biosystems), programmed for one denaturation step at 96 °C for 5 min, followed by 35 cycles at 94 °C for 30s, 60 °C for 30s, and 72 °C for 45s, and a final 10-min extension step at 72 °C. Each 25 μL final volume reaction, contained 5-10 ng of DNA template, 2.5 μL buffer 10x, 2.5 mM of MgCl2, 1.25 units of Taq polymerase (Biotools), 0.5 mM of dNTPs, and 10 pmoles of each primer. Purification of PCR products was performed with the Hi Yield™ Gel/PCR DNA Extraction Kit (Real Genomics™), and both fragment strands were subjected to fluorescent dye-terminator cycle sequencing reactions (ABI Prism® BigDye® Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems), using the same primers above, and run on an ABI 3730 automated sequencer.
2.4. Analyses of generated DNA sequence data
The removal of primer sequences, editing of both forward and reverse strands, and the generation of a consensus sequence for each sample were done using the SEQMAN LASERGENE 7.0 program (DNAStar, Inc.). Sequences were aligned using the default parameters of CLUSTALW2 program (Larkin et al., 2007). Polymorphic sites were identified using MEGA 5 (Tamura et al., 2011). RECON (Bao and Eddy, 2002), and BLASTX (Altschul et al., 1997) were used to identify repeated regions and coding sequences, respectively.
A Bayesian phylogenetic tree for the diagnostic amplicon of 154 R. prolixus and R. robustus s.l. sequences was inferred in BEAST 1.7 (Drummond et al., 2012). Bayesian Information Criterion (BIC) in jMODELTEST (Posada, 2008) was used to elect Jukes-Cantor (Jukes and Cantor, 1969) as the best-fit evolutionary model (delta AIC = 0) for the data set. Tree prior was randomly generated and Yule process of speciation was imposed for all tree reconstructions. Two independent runs were performed for 107 generations, with a burn-in of 106 generations. Convergence of parameters and a proper mixing were confirmed by calculating the Effective Sample Size (ESS) in TRACER 1.5 (Drummond and Rambaut, 2007), excluding the initial 10% (burn-in) of each run. All considered parameters showed ESS values above 104. Runs were combined using LOGCOMBINER, and a maximum credibility tree based on the 20,000 trees generated (burn-in = 2,000) and a posterior probability limit of 0.6 was produced using TREE ANNOTATOR (both programs part of the BEAST package). Statistical support for clades was assessed by the posterior probability method, and trees were visualized in FIGTREE v.1.0 (http://tree.bio.ed.ac.uk/software/figtree/).
Genealogy of the DNA sequences generated for the selected diagnostic amplicon was also inferred with NETWORK 4.6 (Fluxus-Engineering), using the Median-Joining network method (Bandelt et al., 1999), with a Maximum-Parsimony (MP) calculation in the post-processing. Character weights and epsilon values were given a value of 10 (transition/transversion weighting was 1:1, since we analyzed a non-coding region). Also the Maximum-Composite Likelihood (M-CL) algorithm (Tamura et al., 2004) with 1000 bootstrap replicates of variance, available in MEGA 5 (Tamura et al., 2011), was used to construct intra and interespecific distance matrix.
2.5. Genomic search of selected amplicon
Using the MEGABLAST web service (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi), the diagnostic amplicon was searched for in the following 40 complete or partial arthropod genomes: Acromyrmex echinatior, Acyrthosiphon pisum, Aedes aegypti, Anopheles gambiae, An. darlingi, Apis mellifera, A. florea, Atta cephalotes, Bombus impatiens, B. terrestris, Bombyx mori, Camponotus floridanus, Culex quinquefasciatus, Daphnia pulex, Drosophila melanogaster, D. pseudoobscura pseudoobscura, D. simulans, D. yakuba, D. ananassae, D. erecta, D. grimshawi, D. mojavensis, D. persimilis, D. sechellia, D. virilis, D. willistoni, Harpegnathos saltator, Ixodes scapularis, Lepeophtheirus salmonis, Mayetiola destructor, Nasonia giraulti, N. longicornis, Pediculus humanus corporis, Pogonomyrmex barbatus, Rhipicephalus microplus, Rhodnius prolixus, Solenopsis invicta, Varroa destructor, Nasonia vitripennis, and Tribolium castaneum. A second search was done in the BLASTN web service (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&BLAST_PROGRAMS=megaBlast&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome), with standard parameters of word size=7 and e-value=10, and the NCBI nucleotide (nt) database arthropod (taxid 6656) subset.
