

The survival motor neuron (SMN) is known to be involved in the biogenesis of small nuclear RNPs. This study provides evidence that the SMN may also be involved in axonal trafficking of mRNPs. Specifically, in situ hybridization and immuno-fluorescence experiments indicate that many SMN-associated mRNPs are found in neurites and axons, suggesting that SMN deficiency might result in mislocalization of mRNAs required for axonogenesis.
Keywords: spinal muscular atrophy, SMN, mRNA localization, axonal mRNAs
Abstract
Spinal muscular atrophy is a neuromuscular disease resulting from mutations in the SMN1 gene, which encodes the survival motor neuron (SMN) protein. SMN is part of a large complex that is essential for the biogenesis of spliceosomal small nuclear RNPs. SMN also colocalizes with mRNAs in granules that are actively transported in neuronal processes, supporting the hypothesis that SMN is involved in axonal trafficking of mRNPs. Here, we have performed a genome-wide analysis of RNAs present in complexes containing the SMN protein and identified more than 200 mRNAs associated with SMN in differentiated NSC-34 motor neuron-like cells. Remarkably, ∼30% are described to localize in axons of different neuron types. In situ hybridization and immuno-fluorescence experiments performed on several candidates indicate that these mRNAs colocalize with the SMN protein in neurites and axons of differentiated NSC-34 cells. Moreover, they localize in cell processes in an SMN-dependent manner. Thus, low SMN levels might result in localization deficiencies of mRNAs required for axonogenesis.
INTRODUCTION
Spinal muscular atrophy is a neuromuscular disease characterized by the selective degeneration of motor neurons in the anterior horns of the spinal cord (Van Meerbeke and Sumner 2011). With a prevalence of one in 6000 live births, SMA is a prominent autosomal-recessive disorder and a frequent cause of infant mortality (Sendtner 2001). Most SMA cases (>96%) are due to mutations in the SMN1 gene, which encodes the survival motor neuron protein (Lefebvre et al. 1995). Humans have a second copy of this gene, termed SMN2, which is unaffected in SMA patients but cannot generally compensate for the SMN1 mutations because ∼80% of the SMN2 transcripts lack exon 7 and encode a protein isoform that is rapidly degraded (Cho and Dreyfuss 2010). Some SMA patients have additional copies of the SMN2 gene and display less severe symptoms, indicating that the severity of the disease directly correlates with the amount of functional SMN protein (Lefebvre et al. 1997).
The SMN protein is part of a large complex comprising Unrip and seven Gemin proteins (Gemins 2–8) that is essential for the biogenesis of small nuclear ribonucleoprotein particles (snRNPs) (Fischer et al. 1997; Pellizzoni et al. 1998; Meister et al. 2000; for review, see Coady and Lorson 2011; Workman et al. 2012). These particles are major components of the spliceosome, the machinery that carries out pre-mRNA splicing (Wahl et al. 2009). It has been shown that extracts from SMA patient cells have a lower snRNP assembly capacity (Wan et al. 2005), and we have previously reported that the level of the tri-snRNP involved in the splicing of minor introns is dramatically decreased in lymphoblasts from a type I SMA patient (Boulisfane et al. 2011). In support of a role of minor splicing alterations in SMA, an SMN-dependent U12 splicing event was recently shown to be essential for motor circuit function in Drosophila and zebrafish (Lotti et al. 2012).
Overall, these observations suggest a causal relationship between disrupted assembly of snRNPs, splicing alterations, and SMA. Using SMA animal models, it has been reported that SMN deficiency, similar to that occurring in severe SMA, alters the stoichiometry of snRNAs and causes widespread pre-mRNA splicing defects in numerous transcripts of diverse genes, preferentially those containing a large number of introns (Gabanella et al. 2007; Zhang et al. 2008). In agreement with the observations that SMN is ubiquitously expressed in all tissues and is essential for viability in diverse eukaryotic organisms (Wang and Dreyfuss 2001; Paushkin et al. 2002; Campion et al. 2010), such defects were observed in different tissues, suggesting that SMA is a general splicing disease. However, most of the splicing defects were recently reported to occur late in SMA and to represent a consequence of cell injury (Bäumer et al. 2009). Whether the specific degeneration of motor neurons is caused by one or more aberrantly spliced transcripts or by the cumulative effect of numerous splicing alterations remains to be determined.
SMN has also been proposed to exert a neuron-specific role in axonal mRNA transport. The first indication for this activity is the finding that SMN interacts with the different hnRNP Q protein isoforms and, at a lower extent, with the hnRNP R protein that binds to the 3′-untranslated region of the β-actin mRNA (Mourelatos et al. 2001; Rossoll et al. 2002). Moreover, motor neurons isolated from an SMA mouse model show reduced levels of β-actin mRNA at distal axons and growth cones, suggesting that SMN and hnRNP R modulate axonal mRNA transport (Rossoll et al. 2003). In support of this, SMN has been detected in cytoplasmic granules exhibiting rapid and bidirectional movements along axons of primary neurons (Zhang et al. 2003). More recently, the candidate plasticity-related gene 15 (Cpg15) mRNA that encodes neuritin has been reported to colocalize with SMN in motor neuron axons, and, as already observed for β-actin mRNA, Cpg15 mRNA levels are reduced in neurites of SMN-depleted cortical neurons (Akten et al. 2011).
Further evidence supporting an important role of SMN in axonal trafficking comes from numerous studies reporting its association with a growing number of proteins involved in the regulation of mRNA transport, stability, and/or local translation (Fallini et al. 2012). Some SMN-interacting RNA-binding proteins (RBPs), namely, hnRNP U, hnRNP Q, FMRP, and EWS have been identified in RNA transport granules (Kanai et al. 2004), while others, such as KSRP, HuD, and TIA-R, regulate the stability of ARE-containing transcripts and are components of cytoplasmic stress granules (Von Roretz et al. 2011). Based on genome-wide studies identifying numerous mRNA targets of SMN-interacting RBPs, such as HuD (Bolognani and Perrone-Bizzozero 2008), FMRP (Darnell et al. 2005), TIA-R (Kim et al. 2007), or TDP-43 (Sephton et al. 2011), and studies revealing that SMN knockdown results in a reduction of poly(A) mRNA levels in the axonal compartment of primary motor neurons (Fallini et al. 2011), SMN-containing complexes have been proposed to participate in the axonal localization of a large number of mRNAs. However, the full repertoire of SMN-associated RNAs remains to be characterized.
Here, we have performed a genome-wide study of RNA species present in mRNP complexes containing the SMN protein (SMN) using murine motor neuron-like NSC-34 cells and RNA immunoprecipitation experiments coupled to microarray analyses (RIP-Chip) (Keene et al. 2006). This approach identified a subset of mRNAs already described for their localization in axons of different neuron types. Fluorescent in situ hybridization (FISH) and immunofluorescence (IF) experiments indicated that several mRNAs colocalize with the SMN protein in neurites and axons of differentiated NSC-34 cells. Interestingly, we show that axonal localization of some of these mRNAs is altered in SMN-depleted cells, suggesting that SMN deficiency could result in the mislocalization of numerous mRNPs required for axon growth, as well as for neuromuscular junction maturation and/or maintenance.
