

Begun as a desperate attempt to treat refractory ascending cholangitis in infants with biliary atresia (BA) following hepatic portoenterostomy (HPE),1 corticosteroids (steroids) have evolved into a commonly used post-HPE therapy believed to improve clinical outcomes in BA. The original basis for steroid use was its choleretic2 and anti-inflammatory effects, which were proposed to reduce bacterial colonization of the biliary tree by increasing bile flow and reducing periductal inflammation and edema.1 Indeed, short courses of large boluses of intravenous steroids appeared to reduce serum bilirubin, increase bilirubin excretion in the HPE rouxen-Y effluent, resolve fever, and allow resolution of cholangitis during a subsequent course of antibiotics. Over the past 2 decades, this initial observation has evolved into the commonly held belief that a more prolonged course of steroids used routinely following HPE will improve survival of the infant with his or her own liver and delay or prevent the need for liver transplantation. How did acceptance of this treatment paradigm occur? What is the evidence for its effectiveness and safety? Is there a scientific justification for its proposed beneficial effects? A brief review of the emerging themes of the pathophysiology of BA will be necessary to fully understand this evolution in treatment strategy.
BA is the progressive inflammatory obstruction and fibro-obliteration of all or part of the extrahepatic biliary tree and the intrahepatic bile ducts and has its onset exclusively within the first several months of life.3 Surgical HPE re-establishes bile drainage from the liver in up to 60%-80% of patients over the short term, with best results if performed in the first 30-60 days of life.3 Without HPE, the disease is uniformly fatal by 2 years of age. Even following HPE, survival without liver transplantation is only about 30% at age 10 years and 20% at 20 years in the best centers.3 Thus, there is a critical need for designing and testing new therapies. The currently held view is that BA is not a single disease but rather a clinical phenotype resulting from a number of distinct, yet unproven, etiologies.4 Approximately 20% of patients are classified as having the embryonic form, with evidence of other congenital anomalies (for example, polysplenia and asplenia) that are determined at the same embryologic stage as bile duct development. In these infants, presumably there was defective morphogenesis of the extrahepatic biliary tree caused by genetic or epigenetic factors or by an early intrauterine insult (for example, vascular or viral). The remainder of patients have the so-called perinatal or acquired form, in which a perinatal insult (for example, viral infection) has been proposed to trigger bile duct injury and cholangiocyte apoptosis, periductal inflammation, and subsequent obstruction and fibrosis of some portion of the extrahepatic bile duct (Fig. 1).5 In the liver and bile ducts of infants with BA, a T helper 1 T-cell response is the predominant inflammatory mechanism,5 with interferon-γ and tumor necrosis factor-α playing critical roles. There is evidence supporting both CD-4 and CD-8 T-cell–mediated bile duct injury.6 A similar T helper 1 response is replicated in the most promising animal model of BA, the rhesus rotavirus–infected newborn mouse model.7-9 The innate immune response is also activated in the human BA liver, as demonstrated by the later appearance of cells of the CD68+ macrophage/monocyte family in portal tracts and evidence of chemokine and cytokine release by these and related cells.7 Abnormal expression of cellular adhesion molecules (intercellular cell adhesion molecule, CD54, vascular cell adhesion molecule, CD62E) on biliary epithelium and vascular endothelium and elevated circulating levels of soluble adhesion molecules have also been observed.10 Furthermore, a variety of other cytokines, chemokines, and growth factors are released that stimulate downstream amplification of the inflammatory cascade and induction of fibrogenesis (Fig. 1). Recent data from the King's College group10 suggest that circulating inflammatory biomarkers (such as soluble intercellular cell adhesion molecule) may also be predictive of clinical response to HPE (need for liver transplant at 1 year of age). Thus, there appears to be a scientific justification for pursuing novel therapies for BA that would target critical inflammatory pathways.
Fig. 1.