2.6. Selected amplicon genomic contig localization and chromosome synteny
2.6.1. Databases
The Rhodnius prolixus genome assembly v.3.0 and the hard-masked contig GL563069 (“N” masked transposons, repetitive and low complexity regions) were downloaded from VectorBase (http://www.vectorbase.org). Transposable elements and Rhodnius EST databases were kindly provided by Dr. J. M. Ribeiro. A. mellifera, B. terrestris and T. castaneum protein sequences were extracted from the NCBI non-redundant (nr) database using a Perl script that selected and extracted all sequences related to any NCBI taxonomic index point (taxids used were respectively 7460, 30195 and 7070), using taxdump and gi-taxid-prot NCBI relational datasets (ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz, and ftp://ftp.ncbi.nih.gov/pub/taxonomy/gi_taxid_prot.dmp.gz). The T. castaneum genome assembly version 3.0 was downloaded from BeetleBase (ftp://bioinformatics.ksu.edu/pub/BeetleBase/3.0/Tribolium_genome_sequence.fasta). Rhodnius prolixus genes, transcripts and peptides belonging to prediction version C1 (VectorBase gene prediction pipeline using assembly v3.0) were also downloaded from VectorBase (named VectorBase prediction). Alternative gene prediction version Lagerblad 3.0 was done using GENEID v1.3 (Blanco et al., 2007) trained with a large R. prolixus EST database in genome assembly v3.0 (named Lagerblad and available in R. prolixus genomic browser at VectorBase). Transposable elements were identified in contig GL563069 using local RPSBLAST, BLASTN and TBLASTX (Tatusova, 2010), and transposable element databases.
2.6.2. Selected amplicon localization in genomic contigs
The selected amplicon region was searched on R. prolixus genomic contigs using BLASTN (Tatusova, 2010).
2.6.3 Genes and transposons curation in amplicon flanking regions
Genes from both predictions flanking the diagnostic amplicon region were searched for chimeras using BLASTP, NCBI nr protein, UNIPROT databases, and TBLASTN of Rhodnius EST database. Chimera genes were split manually to allow gene synteny analysis. Gene prediction revision was done manually on ARTEMIS (Rutherford et al., 2000) based on EST database hits and GENEWISE (Birney et al., 2004) analysis using contig GL563069 and coded proteins best hit in nr and UNIPROT databases. Transposable elements revision was also carried out in GENEWISE with BLASTN and TBLASTX hits, using TE database.
2.6.4. Gene synteny
Reciprocal BLASTP best-hit strategy (Tatusov et al., 1997) was used to identify homologs at the protein level for 14 genes in the diagnostic amplicon flanking region. We used in the comparison species with chromosome-mapped genomes (A. mellifera, B. terrestris and T. castaneum). The R. prolixus database included predicted proteins from both VectorBase and Lagerblad predictions. Bee and beetle homolog proteins GIs from the NCBI protein database (http://www.ncbi.nlm.nih.gov/protein) were used to identify coding genes and their chromosome location. Transposable elements were not considered in this analysis.
2.6.5 Whole contig synteny
R. prolixus hard-masked contig GL563069 was compared with all T. castaneum chromosomes separately using TBLASTX with a 10-10 e-value cutoff. The results were loaded in ACT (Rutherford et al., 2000) and limited by 50 or 70% identity cutoff when confirming selected gene homolog location, and 50% identity in GL563069 open reading frames (ORFs) larger than 100bp when analyzing entire contig synteny.
3. Results
3.1. Amplicon selection
Seven amplicons were selected based on reproducibility, variability, and presence of species-specific sites across all taxa tested. In this first screening, 27 R. prolixus, two R. robustus I, four of each R. robustus II and III, and six R. robustus IV samples PCR-amplified and sequenced for these seven nuclear loci yielded 13 polymorphic sites, nine of which are SNPs (Table A.2). One such SNP located on the 280th site of the 364-bp region amplified with primers AmpGF, 5’ - GAG AGC TGA AGA TAG GCA AGC G, and AmpGR, 5’ - TGA TAA CTG GAT TAG GCG CAG C was diagnostic for R. prolixus (i.e. all R. prolixus have an adenine, instead of a guanine, on that particular site). To further evaluate the reliability and usefulness of this particular SNP to serve as an important diagnostic marker for the identification of R. prolixus, an extensive effort was made to obtain the greatest amount of R. prolixus and R. robustus s.l. samples aimed at covering the widest geographical distribution for the species. A total of 154 specimens, 46 R. prolixus, two R. robustus I, 83 R. robustus II, 19 R. robustus III, and four R. robustus IV, were sequenced for the 364-bp AmpG region. The 280th site still proved to be diagnostic. This autapomorphic character of R. prolixus is preceded by 5’ – GTCATAAGA and followed by 5’ – TCGTTGGTAG conserved sequences for all species analyzed.