RESULTS
SMN complexes predominantly contain mRNA species
To identify RNA species associated with SMN-containing complexes, we constructed an NSC-34 stable cell line expressing a 3xFlag-SMN fusion protein under control of a doxycycline-inducible promoter. Murine NSC-34 cells can be induced to differentiate in motor neuron-like cells by various stimuli and express properties of motor neurons, such as generation of action potentials, expression of neurofilament triplet proteins, and acetylcholine synthesis (Cashman et al. 1992). After differentiation for 48 h in the presence of low serum concentration to allow outgrowth of neurites, 3xFlag-SMN and mock-transfected NSC-34 cells were induced with doxycycline for 3 d (Fig. 1A). Western blot analysis showed that the doxycycline treatment did not induce a massive overexpression of the 3xFlag-SMN protein since the amounts of exogenous and endogenous SMN proteins levels were very similar (Fig. 1B). Following in vivo UV cross-linking (Ule et al. 2003) and immunoprecipitation (IP) with an anti-Flag antibody, complexes were eluted with a 3xFlag peptide and used for Western blot analyses and purification of associated RNA species (Fig. 1A).
FIGURE 1.

Identification of SMN-associated RNAs. (A) Flow chart showing the sequential steps used for identification of SMN-associated RNAs at genome-wide scale. (B) Western blot analysis of endogenous (Endo-SMN) and exogenous (Flag-SMN) SMN expression levels in noninduced (NI) and induced (I) 3xFlag-SMN NSC-34 cells. (C) Western blot analysis of SMN-interacting hnRNP Q/R proteins in total extracts and immunoprecipitates from NSC-34 cells expressing 3xFlag-SMN (SMN) or no 3xFlag construct (Ctl). Total corresponds to ∼1% of the input used for IP. (D) RT-PCR analysis of β-actin (Actb) and Cpg15 (Nrn1) mRNAs purified from IP samples.
To validate the IP step, we tested for the SMN-interacting RBPs hnRNP Q and hnRNP R proteins (Mourelatos et al. 2001; Rossoll et al. 2002) in the eluted complexes. As shown in Figure 1C, hnRNP Q isoforms were co-IP with 3xFlag-SMN protein, while hnRNP R was not, suggesting its weaker interaction with SMN. Consistent with this, it was previously shown that hnRNP Q isoforms, but not hnRNP R, bind directly to SMN (Mourelatos et al. 2001). RT-PCR experiments on RNA purified from the IP complexes measured the enrichment of two mRNAs, β-actin and Cpg15, that colocalize with SMN in motor neuron axons (Mourelatos et al. 2001; Rossoll et al. 2002; Akten et al. 2011). As shown in Figure 1D, Cpg15 (Nrn1 in mouse) transcripts were significantly enriched in the SMN IP, whereas the signal corresponding to β-actin mRNAs was only slightly higher in SMN IP than in the control. This observation confirms previous results showing that β-actin mRNAs are only weakly enriched in SMN IPs relative to Cpg15 (Akten et al. 2011).
RNA species purified from independent SMN and control IPs were then used to hybridize Mouse GeneChip 1.0 ST Arrays. Following normalization of probe intensity levels, Z-scores, Z-ratio, and associated P-values were determined as described in Materials and Methods. Analysis of the RNA species enriched in the 3xFlag-SMN IPs revealed that 537 probe sets, corresponding to 411 known genes, had a Z-ratio >2.038 (associated P-value < 0.05). The complete list of the 537 probe sets is shown in Supplemental Data Set S1.
These RNA species belong to different classes, including small and long noncoding RNAs, as well as mRNAs (Fig. 2A). The presence of noncoding RNAs, e.g., snRNAs, snoRNAs, scaRNAs, and rRNAs, was anticipated, given the well-established role of SMN in snRNP biogenesis and in the formation of Cajal bodies, along with numerous studies reporting interactions of the SMN protein with proteins of snoRNPs or its localization in specific subnucleolar structures (Terns and Terns 2001; Cioce and Lamond 2005; Chari et al. 2009; Coady and Lorson 2011). The association of tRNAs and miRNAs with SMN is likely explained by the observations that components of the SMN complex, such as Gemin4 and Gemin5, are present in different complexes containing numerous miRNAs (Mourelatos et al. 2002) and involved in the modulation of translation activity (Pacheco et al. 2009), respectively.
FIGURE 2.

Classification of SMN-associated RNAs and KEGG pathways enrichment analyses. (A) SMN-associated RNAs identified by microarray analyses were manually classified into different classes based on NCBI annotations. The pie charts represent the classification of the 411 SMN-associated RNAs. (B) Enriched KEGG pathways for SMN-associated mRNAs. The graphs show the number of corresponding genes observed in each pathway (light gray) and the expected number in the genome (dark gray). All the pathways presented in the graphs have P-values <0.01.
Interestingly, mRNAs constituted >50% of the RNAs associated with the SMN protein. The 50 probe sets corresponding to mRNAs with the highest Z-ratios are shown in Table 1. We used the links to the Kyoto Encyclopedia of Genes and Genomes (KEGG) to identify the biological pathways represented by the SMN-associated mRNAs. As shown in Figure 2B, the “ribosome” pathway contains the highest number of SMN-associated mRNAs with an observed/expected ratio of ∼80. Pathways related to carbohydrate and amino acid metabolism were also found to be significantly enriched in SMN-associated mRNAs. Interestingly, Gene Ontology analyses of different axonal mRNA repertoires have revealed that transcripts coding for proteins involved in protein synthesis and metabolism are also enriched in axons (for review, see Jung et al. 2011).
TABLE 1.
Top 50 SMN-associated mRNAs
To validate our microarray data, we performed semi-quantitative RT-PCR analyses with primers specific for a subset of mRNAs chosen among the lists shown in Table 1 and Supplemental Data Set S1. As a negative control, we analyzed the enrichment of the Cyp51 mRNA, which exhibits a negative Z-ratio (−3.61) and is therefore unlikely to be associated with SMN. As shown in Figure 3A, the mRNAs analyzed from Supplemental Data Set S1 were found to be enriched in SMN IPs, although at different levels. Importantly, no significant enrichment was observed for Cyp51 mRNA. Real-time PCR analyses performed on RNA purified from three independent IPs (Fig. 3B) confirmed that enrichment levels of Anxa2, Pfdn1, and Vim transcripts correlated very well to their respective Z-ratios (6.18, 2.55, and 1.66), demonstrating that a large number of mRNAs interact with SMN-containing complexes.
FIGURE 3.

RT-PCR validation of microarray data. (A) RT-PCR analysis of SMN-associated mRNAs. The Cyp51 mRNA is used as a negative control for enrichment in SMN IPs. (B) Q-PCR validation of SMN-associated mRNAs exhibiting decreasing Z-ratios. Fold enrichment corresponds to 2(Ct IP Ctl − Ct IP SMN) ± SEM (n = 3).