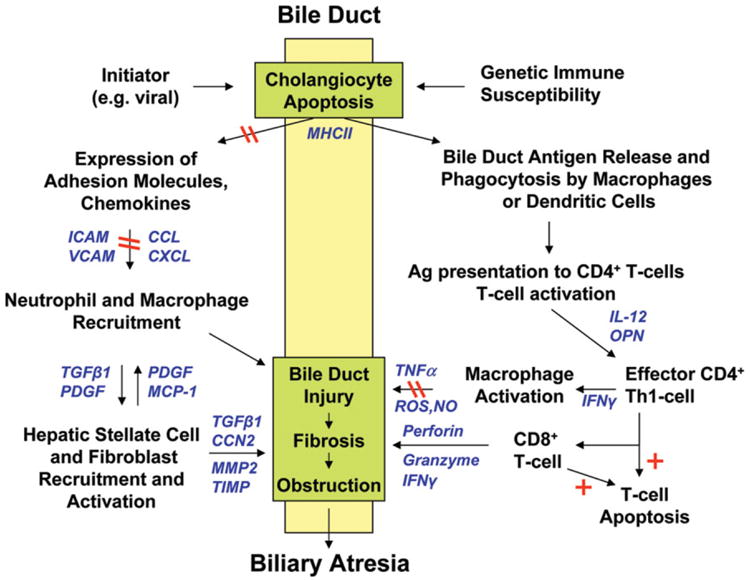
Proposed model of pathogenesis of perinatal (acquired) biliary atresia, incorporating elements from human and murine model investigations, and candidate sites for action of corticosteroids. A perinatal initiating event (for example, viral or other insult) triggers cholangiocyte apoptosis and aberrant major histocompatibility complex class II expression in extrahepatic and intrahepatic bile ducts in an immunologically susceptible host. Viral and/or native or altered bile duct antigens are phagocytosed by macrophages or dendritic cells and presented to naïve T cells in local lymph nodes in which virus/bile duct-specific CD4+ T cells are activated and proliferate, stimulated by IL-2 (right side of the figure). These activated CD4+ T cells (which may be autoreactive) home back to the original site of antigen exposure and elicit T-cell effector functions, including IFNγ-induced macrophage stimulation and activation of cytotoxic CD8+ T cells. Release of TNFα, NO, and ROS by macrophages and release of granzyme, perforin, and IFNγ by CD8+ T cells produce further cholangiocyte injury through apoptotic or necrotic pathways. Simultaneously, cholangiocytes and vascular endothelial cells up-regulate expression of adhesion molecules and secrete chemokines to recruit neutrophils and macrophages to the site of bile duct injury (left side of the figure). Through release of soluble mediators, these cells then recruit and activate hepatic stellate cells (myofibroblasts) and fibroblasts, which initiate extracellular matrix deposition and fibrosis of injured bile ducts. The resulting inflammation, cholangiocyte injury, and fibrosis lead to complete bile duct obstruction and the phenotype of biliary atresia. Pathways that corticosteroids can potentially block are noted by double red lines; those stimulated by corticosteroids are noted by red plus signs. Soluble inflammatory mediators in these pathways are shown in blue. CCN2 indicates connective tissue growth factor; CXCL, chemokine of CXC family, subtype L; CCL, chemokine of CC family, subtype L; ICAM, intercellular cell adhesion molecule; IFNγ, interferon-γ; IL-12, interleukin-12; MCP-1, monocyte chemotactic protein-1; MMP2, matrix metalloproteinase-2; NO, nitric oxide; OPN, osteopontin; PDGF, platelet-derived growth factor; ROS, reactive oxygen species; TGFβ1, transforming growth factor-β1; Th1, T helper 1; TIMP, tissue inhibitor of metalloproteinase; TNFα, tumor necrosis factor-α; and VCAM, vascular cell adhesion molecule.
The potential beneficial effects of steroid therapy following HPE in BA may be related to the drug's induction of Na+K+ATPase, which increases canalicular electrolyte transport and stimulates bile salt–independent bile flow.2 More recently, corticosteroids have been shown to increase the sodium hydrogen exchanger (NHE) isoform of the Na+/H+ exchanger and the CL−/HCO3− exchanger, producing a more bicarbonate-enriched bile.11 Moreover, there are a variety of well-described anti-inflammatory and immunomodulatory properties of corticosteroids (Fig. 1) that could potentially inhibit migration of lymphocytes and macrophages into injured bile ducts, increase transcription of genes coding for anti-inflammatory proteins, and promote a more anti-inflammatory T helper 2 immune response.12 Hsieh et al.13 reported that steroids down-regulated chemokine expression (interleukin-8 and monocyte chemotactic protein-1) in a rat model of bacterial cholangitis, and this might explain the observation of Karrer and Lilly1 of the utility of steroids in treating cholangitis. Since the fibrosis of the bile duct remnant and of intrahepatic ducts in BA is associated with a robust inflammatory response, it is proposed that by suppressing the immune response, steroids would consequently reduce fibrosis and obstruction of small intrahepatic bile ducts that are anastomosed to the roux-en-Y jejunal loop, thus preserving bile flow. Thus, there is a reasonable scientific justification for the potential benefits of steroid therapy in BA.