Ten polymorphic sites were observed, in which R. robustus I share eight with R. prolixus, and three of those are parsimony-informative sites (positions 174, 270, and 360; Table 2). The paraphyletic assemblage of R. robustus s.l., previously observed with mitochondrial cytochrome b and ribosomal DNA sequencing analyses (Monteiro et al., 2003), is corroborated here, based on a Bayesian phylogenetic tree reconstruction and a Median-Joining network with Maximum-Parsimony post-processing (Figure 2). The R. robustus I clade is more closely related to the R. prolixus clade than to the other R. robustus clades. The R. robustus I and R. prolixus sister-species status is also evidenced by M-CL distance comparisons, as sequence divergence between these species is lower than between R. robustus I and R. robustus II-IV (0.6% in comparison with 1.1-1.7%; Table 3).
Table 2. Polymorphic sites of AmpG sequences (364bp) observed in 154 Rhodnius prolixus and R. robustus I-IV.
The bold column highlights the single-nucleotide polymorphism (SNP) that separates R. prolixus from members of the R. robustus cryptic species complex (site 280).
| Species | 74 | 80 | 167 | 174 | 270 | 280 | 321 | 337 | 341 | 360 |
|---|---|---|---|---|---|---|---|---|---|---|
| R. prolixus | C | G | G | A | C | A | G | C | C | A |
| R. robustus I | S | . | . | . | . | G | . | . | . | . |
| R. robustus II | . | R | R | T | A | G | K | Y | S | T |
| R. robustus III | . | . | . | T | A | G | T | T | G | T |
| R. robustus IV | . | . | . | T | A | G | T | . | . | T |
A dot (.) indicates identity with the consensus nucleotide (in this case, the consensus sequence is from R. prolixus).
Codes: A – adenine; C – cytosine; G – guanine; T – thymine; S – cytosine or guanine; R – guanine or adenine; K – guanine or thymine; Y – cytosine or thymine.
Figure 2. Phylogenetic relationship between Rhodnius prolixus and R. robustus I-IV haplotypes.
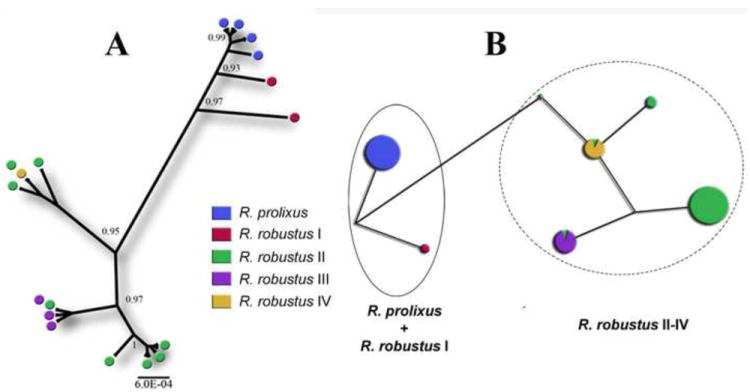
Species are indicated by different colors. (A) Non-rooted 60% majority-rule Bayesian consensus tree obtained from all 154 AmpG sequences of R. prolixus and R. robustus. Posterior probability values higher than 0.9 are indicated near the nodes. Shared genotypes between different R. robustus species is represented by more than one circle in a single terminal branch. Note that R. robustus I specimens (red circles) are more closely related to R. prolixus (blue circles) than to the other R. robustus species, evidencing its paraphyletic assemblage. (B) Median-Joining network with Maximum-Parsimony post-processing. Each node (or circle) represents a unique haplotype. The size of each node is proportional to the number of specimens that shared the same haplotype, and the branch size is proportional to the number of mutational steps. Note that the distance (measured by branch sizes) between R. prolixus and R. robustus I is smaller than between R. robustus I and R. robustus II-IV, confirming their sister-species status. The R. robustus paraphyly is shown by dot and dashed circles, which highlight the R. prolixus + R. robustus I group, and the Amazonian R. robustus (II-IV), respectively.