Enriched mRNAs colocalize with endogenous SMN protein in neurites of NSC-34 cells
The repertoires of axonally localized mRNAs have been identified by microarray or SAGE analyses for different neuronal cell types; their comparison reveals that they are highly similar (Jung and Holt 2011; Jung et al. 2011, 2012). To identify SMN-associated mRNAs in these repertoires, we compared our microarray data with the transcriptome analyses of (1) axons from dorsal root ganglion neurons (Willis et al. 2007), (2) uninjured and regenerating cortical axons (Taylor et al. 2009), and (3) embryonic and adult sensory axons (Gumy et al. 2011). This identified 75 localized mRNAs present in SMN complexes (Fig. 4). We selected mRNAs encoding Annexin A2 (Anxa2) and Selenoprotein W1 (SepW1) for further studies because of their suggested role in neuritogenesis and the prevention of neurodegeneration (Jacovina et al. 2001; Wirth et al. 2010). We also analyzed the colocalization of SMN with Vim transcripts, which were identified in most of the mentioned transcriptome studies, and Cox4i2 mRNAs, since mRNAs encoding other subunits of cytochrome c oxidase were detected in axons (Taylor et al. 2009; Gumy et al. 2011) and because of their high enrichment in SMN-containing complexes (Z-ratio 3.24) (Fig. 3A).
FIGURE 4.

SMN interacts with a subset of mRNAs known to be localized in axons. (A) Venn diagram (http://bioinfogp.cnb.csic.es/tools/venny/) showing overlap between SMN-associated mRNAs and transcripts identified in transcriptome studies of axons from dorsal root ganglion neurons (Willis et al. 2007) and uninjured and regenerating cortical axons (Taylor et al. 2009), as well as embryonic and adult sensory axons (Gumy et al. 2011). (B) List of SMN-associated mRNAs already identified in axons.
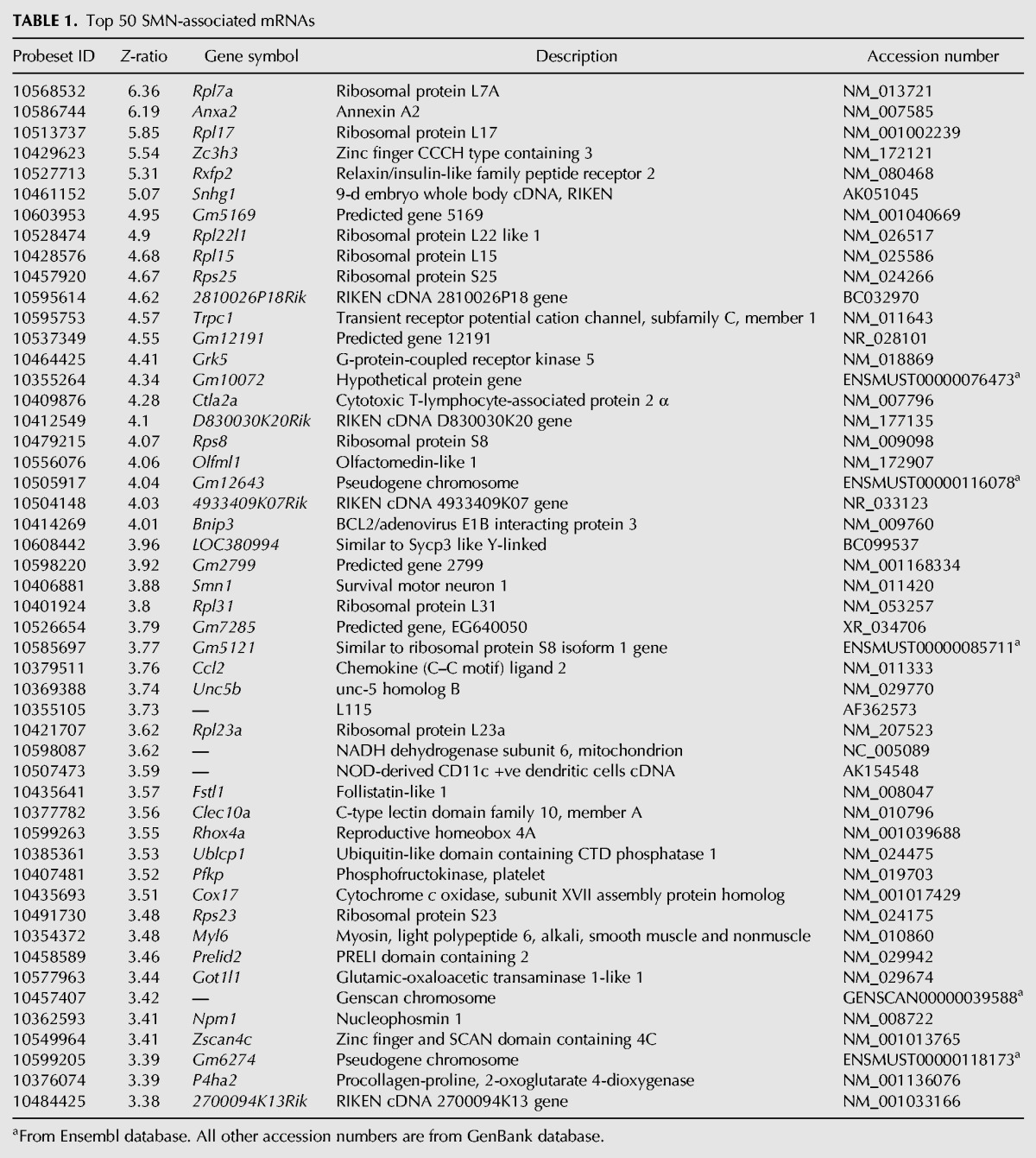
We first performed FISH experiments coupled to IF analysis of the endogenous SMN protein in differentiated NSC-34 cells. As shown in Figure 5A, punctate staining (about 200 foci per neurite) along neuritic processes was observed for each of the four mRNAs, indicating that they are efficiently transported in NSC-34 neurites. Under the same experimental conditions, only a few spots (about 10 per neurite) (data not shown) were observed with probes specific for H1f0 transcripts, whose localization is restricted to the cell body (Brittis et al. 2002). Variable fractions of the RNA spots were found to colocalize with the SMN protein. Quantitative analysis was performed with the Imaris software as described in Materials and Methods. Results showed that the colocalization with SMN was statistically nonrandom with mean values of 22.1% ± 1.9% (mean ± SEM; n = 9) for Anxa2, 19.0% ± 2.3% (n = 5) for Cox4i2, 16.4% ± 0.8% (n = 11) for SepW1, and 11.8% ± 1.6% (n = 5) for Vim mRNAs (Fig. 5B). This indicates that SMN-containing complexes may assist the assembly of specific mRNAs into mRNP particles and their targeting to the neuronal transport system along neurites.
FIGURE 5.
Differential colocalization of Anxa2, Cox4i2, SepW1, and Vim mRNAs with endogenous or exogenous SMN proteins in neurites of differentiated NSC-34 cells. (A) Endogenous SMN protein (green spots) colocalizes with variable fractions of spots corresponding to the indicated mRNAs (red spots). Images generated by the Imaris software following removal of noncolocalized spots (filter) are shown. Scale bar, 10 μm. (B) Histograms showing the percentage ± SEM of mRNA spots colocalizing with SMN spots. (C) Histograms showing the percentage ± SEM of mRNA spots colocalizing with 3xFlag-SMN spots. (*) P-value <0.05; (**) P-value <0.01.