The clinical use of steroids in BA has exploded in recent years following the publication of a series of reports14-20 attesting to better outcomes with routine post-HPE steroid therapy compared to a variety of historical or concurrent control groups. Each report used a different maximum dose of steroids (1-10 mg/kg/day), had its own unique dose tapering schedule, and was of variable treatment duration (generally 1-3 months). It should be noted that none of these reports were of a prospective, randomized, placebo-controlled design; that there were other variables (for example, use of intravenous antibiotics, bile acid therapy, and surgery performed by different surgeons with varied experience) that could also account for differences in outcome in the steroid group; and that virtually all publications concluded with a plea for a prospective, controlled randomized trial to determine if steroids were truly effective. The extent of use of steroid therapy in BA was quantified in 2002-2003 by a nationwide survey in Japan.21 Among 54 institutions responding to the survey, only 2 did not use steroids, it was used routinely for every patient in 43 and used selectively at 9 centers. From 1997 to 2000, 208 patients with type III BA received steroids, and only 14 did not. Similar data are not available in the United States; however, the impression is that the majority of pediatric surgeons feel compelled to treat BA with steroids.
The determination of whether steroid therapy should be used in BA should take into account not only its potential efficacy but also adverse effects of the drug in order to make a reasonable risk-benefit determination. Unfortunately, the safety of steroid treatment in infants with BA has not previously been prospectively assessed. In fact, few if any adverse events have been reported in the many published reports, and this is not unexpected given the retrospective nature of the data collection. In the Japanese national survey,21 five complications were reported, including wound dehiscence, gastrointestinal hemorrhage and perforation, anastomotic failure, and candida infection. However, this report also lacked an adequate control group, so causation cannot be accurately assessed. Is there reason to be concerned about potential side effects in this age group? It is now apparent that a variety of adult diseases are determined by prenatal and postnatal events that heretofore were not considered linked (for example, intrauterine growth retardation and adult hypertension).22 Could early steroid therapy imprint an infant's immune system to behave differently later in life? We do not know. Is there risk for cataracts or impaired response to childhood immunizations given around the time of the steroid therapy? Moreover, several adverse effects of high-dose dexamethasone in extremely low-birth-weight preterm infants have now been identified by careful prospective study (for example, spontaneous gastrointestinal perforations, smaller head circumference, and delayed cognitive development).23,24 It is not believed that similar effects would occur in the older infant with BA treated with a different form of steroids; however, only through prospective study will the safety of steroid therapy be established.
Given this background, the prospective, placebo-controlled randomized trial by Davenport et al.25 published in this issue of Hepatology is a welcome addition to the medical literature. Seventy-three post-HPE BA patients were randomly assigned to oral prednisolone (n = 36) or placebo (n = 37) for a 21-day course (2 mg/kg/day days 7-21 post-HPE and 1 mg/kg/day days 22-28) and followed prospectively for up to 7 years. Other therapies were standardized, and surgery was performed by only two surgeons. The groups were well matched at baseline. Clearance of jaundice at 6 and 12 months (48% and 44%) and need for liver transplantation at 12 months (35% placebo and 26% steroids) did not differ between the two groups. There was a trend to a lower mean serum bilirubin in the steroid group at 1 month in those who had not been transplanted; however, no other laboratory tests differed through the 12-month study. Kaplan-Meier curves of survival without liver transplant carried out to 7 years were identical between the two groups. Among those infants < 70 days at HPE who might be expected to have the best response to steroids, median bilirubin 1 month after HPE was significantly lower in the steroid group, although this difference disappeared by 6 months, and survival without liver transplantation was similar in both groups. There were no adverse effects that were attributable to steroids, although a listing of adverse events in both groups was not provided. The authors correctly conclude that the steroids were of no benefit in their trial.
How do we put this study in the context of the other retrospective reports of benefit? Does this report provide conclusive evidence that steroids are of no benefit in BA? Davenport et al.25 powered their study to detect a 30% improvement in outcome. Since this group has previously reported achieving survival with native liver at 1 year exceeding 60% without the use of steroids, is it reasonable to believe that 90% survival could have been achieved with the addition of steroid therapy? A more reasonable target for improvement in survival might have been in the range of 10%-20%, providing for survival with native liver of 70%-80%. As noted by the authors, this would have required a much larger number of study participants and would have required a multicentered study design. Another key point to note is that the authors used an admittedly modest dose of prednisolone for only 3 weeks' duration. In most published retrospective reports, the dosing and duration of steroids were almost double that used in this trial. Perhaps a higher dose, initially given intravenously and for a more sustained period of time, would have produced better results. Thus, further study of this potentially important treatment for this devastating disease is certainly needed.