Table 3. Maximum-Composite Likelihood (M-CL) estimates of intraspecific (in italics) and interspecific evolutionary divergences distance matrix.
The analysis involved all 154 R. prolixus and R. robustus s.l. AmpG sequences. In parentheses, standard deviations calculated after 1000 bootstrap replications.
| Species | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1. R. prolixus | 0.000 | ||||
| 2. R. robustus I | 0.006 (±0.003) | 0.005 (±0.003) | |||
| 3. R. robustus II | 0.012 (±0.007) | 0.015 (±0.006) | 0.003 (±0.001) | ||
| 4. R. robustus III | 0.020 (±0.007) | 0.017 (±0.007) | 0.006 (±0.003) | 0.000 | |
| 5. R. robustus IV | 0.014 (±0.006) | 0.011 (±0.004) | 0.004 (±0.003) | 0.006 (±0.004) | 0.000 |
Note that all comparisons between R. robustus I and R. robustus II-IV resulted in divergence levels higher than between R. robustus I and R. prolixus.
3.2. Amplicon G genomic contig and flanking region
The AmpG region is located in the Rhodnius prolixus genomic contig GL563069 from 1116977 to 1117340 positions. It was not found in any other genome searched, not even when high e-values and smaller word sizes were used (allowing more dissimilar matches), which suggests that this region is unique.
Genes in the AmpG flanking region (contig GL563069 from 925995 to 1319022 positions) were curated (Figure 3B) to remove chimera and wrong predictions. Lagerblad genes 17948_72 and 17948_73 were joined (Src tyrosine kinase) and 17948_78 was split into two genes (X11Lbeta and Transmembrane protein 165). Genes 17948_79 and 17948_84 were wrong predictions and then were removed (not shown). No chimera or false positive gene was found in the VectorBase prediction, but four genes were absent (17948_70 – Ephrin, 17948_75 – Sarcoplasmic calcium-binding protein, 17948_81 – Interleukin 16 and 17948_88 – KN motif and Ankyrin repeat domain-containing protein). Final genes in the AmpG flanking regions were: Ephrin, Ribonuclease, Src tyrosin kinase, Odorant receptor, Sarcoplasmic calcium binding protein, Small glutamine-rich tetratricopeptide repeat-containing protein alpha, X11Lbeta, Transmembrane protein 165, Interlukin 16, WNT4 protein, UNC-112 related protein, NADH dehydrogenase, KN motif, and ankyrin repeat domain, containing protein and Band 4.1-like protein 4 (Figure 3B). The detailed information about this region was included in Table A.3.
Figure 3. AmpG neighborhood.
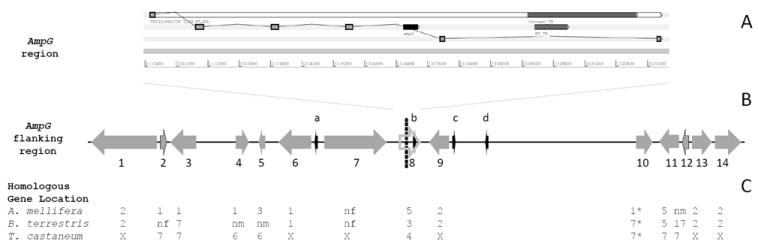
Rhodnius prolixus contig GL563069 (region from 1110523-1123517) that contains Transmembrane protein 165 (TP165). Exons were shown as gray rectangles with black outlines linked together. AmpG region (1116877-1117340) is inside the fourth gene intron. Outcast_TE and RT_TE are in the fifth intron and represent an outcast class I transposable element and its reverse transcriptase (A). Rhodnius prolixus contig GL563069 (region from 925000 to 1319500) contains 14 genes and four transposons. AmpG region is shown as a black vertical dashed line. Genes were represented as gray arrows, and EST-supported genes as gray arrows with black outlines. Gene 8 had fully manual curation and EST support, and was represented as a gray outlined arrow. (1- ephrin; 2- ribonuclease; 3- src tyrosine kinase; 4- odorant receptor; 5- sarcoplasmic Ca-binding protein; 6- small Glu-rich tetratricopeptide repeat-containing protein alpha; 7- X11Lbeta; 8- transmembrane protein 165; 9- interleukin 16; 10- WNT4 protein; 11- UNC112 related protein; 12- NADH dehydrogenase; 13- KN motif and ankyrin repeat domain-containing protein; 14- band 4.1-like protein 4). Transposable elements were represented as black arrows (a- hAT – class II; b- outcast – class I; c- mariner – class II; d- L1 – class I) (B). Homologous genes were identified at the protein level using reciprocal best hit strategy (Tatusov et al., 1997). Numbers mean chromosome number. X= chromosome; X. nf= homologs not found; nm= homologs not mapped; *= not homologous, only a similar gene (C).