To determine whether similar colocalization also occurred with the ectopic SMN protein, we also performed FISH and IF analyses in differentiated NSC-34 cells expressing the 3xFlag-SMN protein (Fig. 5C). Quantitative analysis indicated that 13.9% ± 1.9% (n = 3) of Anxa2, 15.8% ± 2.4% (n = 3) of Cox4i2, 7.8% ± 2.2% (n = 6) of SepW1, and 6.8% ± 1.5% (n = 3) of Vim mRNA spots were colocalized with the exogenous protein along neurites. Except for Cox4i2, these values were significantly lower than those observed with the endogenous SMN protein (Fig. 5B). This suggests that mRNAs initially associated with the endogenous protein likely redistribute between the endogenous and the ectopic 3xFlag-SMN proteins. Such redistribution could result from limiting amounts of mRNA and/or RNA binding proteins that associate with SMN in mRNPs.
Localization of some, but not all, SMN-associated mRNAs is reduced in neurites of SMN-depleted NSC-34 cells
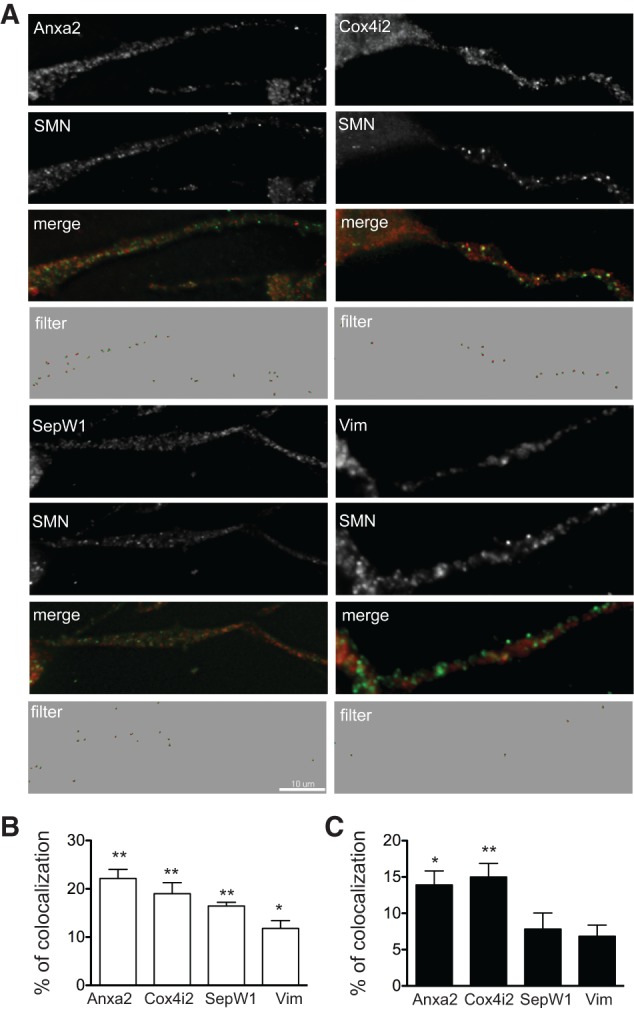
We then determined the impact of SMN depletion on the localization of SMN-associated mRNAs. In differentiated shSMN-NSC-34 cells, levels of the endogenous SMN protein can be reduced by 50%–60% following 3 d of doxycycline-induced expression of shRNAs (Fig. 6A). Using FISH experiments coupled to IF analysis, the numbers of spots corresponding to the SMN protein and the different mRNAs were compared in neurites of either induced or noninduced cells. As shown in Figure 6, B and C, induction of shRNA expression for 72 h resulted in a 45%–73% decrease of the number of SMN spots in neurites of shSMN-NSC-34 cells. Strikingly, shRNA expression also resulted in a dramatic decrease of the number of Anxa2 and Cox4i2 mRNA spots (73% and 78%, respectively) while only moderately diminishing the number of SepW1 and Vim mRNA spots (17% and 16.7% decrease). Since this could reflect a global reduction of mRNA in SMN-depleted cells, we performed RT-PCR analyses of total RNA purified from induced and noninduced shSMN-NSC-34 cells. As shown in Figure 6D, the reduction of SMN protein level was correlated with an increase of Anxa2 and SepW1 mRNA levels (50% and 25%, respectively) and with a 25% and 12% decrease of Cox4i2 and Vim mRNA levels.
FIGURE 6.

Localization of distinct mRNAs in neurites of differentiated shSMN-NSC-34 cells is not affected at the same level upon SMN deficiency. (A) RT-PCR (left panel) and Western blot analysis (right panel) of SMN mRNA and protein levels in noninduced and induced differentiated shSMN-NSC-34 cells. (B) Spots corresponding to the indicated mRNAs (red) and colocalizing with the endogenous SMN protein (green spots) in neurites of noninduced (NI) or induced (I) shSMN-NSC-34 cells are shown (merge). Imaris-generated images (red filter) show the amounts of RNA spots localized in neurites before and following SMN depletion. Scale bar, 10 μm. (C) Histograms showing the average number ± SEM of SMN and RNA spots in neurites of noninduced (NI) or induced (I) shSMN-NSC-34 cells. (*) P-value <0.05; (**) P-value <0.01; (ns) nonstatistically significant. (D) RT-PCR analysis of Anxa2, Cox4i2, SepW1, and Vim mRNA levels in SMN-depleted NSC-34 cells. Histograms showing the percentage ± SEM (n = 3) of variation for the indicated mRNA in induced (I) compared with noninduced (NI) cells. (*) P-value <0.05; (**) P-value <0.01.
Thus, the localization of these SMN-associated mRNAs in neurites of NSC-34 cells is differentially affected by the reduction of SMN expression, notably that of Anxa2 and Cox4i2 mRNAs. This indicates that SMN is directly involved in the transport of these transcripts (see Discussion).
Localization of SMN-associated mRNAs is reduced in axons of SMN-depleted NSC-34 cells
Since SMN is required for motor axon development and maintenance (for review, see Fallini et al. 2012), we tested whether the localization of SMN-associated mRNAs is also altered in axons of SMN-depleted NSC-34 cells. We used microfluidic chambers (MFC) in which axons are isolated in a specific compartment and that previously allowed the study of axonally localized mRNA populations (Taylor et al. 2005, 2009; Park et al. 2006). Uninduced shSMN-NSC-34 cells grown in MFCs were used to measure colocalization of endogenous SMN protein with Anxa2 and Cox4i2 mRNAs in axons. As shown in Figure 7, 18.7% ± 4.1% (n = 4) of Anxa2 and 19.3% ± 3.0% (n = 4) of Cox4i2 mRNA spots colocalized with SMN in axonal processes (RNAvsSMN). For the SMN spots, 23.5% ± 3.0% (n = 4) and 24.6% ± 4.5% (n = 4) colocalized with Anxa2 and Cox4i2 mRNA granules (SMNvsRNA). Notably, only a few spots (about five to 10 per axon) were observed for the dendritic marker mRNA Camk2a (Steward and Halpain 1999), indicating that dendrites did not extend into the axonal side of the chamber (data not shown).
FIGURE 7.