In this regard, a randomized, placebo-controlled, double-blinded multicentered clinical trial of high-dose steroids following HPE is currently being conducted by the Biliary Atresia Research Consortium (BARC) in the United States (funded by the National Institute of Diabetes and Digestive and Kidney Diseases with support from the Office of Rare Diseases) and is actively recruiting study participants. The BARC study will not only assess a number of clinical and nutritional outcome measures but is also carefully assessing the safety of steroid therapy. The study is powered to detect a more modest improvement in outcome than that of Davenport et al.25 and uses a higher dose and longer duration of steroid therapy. Details about enrollment criteria and clinical site information can be found at ClinicalTrials.gov (http://clinicaltrials.gov/ct/show/NCT00294684?order=3) and the BARC Web site (http://www.barcnetwork.org/).
Improving clinical outcomes in BA is a challenge for pediatric hepatologists and surgeons. However, therapy should be guided by evidence and not by anecdotes. It is hoped that the BARC steroid trial and similar multicentered studies will provide the needed data to determine if this commonly used therapy is of benefit and not harm to infants affected by BA.
Acknowledgments
Supported in part by grants from the National Institutes of Health (UO1-DK062453, U54 DK078377, and MO1 RR00069).
Abbreviations
- BA
biliary atresia
- BARC
Biliary Atresia Research Consortium
- HPE
hepatic portoenterostomy
- ICAM
intercellular cell adhesion molecule
- IFNγ
interferon-γ
- IL-2
interleukin-2
- MCP-1
monocyte chemotactic protein-1
- MMP2
matrix metalloproteinase-2
- NO
nitric oxide
- ROS
reactive oxygen species
- TGFβ1
transforming growth factor-β1
- Th1
T helper 1
- TNFα
tumor necrosis factor-α
- VCAM
vascular cell adhesion molecule
Footnotes
Potential conflict of interest: Dr. Sokol is an investigator and Chair of the Steering Committee of the Biliary Atresia Research Consortium and is a member of the Scientific Advisory Board of Yasoo Health, Inc.
References
- 1.Karrer FM, Lilly JR. Corticosteroid therapy in biliary atresia. J Pediatr Surg. 1985;20:693–695. doi: 10.1016/s0022-3468(85)80026-9. [DOI] [PubMed] [Google Scholar]
- 2.Miner PB, Jr, Gaito JM. Bile flow in response to pharmacologic agents. Hepatic DNA as a reference standard. Biochem Pharmacol. 1979;28:1063–1066. doi: 10.1016/0006-2952(79)90304-6. [DOI] [PubMed] [Google Scholar]
- 3.Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology. 2007;46:566–581. doi: 10.1002/hep.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perlmutter DH, Shepherd RW. Extrahepatic biliary atresia: a disease or a phenotype? Hepatology. 2002;35:1297–1304. doi: 10.1053/jhep.2002.34170. [DOI] [PubMed] [Google Scholar]
- 5.Mack CL, Sokol RJ. Unraveling the pathogenesis and etiology of biliary atresia. Pediatr Res. 2005;57(pt 2):87R–94R. doi: 10.1203/01.PDR.0000159569.57354.47. [DOI] [PubMed] [Google Scholar]
- 6.Mack CL, Falta MT, Sullivan AK, Karrer F, Sokol RJ, Freed BM, et al. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133:278–287. doi: 10.1053/j.gastro.2007.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mack CL, Tucker RM, Sokol RJ, Kotzin BL. Armed CD4+ Th1 effector cells and activated macrophages participate in bile duct injury in murine biliary atresia. Clin Immunol. 2005;115:200–209. doi: 10.1016/j.clim.2005.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shivakumar P, Sabla G, Mohanty S, McNeal M, Ward R, Stringer K, et al. Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133:268–277. doi: 10.1053/j.gastro.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen C, Kuske M, Bruns E, Biermanns D, Wussow PV, Mildenberger H. Progress in developing animal models for biliary atresia. Eur J Pediatr Surg. 1998;8:137–141. doi: 10.1055/s-2008-1071140. [DOI] [PubMed] [Google Scholar]