The AmpG region was found in the fourth intron of the Transmembrane protein 165 gene. This gene’s original predictions (RPRC07420-RA and 17948_78) were manually curated (Figure 3A), including complete EST support. The last intron of this gene also has an Outcast transposable element (Class I) with a truncated reverse transcriptase (Figure 3A). Other transposable elements identified were in intergenic regions (Figure 3B).
3.3. Amplicon G chromosome localization
Arthropod choice for synteny analysis was based on the following criteria: 1) to be the closest organisms with chromosome-mapped genome; and 2) to have the closest number of chromosomes to Rhodnius prolixus, which was known to be 20 autosomes, plus XY (Panzera et al., 2007). R. prolixus (taxid 13249) taxonomic classification in the NCBI taxonomic web database (http://www.ncbi.nlm.nih.gov/taxonomy) was followed back by tracing the branches of the Insecta phylogenetic tree toward its trunk to search for organisms with chromosome-mapped genomes. Endopterygota Infraclass (taxid 33392), inside Neoptera Subclass (taxid 33340) was the closest taxonomic point that satisfied those two mentioned criteria, where the following 13 species genomes (and their respective number of chromosomes in parenthesis) were found: Anopheles gambiae (4), Drosophila pseudoobscura (7), Apis mellifera (16), Drosophila miranda (6), Bombus terrestris (18), Mayetiola destructor (4), Nasonia vitripennis (5), Drosophila virilis (6), Tribolium castaneum (10), Drosophila simulans (6), Drosophila yakuba (7), Drosophila melanogaster (7) and Aedes aegypti (3). Based on the second criterion, we chose for the analysis two bees, A. mellifera and B. terrestris, and the beetle T. cataneum.
Almost all homolog genes of the AmpG flanking region were found in the three selected genomes, when we used reciprocal best-hit strategy in gene synteny analysis (Tatusov et al., 1997). The only exception was in the T. castaneum analyzed region, where we did not confirm a homolog for the WNT4 protein (GI 91086553; Figure 3C), as the best hit was not reciprocal to R. prolixus. However, this protein has the same chromosomal localization as UNC112 related protein and NADH dehydrogenase (Figure 3C), and thus was not discarded from analysis. X11LBeta homolog searches in bees did not retrieve any similar gene, probably due to a lack of prediction, as the TBLASTN web search showed similar genomic regions in unmapped genomic scaffolds (data not shown). Transmembrane protein 165 gene contained the AmpG region inside its fourth intron, but it was the only protein that exhibited homologs located in different chromosomes in all three compared insects (Figure 3C). Many homolog R. prolixus genes were not found, or their chromosome localization did not suggest a pattern in bee genomes, especially B. terrestris. Considering A. mellifera, four of the first six homolog genes were localized in chromosome 1 (CHR1), but this pattern did not seem to overlap the AmpG region. Moreover, no other pattern was observed until the end of the gene block. T. castaneum homologs showed a mixed localization pattern in CHRX and CHR7 (six and five occurrences, respectively), but the two homologs flanking Transmembrane protein 165 gene are located at the X chromosome (Figure 3C).
Whole contig synteny of contig GL563069 was done with all T. castaneum chromosomes separately. Synteny results from the whole contig have confirmed the localization previously identified in reciprocal best-hit approach for the following genes: Ephrin, Src tyrosin kinase, Small glutamine-rich tetratricopeptide repeat-containing protein alpha, X11Lbeta, Transmembrane protein 165, Interlukin 16, WNT4 protein, UNC-112 related protein, NADH dehydrogenase, KN motif and ankyrin repeat domain containing protein and Band 4.1-like protein 4 (Figure A.1). Despite Ribonuclease gene did not show similarity regions neither in CHR7 or CHRX (using an e-value < 10-10), it homolog had been found in reciprocal best-hit approach (Figure A.1B). Genes Odorant receptor and Sarcoplasmic calcium-binding protein were confirmed to be located in CHR6 (data not shown). The overall synteny of this contig in T. castaneum CHRX, CHR7 and CHR4 showed 161, 158 and 52 hits, respectively (Figure A.2) and rates of 14.6, 7.7, and 3.7 hits/Mbase, respectively. The synteny of all other chromosomes presented between 36 and 6 hits and 3.1 to 0.3 hits/Mbase.