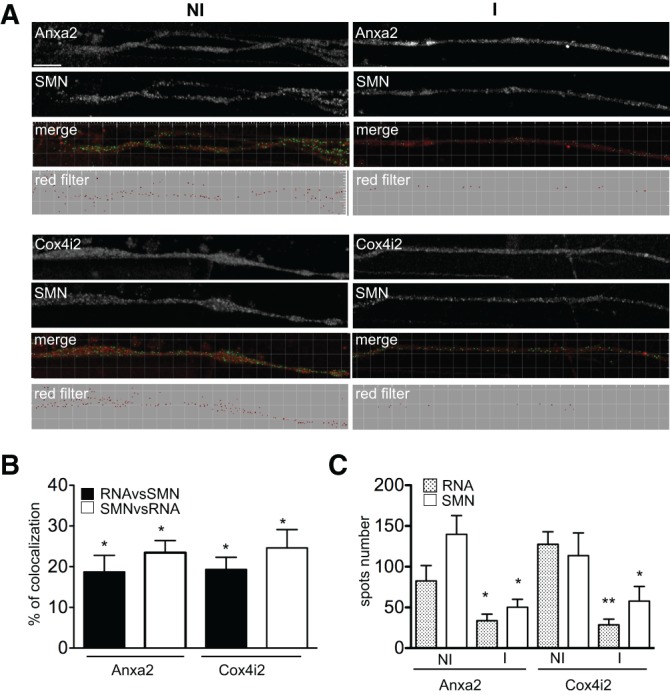
Localization of Anxa2 and Cox4i2 mRNAs in axons of differentiated shSMN-NSC-34 cells is not affected at the same level upon SMN deficiency. (A) Spots corresponding to the indicated mRNAs (red) and colocalizing with the endogenous SMN protein (green spots) in axons of noninduced (NI) or induced (I) shSMN-NSC-34 cells are shown (merge). Imaris-generated images (red filter) show the amounts of RNA spots localized in axons before and following SMN depletion. Scale bar, 10 μm. (B) Histograms showing the percentage ± SEM of mRNA granules colocalizing with SMN spots (black) and of SMN spots colocalizing with mRNA granules (white). (*) P-value <0.05. (C) Histograms showing the average number ± SEM of SMN and RNA spots in axons of noninduced (NI) or induced (I) shSMN-NSC-34 cells. (*) P-value <0.05; (**) P-value <0.01.
A 72-h induction of shRNA expression led to a 54%–64% decrease of the number of SMN spots in the axons of shSMN-NSC-34 cells (Fig. 7A,C). As observed above in neurites (Fig. 6), this decrease correlated with an extensive reduction of the Anxa2 (60%) and Cox4i2 (78%) mRNA spots. This confirms the key role of SMN in the regulation of their axonal levels.
DISCUSSION
It is now well established that mRNA localization and local protein synthesis play key roles in the processes of synaptic plasticity, development, and maintenance (Wang et al. 2010; Swanger and Bassell 2011). Numerous studies showing that SMN granules exhibit rapid and bidirectional movements in axons of primary neurons (Rossoll et al. 2003; Fallini et al. 2010), together with observations that SMN interacts with a growing number of RNA binding proteins, have led to the hypothesis that SMN plays a crucial role in the axonal localization of mRNPs. In support of this, polyadenylated mRNA levels are decreased in axons of SMN-deficient motor neurons (Fallini et al. 2011). To date, only β-actin and Cpg15 mRNAs were found to be associated with SMN in motor neuron axons (Rossoll et al. 2003; Akten et al. 2011).
We present the first genome-wide analysis of SMN-associated RNA species. This approach identified more than 200 mRNAs potentially associated with the SMN protein, but the number of SMN-associated mRNAs could be much greater. For example, neither β-actin nor Cpg15/Nrn1 mRNAs were significantly enriched in SMN-IPs, based on their associated Z-ratio and P-value. The associated Z-ratio is negative for β-actin transcripts, and RT-PCR analysis confirmed their nonenrichment in SMN IPs (Fig. 1D). Interestingly, our Western blot analysis showed that hnRNP Q isoforms, but not hnRNP R, are co-IPed with 3xFlag-SMN proteins (Fig. 1C). Since Glinka et al. (2010) reported that hnRNP R is necessary for axonal β-actin mRNA translocation in motor neurons, this could explain the lack of enrichment of β-actin transcripts in our IP experiments. In the case of Cpg15/Nrn1 and Vim mRNAs, for which the associated Z-ratios (0.40 and 1.66, respectively) are below the threshold retained in our statistical analyses, RT-PCR experiments indicated that these transcripts are enriched in SMN IPs (Fig. 1D). Moreover, we find Vim mRNAs colocalized in endogenous SMN-containing granules along neurites of differentiated NSC-34 cells (Fig. 5).
Interestingly, a significant subset (∼30%) of the SMN-associated mRNAs was already reported to be localized in axons from different neuronal cell types. Using microfluidic chambers, we have shown that two mRNAs (Anxa2 and Cox4i2) colocalize with the SMN protein in axons from differentiated NSC-34 cells and that their axonal localization is dramatically reduced in SMN-deficient cells. Along with previous studies reporting the significant reduction of poly(A) mRNA levels in axons and growth cones of SMN-deficient motor neurons (Fallini et al. 2011), these observations support the notion that the SMN protein is directly involved in the axonal localization of a large number of mRNA species.
Our results raise the interesting question of why the axonal localization of several SMN-associated mRNAs is so highly reduced upon SMN knockdown. Notably, the localization of Anxa2 and Cox4i2 mRNAs is much more strongly affected in SMN-depleted NSC-34 cells than might be expected from our colocalization studies. While the ∼60% decrease of the number of axonal SMN-positive granules correlates with a 60% and 78% reduction of Anxa2 and Cox4i2 mRNA spots, respectively, ∼20% of these mRNAs colocalize with endogenous SMN in noninduced shSMN-NSC-34 cells (Fig. 7). This is similar to Cpg15 mRNA, whose localization in neurites of SMN-depleted cortical neurons is reduced by ∼60%, whereas ∼37% of Cpg15 mRNA spots are colocalized with SMN granules in embryonic mouse motor neuron axons (Akten et al. 2011). However, Cpg15 transcript levels are also decreased by 60% in cell bodies of SMN-depleted cortical neurons, raising the possibility that a global reduction of Cpg15 expression accounts for the observed localization defect. The situation is different for Anxa2 and Cox4i2 mRNAs. The change in their global expression levels seen with SMN depletion (50% increase for Anxa2 mRNA; 25% decrease for Cox4i2 mRNA) (Fig. 6D) cannot account for the dramatic reduction in their axonal localization. One possible explanation is that SMN transiently interacts with the mRNPs during the transport process and/or dissociates from the complex after it has been correctly localized. In addition, an indirect mechanism could also impair the axonal localization of mRNPs. Indeed, SMN-associated RBPs exhibit reduced protein levels upon SMN deficiency. The expression levels of hnRNP Q, as well as that of Gemins 2 and 3, which colocalize with SMN in growth cones of motor neurons (Zhang et al. 2006), are significantly reduced in cells from SMA patients, this notably in correlation to the clinical severity of the disease (Helmken et al. 2003). A similar reduction was also noted for the KSRP protein in an SMN-depleted neuronal cell line and in spinal cords of a SMA mouse model (Tadesse et al. 2008), as well as for HuD in axons of primary motor neurons (Fallini et al. 2011). Interestingly, HuD overexpression has been reported to rescue SMA-like neuronal defects in a Smn-knockdown MN-1 cell line (Hubers et al. 2011). Thus, SMN deficiency can dramatically impair mRNA axonal localization through different synergistic mechanisms: (1) global down-regulation of mRNA levels, possibly resulting from splicing alterations, nuclear retention, and subsequent degradation); (2) defects in assembly and/or transport of mRNP particles; and (3) down-regulation of the level of SMN-associated RNA binding proteins also involved in mRNA axonal transport.
Alterations in axonal mRNA transport could be highly relevant to the SMA disease, since the defects in axonal outgrowth and branching observed in a SMA zebrafish model (Winkler et al. 2005) are not rescued by SMN mutants competent for snRNP assembly (Carrel et al. 2006). In the light of the morphological and functional defects of neuromuscular junctions (NMJs) in different SMA mouse models (Cifuentes-Diaz et al. 2002; Kariya et al. 2008; Murray et al. 2008; Kong et al. 2009; Bowerman et al. 2012), it is also tempting to propose that alterations of axonal mRNA transport and local translation at the synapses would affect NMJ maturation and/or maintenance. Consistent with this, about 30 of the SMN-associated mRNAs identified in the present study have been reported to be localized in growth cones of retinal ganglion cell (RGC) axons, some of them such as Vim (encoding Vimentin), Tmsb10 (encoding Thymosin β10), and Ptma (encoding Prothymosin α) being involved in axon guidance and/or synaptogenesis (Zivraj et al. 2010). Further characterization of the role of SMN-associated mRNAs in axonal growth and/or synaptogenesis will bring further insights into the role of SMN-mediated axonal transport of mRNPs in SMA.
MATERIALS AND METHODS
SMN and shSMN expression vectors
The SMN cDNA was obtained following RT-PCR amplification of total RNA purified from spinal cord of E15 mouse embryos. SMN sequences were first cloned in the p3xFlag-CMV-10 expression vector (Sigma-Aldrich), and the resulting FLAG-SMN fusion was then transferred in the pTRE-Tight-Puro response plasmid to generate the pTRE-3xFlag-SMN vector.
The shSMN sequences cloned in the pGIPZ vector (Open Biosystems; Oligo ID: V2LMM_6035) were first subcloned into the pTRIPZ plasmid to allow doxycycline-inducible expression. Turbo-RFP sequences were then removed from the resulting construct to generate the pTRIPZ-shSMN-ΔRFP plasmid in order to permit ISH-IF studies in doxycycline-induced cells. Cloning strategies are available upon request.
Cell culture conditions and construction of SMN-inducible cell lines
NSC-34 cells were maintained in DMEM supplemented with 10% fetal bovine serum (tetracycline-free, Biowest). Differentiation of the cells was obtained by growing them in the presence of low serum concentration (1%) for 2 d. To obtain SMN-inducible cell lines, NSC-34 cells were first transfected with the pUHD172-1-neo expressing the rtTA gene under the control of the CMV promoter. Following G418 selection (0.7 mg/mL), independent clones were expanded and transiently transfected with a pTRE-Tight-Puro plasmid expressing GFP sequences. The transactivation efficiency in response to induction with various doxycycline concentrations (0.1–3 μg/mL, Sigma-Aldrich) was determined by fluorescence microscopy. Cells showing the most efficient induction were then transfected with the pTRE-3xFlag-SMN vectors, and independent clones were selected in the presence of G418 and puromycin (3 μg/mL). For the construction of shSMN-inducible cell lines, NSC-34 cells were transfected with the pTRIPZ-shSMN-ΔRFP plasmid and selected in the presence of puromycin (3 μg/mL).
CLIP assays
NSC-34 cells containing the 3xFlag-SMN expression vector and NSC-34 control cells were induced to differentiate for 48 h and then incubated in the presence of doxycycline (3 μg/mL) for 3 d. Treated cells were resuspended in PBS, placed in a UV Crosslinker (Amersham Life Science), and irradiated one time for 400 mJ/cm2. Following lysis of the cells in the recommended buffer, immunoprecipitation was performed with anti-Flag M2 magnetic beads (Sigma-Aldrich) according to the manufacturer's instructions. Immunoprecipitated proteins were eluted with 3X-Flag peptide (Sigma-Aldrich) and used directly for Western blot analyses or digested with proteinase K before purification of coimmunoprecipitated RNAs by phenol:chloroform extraction.
Western blot analysis
Total extracts or immunoprecipitated proteins were separated on 10%–12% SDS-PAGE and blotted onto nitrocellulose membrane (Protran, Whatman). Membranes were probed with anti-hnRNP Q/R antibodies 18E4 (Abcam) at 1:500 dilution, anti-SMN (BD Transduction Laboratories) at 1:5000 dilution, anti-Flag M2 (Sigma-Aldrich) at 1:1000 dilution, and anti-tubulin YOL1/34 (Serotec) at 1:10 000 dilution. Following incubation with anti-mouse (Sigma-Aldrich), anti-rat (Santa Cruz Biotechnology), or Mouse true blot ultra IgHRP (Ebioscience) secondary antibodies, membranes were developed using the ECL kit (Roche).
RT-PCR analyses
Purified coimmunoprecipitated RNA was treated with RQ1 RNase free DNase (Promega), and cDNA was synthesized from 0.5 μg of RNA and pd(N)6 random oligonucleotides with a First strand cDNA synthesis kit (GE-Healthcare). For semi-quantitative RT–PCR analyses, one-tenth of the reaction was amplified for 25–35 cycles with GoTaq polymerase (Promega) according to the supplier's recommendations. The PCR products were separated on 1.5%–2.5% agarose gels containing ethidium bromide and visualized under UV light. The gel images were digitally captured and analyzed using the Image J software. Quantitative RT-PCR analyses were performed with one-twentieth of the cDNA synthesis reaction using the LightCycler 480 SYBR Green I Master kit (Roche) and the LightCycler 480 system. Primer sequences and PCR regimes are available upon request.
Microarray hybridizations and bioinformatic analyses
RNA was purified as mentioned above from independent IPs (three for 3xFlag-SMN and two for mock-transfected NSC-34 cells). Probe labeling was done using the GeneChip WT Double-Stranded cDNA Synthesis Kit (Affymetrix) following the manufacturer's protocol. Labeled probes were hybridized to Mouse GeneChip 1.0 ST Arrays (Affymetrix) using the manufacturer's protocol. Scanning and data collection were done by GeneChip Scanner 3000 7G (Affymetrix) and GeneChip Operating Software. Microarrays were analyzed with the specific R script (http://www.R-project.org/), which includes methods from the Bioconductor (Gentleman et al. 2004) tools package. Probes intensities were extracted from CEL files with the “expresso” function. According to Bolstad et al. (2003), this method has been designed to only correct background noise bias with Robust Multichip Average (RMA) (Irizarry et al. 2003) adjustments. Z-scores and ratios with associated P-values were calculated as described in Cheadle et al. (2003). Probe sets with Z-ratios >2.038 (associated P-values <0.05) were scored as potential SMN-associated mRNAs.