4. Discussion
The R. prolixus diagnostic SNP was identified during the initial phase of the Rhodnius prolixus genome project, known as genome survey sequencing (GSS). GSS sequences will be used, amongst other things, as a framework for the mapping and sequencing of genome size pieces, and this step is required for the assessment of genetic variability values, as high heterozygosity levels could lead to difficulties during genome assembly. This task is accomplished through the identification of variable non-coding single-copy nuclear loci that are then used as markers to estimate colony heterozygosity. In the particular case of the R. prolixus genome project, it was also used with the purpose of identifying a colony that was indeed R. prolixus, and not R. robustus, or a mixture of the two species. The amplicon G (AmpG) was the only locus to display a derived character unique to (and thus distinctive of) the R. prolixus lineage: an adenine, instead of a guanine, on the 280th position. Due to its usefulness in the colony-screening phase of the project, we decided to evaluate its applicability over a larger sample and geographic area. Our results are very encouraging as they show proven reliability after being tested on an extensive collection of specimens spanning a significant geographical area. Our sampling is evidently not ideal as there are still large areas from where we were unable to obtain specimens. Needless to say, some of these regions are very remote and difficult to access. With respect to the sampling of R. robustus I, it is worth mentioning that this species is rare and of restricted geographic distribution. To date, it has only been found in the state of Trujillo, Venezuela.
The ribosomal second internal transcribed spacer (ITS-2) is frequently used for solving taxonomical problems in triatomines. This region seems to be in concerted evolution in some organisms, a molecular process of homogenization between different loci in gene families, as a consequence of gene conversion and unequal recombination (Liao, 1999). This homogenization can be completed, as it seems to be the case of Triatoma species (Bargues et al., 2006; 2002), or reveal intragenomic and intraspecific variation, as showed in other insects as Culex and Lutzia species (Diptera: Culicidae) (Vesgueiro et al., 2011). As Rhodnius species seem to fit the latter case (CL, MGP, and FAM, unpublished), ITS-2 copies must be cloned and sequenced, which makes its use more expensive and time-consuming.
Although this methodology entails the sequencing of PCR products, as opposed to multiplex-PCR methods (Pavan and Monteiro, 2007), no cloning is required (such as with most rDNA targets). It is also simple and straightforward, because it only involves the identification of a single nucleotide site. We hope that this SNP will be of use for a number of professionals with a broad variety of interests regarding this particular group of insects, however, the applicability of this marker needs to be performed over larger samples of R. robustus I and R. robustus IV is desirable. The validation of its potential awaits further usage by the research community that will put it to the test.
In order to determine its genome localization, the AmpG region was searched for in all available partial or complete arthropod genomes (40 in total), but it was only located in the genome of R. prolixus itself, in contig GL563069, inside the fourth intron of Transmembrane protein 165 gene (TP165). Fourteen genes flanking the AmpG region were annotated and their homologs searched at the protein level, using reciprocal best-hit strategy (Tatusov et al., 1997) in A. mellifera, B. terrestris and T. castaneum. These were the three arthropods that have shown the closest chromosome number and phylogenetic relationship to R. prolixus. Despite the detection of the fourth TPS165 intron as the location of the AmpG region, this protein was the only one of the R. prolixus contig that its homologs were located on different chromosomes in all three species searched, and thus could not be used to infer AmpG localization. Considering all 14 gene homologs’ localization in all three species analyzed, T. castaneum showed the most homogenous pattern concerning genes location, with six and five occurrences in X and 7 chromosomes (CHRX and CHR7), respectively; notwithstanding, both AmpG flanking genes homologs were located in CHRX.
The whole contig synteny approach represents an overall and more reliable look at genomic similarities, as DNA sequences are compared along the complete extension of each contig. R. prolixus contig GL 563069 synteny in comparison with T. castaneum contig also pointed CHRX and CHR7 with almost equal number of hits (161 and 158) as the possible chromosome location, even though their lengths are very different. The frequency of hits in CHRX was two-times higher than in CHR7 (14,6 to 7,7 hits/Mbase, respectively) and about ten times higher than all other chromosomes, showing that contig GL563069 shares more potential gene content with T. castaneum CHRX. Thus, our results suggest that this contig could be part of R. prolixus X chromosome.
DNA sequencing analysis of the AmpG region of R. prolixus and R. robustus s.l. samples shows that R. prolixus and R. robustus I share eight polymorphic sites, three of these derived nucleotides (A, C, and A, at positions 174, 270, and 360, respectively; Table 2), and hence Maximum-Composite Likelihood divergence between these species reached 0.6% in contrast to 1.1-1.7% between R. robustus I and R. robustus II-IV. Bayesian maximum credibility tree and a Median-Joining network with Maximum-Parsimony post-processing further support the paraphyletic nature of R. robustus (Figure 2), previously observed with cyt b and rDNA sequences (Monteiro et al., 2003).
In addition to its utility in taxonomy, this marker could be used in combination with the cyt b multiplex-PCR (Pavan and Monteiro, 2007) for the detection of natural crossbreeding and introgression of mitochondrial DNA along putative hybrid zones, as reported by Fitzpatrick and collaborators (2008) in Venezuela. The presence of an adenine/guanine double-peak in electropherograms could be indicative of a recent hybridization event. In addition, as R. prolixus has long been used as a model for insect physiology and biochemistry (e.g. Ribeiro et al. (1993; 1957) and Wigglesworth (1957)), several laboratories worldwide currently maintain colonies of this species. There is evidence, though, that some of these colonies have been contaminated with, or misidentified as, R. robustus s.l. (MGP, CL, EMD, FAM, unpublished). The SNP here described could also be used to evaluate the identity and purity of R. prolixus colonies. However, although the possibility of using this SNP as a diagnostic marker for this important vector species seems very promising, its “universal” applicability remains to be evaluated by the research community.
Supplementary Material
-
*
Accurate species identification is key for effective vector control of Chagas disease
-
*
Rhodnius prolixus is an efficient domestic vector and R. robustus s.l. is sylvatic
-
*
These species are cryptic, can co-occur and occasionally hybridize in nature
-
*
We describe a SNP in a scnDNA that separates R. prolixus from R. robustus s.l.
-
*
This marker will allow for the detection of crossbreeding and mtDNA introgression.
Acknowledgments
To John Spieth at the Genome Institute at Washington University School of Medicine in St. Louis for the assistance with library construction and identification of nuclear loci. To Sebastião Aldo Valente, Luis Herman S. Gil, Toby Barrett, José R. Coura, Christine Aznar, and Celia Cordon-Rosales, for kindly providing specimens. We thank José Marcos Ribeiro for providing the Transposon database. The authors are also grateful to the comments of two anonymous referees that helped improve the original manuscript. We also thank the PDTIS-FIOCRUZ DNA sequencing core for running sequencing reactions.
Role of the funding source
This work was supported by the Brazilian Research Council (CNPq), the Brazilian Ministry of Science, Technology and Innovation (MCTI), and NIH grant number U54 HG003079. The sponsors did not take part nor influenced the any aspect of this study.
Footnotes
Disclosure Statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abad-Franch F, Ferraz G, Campos C, Palomeque FS, Grijalva MJ, Aguilar HM, Miles MA. Modeling disease vector occurrence when detection is imperfect: infestation of Amazonian palm trees by triatomine bugs at three spatial scales. PLoS Negl Trop Dis. 2010;4:e620. doi: 10.1371/journal.pntd.0000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad-Franch F, Monteiro F. Molecular research and the control of Chagas disease vectors. Anais da Academia Brasileira de Ciências. 2005;77:437–454. doi: 10.1590/s0001-37652005000300007. [DOI] [PubMed] [Google Scholar]
- Abubucker S, Martin J, Yin Y, Fulton L, Yang SP, Hallsworth-Pepin K, Johnston JS, Hawdon J, McCarter JP, Wilson RK, Mitreva M. The canine hookworm genome: analysis and classification of Ancylostoma caninum survey sequences. Mol Biochem Parasitol. 2008;157:187–192. doi: 10.1016/j.molbiopara.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargues MD, Klisiowicz DR, Panzera F, Noireau F, Marcilla A, Perez R, Rojas MG, O’Connor JE, Gonzalez-Candelas F, Galvao C, Jurberg J, Carcavallo RU, Dujardin JP, Mas-Coma S. Origin and phylogeography of the Chagas disease main vector Triatoma infestans based on nuclear rDNA sequences and genome size. Infect Genet Evol. 2006;6:46–62. doi: 10.1016/j.meegid.