The full list of probe sets significantly enriched in 3xFlag-SMN IPs is shown in Supplemental Data Set S1.
Gene Ontology analyses
The WebGestalt program (http://bioinfo.vanderbilt.edu/webgestalt) (Zhang et al. 2005) was used to determine the frequencies and numbers of genes corresponding to SMN-associated mRNAs in the different KEGG biological pathways and to compare them with those in the mouse genome. A hypergeometric distribution test was used to determine the statistical significance of the observed over the expected frequency. P-values <0.01 were considered statistically significant, and only Gene Ontology categories with more than three genes in the SMN-associated mRNA data sets were considered.
FISH and IF experiments
Amine-modified oligonucleotides specific for the mRNA coding region were labeled with amine-reactive compound (Cy3, Invitrogen, Molecular Probes), a mix of six probes being used for each in situ hybridization. Cells were fixed with 4% paraformaldehyde in 1× PBS and then permeabilized with 70% ethanol overnight at 4°C. NSC-34 cells were hybridized at 37°C (40% formamide, 10% dextran sulfate, 10 µg of yeast tRNA, 1× SSC, 0.2% BSA, 0.2% VRC, and 10 ng of each probe) overnight and washed twice in 40% formamide, 1× SSC for 1 h and then in 1× SSC for 30 min. For immunofluorescence studies, cells were washed twice in B1 buffer (0.1 M Tris-HCl at pH 7.5, 0.15 M NaCl), permeabilized in B1 buffer containing 0.1% Triton X-100 for 10 min, and then blocked in PBS containing 1% Blocking Reagent (ROCHE), 0.1% Triton X-100 for 30 min at room temperature. Cells were incubated with the primary antibodies in the blocking buffer overnight at 4°C. Secondary antibodies were labeled with FITC or Cy3 and used at 1:600 dilution. Mouse monoclonal anti-SMN (BD) and mouse anti-Flag (Sigma-Aldrich) antibodies were used at 1:200 and 1:1000 dilution, respectively. Coverslips were mounted with Vectashield containing DAPI and then analyzed by fluorescence microscopy.
Image acquisition and processing
Images were acquired on a DMRA microscope equipped for epifluorescence, and with a 100× PlanApo objective. Digital images were recorded with a 12-bit C4795-NR CCD camera (Hamamatsu). The camera and microscope were controlled by MetaMorph software. Three-dimensional images were deconvolved with Huygens 2.3 software (Scientific Volume Imaging) using an MLE algorithm. Maximal image projections of the resulting stacks were then converted to 8-bit images and colorized with PhotoShop (Adobe).
For quantitative analyses of the RNA granules and SMN protein colocalization, we used the Spot function of Imaris (Bitplane), which allows us to consider only the signal present in foci on three-dimensional images. This software first applies a Gaussian filter of a given size (0.4 μm in this case) to remove small objects and then identifies local maxima above a given threshold (the same threshold was used for all images of a given data set). A distance of 0.4 μm between two spots was arbitrarily selected, and the Spot function was run in each channel. The spots that colocalized or did not colocalize were then counted. Each colocalization analysis was performed on a total of six to 15 neurites representing 60–145 spots. The significance of the colocalization data was determined with the unpaired student t-test (Prism software). Image acquisition and analysis were performed on workstations of the Montpellier RIO Imaging facility of the CNRS campus.
Microfluidic chambers
The AXIS (AXon ISolation device, Millipore) microfluidic chamber consisting of two compartments separated by a set of microgrooves (length 150 μm) was used as recommended by the manufacturer, with slight modifications. Briefly, glass-bottom dishes were washed once with 70% ethanol and rinsed with sterile water. Then plates were coated with 0.5 mg/mL poly-L-ornithine for 2 h at 37°C or overnight at 4°C. After PBS washes and plasma oven treatment, the silicon device was carefully stuck to the glass plate with the imprinted surface placed face down. Precoating with medium containing 1% serum was performed before seeding cells. The cell suspension (2 × 105 cells in 10 μL) was loaded directly into one compartment of the chamber. The AXIS device was placed into the incubator for 15 min, allowing cells to attach to the coated glass surface. Then, 400 μL and 200 μL of differentiated medium were added into the cellular and axonal compartments, respectively, in order to allow a fluidic gradient for the development of the axon through the microgrooves. Brain-derived neurotrophic factor (BDNF, 20 ng/mL) was added to the axonal compartment to attract developing axons. Eight days later, cells were treated with doxycycline for 72 h to induce shRNA expression.
DATA DEPOSITION
Microarray data have been deposited in the GEO database and are available through the series accession number GSE40197.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the CNRS and by grants of the AFM to J.S., SMA Europe to R.B., and ANR RNAtrans (ANR-10-Blanc Inter SVSE 4-001) to E.B. and F.R. N.B. was supported by a fellowship of the French Research Ministry. Thanks are due to Dr. S. Salinas for precious help with microfluidic chambers and to Dr. R.A. Hipskind for critical reading of the manuscript. Affymetrix microarrays were processed in the Microarray Core Facility of the Institute of Research on Biotherapy, CHRU-INSERM-UM1 Montpellier (http://irb.chu-montpellier.fr/). This work is dedicated to the memory of our friend and colleague Henry Neel.