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Bargues MD, Marcilla A, Dujardin JP, Mas-Coma S. Triatomine vectors of Trypanosoma cruzi: a molecular perspective based on nuclear ribosomal DNA markers. Trans R Soc Trop Med Hyg. 2002;96(Suppl 1):S159–164. doi: 10.1016/s0035-9203(02)90069-6. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protocols in Bioinformatics. 2007;4(Unit 4.3) doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- Coura JR. Chagas disease: what is known and what is needed--a background article. Mem Inst Oswaldo Cruz. 2007;102(Suppl 1):113–122. doi: 10.1590/s0074-02762007000900018. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick S, Feliciangeli MD, Sanchez-Martin MJ, Monteiro FA, Miles MA. Molecular genetics reveal that silvatic Rhodnius prolixus do colonise rural houses. PLoS Negl Trop Dis. 2008;2:e210. doi: 10.1371/journal.pntd.0000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011;39:D141–145. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guhl F, Pinto N, Aguilera G. Sylvatic triatominae: a new challenge in vector control transmission. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):71–75. doi: 10.1590/s0074-02762009000900012. [DOI] [PubMed] [Google Scholar]
- Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, Periago MR. The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl Trop Dis. 2008;2:e300. doi: 10.1371/journal.pntd.0000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. Academic Press; New York: 1969. pp. 21–123. [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Liao D. Concerted evolution: molecular mechanism and biological implications. Am J Hum Genet. 1999;64:24–30. doi: 10.1086/302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez DC, Jaramillo C, Guhl F. Population structure and genetic variability of Rhodnius prolixus (Hemiptera: reduviidae) from different geographic areas of Colombia. Biomedica. 2007;27(Suppl 1):28–39. [PubMed] [Google Scholar]
- Marcilla A, Bargues MD, Ramsey JM, Magallon-Gastelum E, Salazar-Schettino PM, Abad-Franch F, Dujardin JP, Schofield CJ, Mas-Coma S. The ITS-2 of the nuclear rDNA as a molecular marker for populations, species, and phylogenetic relationships in Triatominae (Hemiptera: Reduviidae), vectors of Chagas disease. Mol Phylogenet Evol. 2001;18:136–142. doi: 10.1006/mpev.2000.0864. [DOI] [PubMed] [Google Scholar]
- Monteiro FA, Barrett TV, Fitzpatrick S, Cordon-Rosales C, Feliciangeli D, Beard CB. Molecular phylogeography of the Amazonian Chagas disease vectors Rhodnius prolixus and R. robustus. Mol Ecol. 2003;12:997–1006. doi: 10.1046/j.1365-294x.2003.01802.x. [DOI] [PubMed] [Google Scholar]
- Panzera F, Ferrandis I, Ramsey J, Salazar-Schettino PM, Cabrera M, Monroy C, Bargues MD, Mas-Coma S, O’Connor JE, Angulo VM, Jaramillo N, Perez R. Genome size determination in chagas disease transmitting bugs (hemiptera-triatominae) by flow cytometry. The American journal of tropical medicine and hygiene. 2007;76:516–521. [PubMed] [Google Scholar]
- Pavan MG, Monteiro FA. A multiplex PCR assay that separates Rhodnius prolixus from members of the Rhodnius robustus cryptic species complex (Hemiptera: Reduviidae) Trop Med Int Health. 2007;12:751–758. doi: 10.1111/j.1365-3156.2007.01845.x. [DOI] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Ribeiro JM, Hazzard JM, Nussenzveig RH, Champagne DE, Walker FA. Reversible binding of nitric oxide by a salivary heme protein from a bloodsucking insect. Science. 1993;260:539–541. doi: 10.1126/science.8386393. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Senior K. Chagas disease: moving towards global elimination. Lancet Infect Dis. 2007;7:572. doi: 10.1016/s1473-3099(07)70194-9. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tatusova T. Genomic databases and resources at the National Center for Biotechnology Information. Methods Mol Biol. 2010;609:17–44. doi: 10.1007/978-1-60327-241-4_2. [DOI] [PubMed] [Google Scholar]
- Vesgueiro FT, Demari-Silva B, Malafronte Rdos S, Sallum MA, Marrelli MT. Intragenomic variation in the second internal transcribed spacer of the ribosomal DNA of species of the genera Culex and Lutzia (Diptera: Culicidae) Mem Inst Oswaldo Cruz. 2011;106:1–8. doi: 10.1590/s0074-02762011000100001. [DOI] [PubMed] [Google Scholar]
- Wigglesworth V. The physiology of insect cuticle. Annual Reviews in Entomology. 1957;2:37–54. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.