REFERENCES
- Akten B, Kye MJ, Hao LT, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, et al. 2011. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA Cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci 108: 10337–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer D, Lee S, Nicholson G, Davies JL, Parkinson NJ, Murray LM, Gillingwater TH, Ansorge O, Davies KE, Talbot K. 2009. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 5: e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognani F, Perrone-Bizzozero NI. 2008. RNA–protein interactions and control of mRNA stability in neurons. J Neurosci Res 86: 481–489. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193. [DOI] [PubMed] [Google Scholar]
- Boulisfane N, Choleza M, Rage F, Neel H, Soret J, Bordonné R. 2011. Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum Mol Genet 20: 641–648. [DOI] [PubMed] [Google Scholar]
- Bowerman M, Murray LM, Beauvais A, Pinheiro B, Kothary R. 2012. A critical SMN threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul Disord 22: 263–276. [DOI] [PubMed] [Google Scholar]
- Brittis PA, Lu Q, Flanagan JG. 2002. Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell 110: 223–235. [DOI] [PubMed] [Google Scholar]
- Campion Y, Neel H, Gostan T, Soret J, Bordonné R. 2010. Specific splicing defects in S. pombe carrying a degron allele of the Survival of Motor Neuron gene. EMBO J 29: 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel TL, McWhorter ML, Workman E, Zhang H, Wolstencroft EC, Lorson C, Bassell GJ, Burghes AHM, Beattie CE. 2006. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci 26: 11014–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. 1992. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 194: 209–221. [DOI] [PubMed] [Google Scholar]
- Chari A, Paknia E, Fischer U. 2009. The role of RNP biogenesis in spinal muscular atrophy. Curr Opin Cell Biol 21: 387–393. [DOI] [PubMed] [Google Scholar]
- Cheadle C, Cho-Chung YS, Becker KG, Vawter MP. 2003. Application of z-score transformation to Affymetrix data. Appl Bioinformatics 2: 209–217. [PubMed] [Google Scholar]
- Cho S, Dreyfuss G. 2010. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev 24: 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J. 2002. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet 11: 1439–1447. [DOI] [PubMed] [Google Scholar]
- Cioce M, Lamond AI. 2005. Cajal bodies: A long history of discovery. Annu Rev Cell Dev Biol 21: 105–131. [DOI] [PubMed] [Google Scholar]
- Coady TH, Lorson CL. 2011. SMN in spinal muscular atrophy and snRNP biogenesis. Wiley Interdiscip Rev RNA 2: 546–564. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Mostovetsky O, Darnell RB. 2005. FMRP RNA targets: Identification and validation. Genes Brain Behav 4: 341–349. [DOI] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. 2010. High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol Neurodegener 5: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Zhang H, Su Y, Silani V, Singer RH, Rossoll W, Bassell GJ. 2011. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci 31: 3914–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. 2012. Spinal muscular atrophy: The role of SMN in axonal mRNA regulation. Brain Res 1462: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Liu Q, Dreyfuss G. 1997. The SMN–SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90: 1023–1029. [DOI] [PubMed] [Google Scholar]
- Gabanella F, Butchbach MER, Saieva L, Carissimi C, Burghes AHM, Pellizzoni L. 2007. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2: e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka M, Herrmann T, Funk N, Havlicek S, Rossoll W, Winkler C, Sendtner M. 2010. The heterogeneous nuclear ribonucleoprotein-R is necessary for axonal β-actin mRNA translocation in spinal motor neurons. Hum Mol Genet 19: 1951–1966. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Yeo GSH, Tung YCL, Zivraj KH, Willis D, Coppola G, Lam BYH, Twiss JL, Holt CE, Fawcett JW. 2011. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17: 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schöneborn S, Zerres K, Wirth B. 2003. Evidence for a modifying pathway in SMA discordant families: Reduced SMN level decreases the amount of its interacting partners and Htra2-β1. Hum Genet 114: 11–21. [DOI] [PubMed] [Google Scholar]
- Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Côté J. 2011. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet 20: 553–579. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacovina AT, Zhong F, Khazanova E, Lev E, Deora AB, Hajjar KA. 2001. Neuritogenesis and the nerve growth factor-induced differentiation of PC-12 cells requires annexin II-mediated plasmin generation. J Biol Chem 276: 49350–49358. [DOI] [PubMed] [Google Scholar]
- Jung H, Holt CE. 2011. Local translation of mRNAs in neural development. Wiley Interdiscip Rev RNA 2: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, O'Hare CM, Holt CE. 2011. Translational regulation in growth cones. Curr Opin Genet Dev 21: 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE. 2012. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, Hirokawa N. 2004. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 43: 513–525. [DOI] [PubMed] [Google Scholar]
- Kariya S, Park G-H, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. 2008. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17: 2552–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, Friedersdorf MB. 2006. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc 1: 302–307. [DOI] [PubMed] [Google Scholar]
- Kim HS, Kuwano Y, Zhan M, Pullmann R Jr, Mazan-Mamczarz K, Li H, Kedersha N, Anderson P, Wilce MCJ, Gorospe M, et al. 2007. Elucidation of a C-rich signature motif in target mRNAs of RNA-binding protein TIAR. Mol Cell Biol 27: 6806–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. 2009. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29: 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M. 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80: 155–165. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. 1997. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16: 265–269. [DOI] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao LT, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al. 2012. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151: 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Bühler D, Laggerbauer B, Zobawa M, Lottspeich F, Fischer U. 2000. Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet 9: 1977–1986. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Abel L, Yong J, Kataoka N, Dreyfuss G. 2001. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J 20: 5443–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. 2002. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16: 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. 2008. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet 17: 949–962. [DOI] [PubMed] [Google Scholar]
- Pacheco A, López de Quinto S, Ramajo J, Fernández N, Martínez-Salas E. 2009. A novel role for Gemin5 in mRNA translation. Nucleic Acids Res 37: 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. 2006. Microfluidic culture platform for neuroscience research. Nat Protoc 1: 2128–2136. [DOI] [PubMed] [Google Scholar]
- Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. 2002. The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol 14: 305–312. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Kataoka N, Charroux B, Dreyfuss G. 1998. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 95: 615–624. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Kröning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. 2002. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: A role for Smn in RNA processing in motor axons? Hum Mol Genet 11: 93–105. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M. 2003. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of β-actin mRNA in growth cones of motoneurons. J Cell Biol 163: 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendtner M. 2001. Molecular mechanisms in spinal muscular atrophy: Models and perspectives. Curr Opin Neurol 14: 629–634. [DOI] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, et al. 2011. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem 286: 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Halpain S. 1999. Lamina-specific synaptic activation causes domain-specific alterations in dendritic immunostaining for MAP2 and CAM kinase II. J Neurosci 19: 7834–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Bassell GJ. 2011. Making and breaking synapses through local mRNA regulation. Curr Opin Genet Dev 21: 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse H, Deschênes-Furry J, Boisvenue S, Côté J. 2008. KH-type splicing regulatory protein interacts with survival motor neuron protein and is misregulated in spinal muscular atrophy. Hum Mol Genet 17: 506–524. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. 2005. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2: 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Li Jeon N, Cotman CW. 2009. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci 29: 4697–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns MP, Terns RM. 2001. Macromolecular complexes: SMN—the master assembler. Curr Biol 11: R862–R864. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215. [DOI] [PubMed] [Google Scholar]
- Van Meerbeke JP, Sumner CJ. 2011. Progress and promise: The current status of spinal muscular atrophy therapeutics. Discov Med 12: 291–305. [PubMed] [Google Scholar]
- Von Roretz C, Di Marco S, Mazroui R, Gallouzi IE. 2011. Turnover of AU-rich-containing mRNAs during stress: A matter of survival. Wiley Interdiscip Rev RNA 2: 336–347. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: Design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G. 2005. The survival of motor neurons protein determines the capacity for snRNP assembly: Biochemical deficiency in spinal muscular atrophy. Mol Cell Biol 25: 5543–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dreyfuss G. 2001. Characterization of functional domains of the SMN protein in vivo. J Biol Chem 276: 45387–45393. [DOI] [PubMed] [Google Scholar]
- Wang DO, Martin KC, Zukin RS. 2010. Spatially restricting gene expression by local translation at synapses. Trends Neurosci 33: 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis DE, van Niekerk EA, Sasaki Y, Mesngon M, Merianda TT, Williams GG, Kendall M, Smith DS, Bassell GJ, Twiss JL. 2007. Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J Cell Biol 178: 965–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U. 2005. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev 19: 2320–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth EK, Conrad M, Winterer J, Wozny C, Carlson BA, Roth S, Schmitz D, Bornkamm GW, Coppola V, Tessarollo L, et al. 2010. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J 24: 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman E, Kolb SJ, Battle DJ. 2012. Spliceosomal small nuclear ribonucleoprotein biogenesis defects and motor neuron selectivity in spinal muscular atrophy. Brain Res 1462: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ. 2003. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci 23: 6627–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. 2005. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33: W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ. 2006. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci 26: 8622–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. 2008. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivraj H, Tung YCL, Piper M, Gumy L, Fawcett JW, Yeo GSH, Holt CE. 2010. Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci 30: 15464–15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.