- 10.Narayanaswamy B, Gonde C, Tredger JM, Hussain M, Vergani D, Davenport M. Serial circulating markers of inflammation in biliary atresia— evolution of the post-operative inflammatory process. Hepatology. 2007;46:180–187. doi: 10.1002/hep.21701. [DOI] [PubMed] [Google Scholar]
- 11.Alvaro D, Gigliozzi A, Marucci L, Alpini G, Barbaro B, Monterubbianesi R, et al. Corticosteroids modulate the secretory processes of the rat intrahepatic biliary epithelium. Gastroenterology. 2002;122:1058–1069. doi: 10.1053/gast.2002.32374. [DOI] [PubMed] [Google Scholar]
- 12.Elenkov IJ. Glucocorticoids and the Th1/Th2 balance. Ann N YAcad Sci. 2004;1024:138–146. doi: 10.1196/annals.1321.010. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh CS, Huang CC, Huang LT, Tsai YJ, Chou MH, Chuang JH. Glucocorticoid treatment down-regulates chemokine expression of bacterial cholangitis in cholestatic rats. J Pediatr Surg. 2004;39:10–15. doi: 10.1016/j.jpedsurg.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Dillon PW, Owings E, Cilley R, Field D, Curnow A, Georgeson K. Immunosuppression as adjuvant therapy for biliary atresia. J Pediatr Surg. 2001;36:80–85. doi: 10.1053/jpsu.2001.20013. [DOI] [PubMed] [Google Scholar]
- 15.Escobar MA, Jay CL, Brooks RM, West KW, Rescorla FJ, Molleston JP, et al. Effect of corticosteroid therapy on outcomes in biliary atresia after Kasai portoenterostomy. J Pediatr Surg. 2006;41:99–103. doi: 10.1016/j.jpedsurg.2005.10.072. discussion 199-103. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi H, Yamataka A, Koga H, Okazaki T, Tamura T, Urao M, et al. Optimum prednisolone usage in patients with biliary atresia postportoenterostomy. J Pediatr Surg. 2005;40:327–330. doi: 10.1016/j.jpedsurg.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Meyers RL, Book LS, O'Gorman MA, Jackson WD, Black RE, Johnson DG, et al. High-dose steroids, ursodeoxycholic acid, and chronic intravenous antibiotics improve bile flow after Kasai procedure in infants with biliary atresia. J Pediatr Surg. 2003;38:406–411. doi: 10.1053/jpsu.2003.50069. [DOI] [PubMed] [Google Scholar]
- 18.Muraji T, Higashimoto Y. The improved outlook for biliary atresia with corticosteroid therapy. J Pediatr Surg. 1997;32:1103–1106. doi: 10.1016/s0022-3468(97)90408-5. discussion 1106-1107. [DOI] [PubMed] [Google Scholar]
- 19.Muraji T, Nishijima E, Higashimoto Y, Tsugawa C. Biliary atresia: current management and outcome. Tohoku J Exp Med. 1997;181:155–160. doi: 10.1620/tjem.181.155. [DOI] [PubMed] [Google Scholar]
- 20.Shimadera S, Iwai N, Deguchi E, Kimura O, Fumino S, Ono S. The significance of steroid therapy after hepatoportoenterostomy in infants with biliary atresia. Eur J Pediatr Surg. 2007;17:100–103. doi: 10.1055/s-2007-965120. [DOI] [PubMed] [Google Scholar]
- 21.Muraji T, Nio M, Ohhama Y, Hashimoto T, Iwanaka T, Takamatsu H, et al. Postoperative corticosteroid therapy for bile drainage in biliary atresia—a nationwide survey. J Pediatr Surg. 2004;39:1803–1805. doi: 10.1016/j.jpedsurg.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 22.Sinclair SK, Lea RG, Rees WD, Young LE. The developmental origins of health and disease: current theories and epigenetic mechanisms. Soc Reprod Fertil Suppl. 2007;64:425–443. doi: 10.5661/rdr-vi-425. [DOI] [PubMed] [Google Scholar]
- 23.Yeh TF, Lin YJ, Lin HC, Huang CC, Hsieh WS, Lin CH, et al. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N Engl J Med. 2004;350:1304–1313. doi: 10.1056/NEJMoa032089. [DOI] [PubMed] [Google Scholar]
- 24.Stark AR, Carlo WA, Tyson JE, Papile LA, Wright LL, Shankaran S, et al. for the National Institute of Child Health and Human Development Neonatal Research Network. Adverse effects of early dexamethasone in extremely-low-birth-weight infants. N Engl J Med. 2001;344:95–101. doi: 10.1056/NEJM200101113440203. [DOI] [PubMed] [Google Scholar]
- 25.Davenport M, Stringer MD, Tizzard SA, McLean P, Mieli-Vergani FG, Hadzic N. Randomized, double-blind placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology. 2007 doi: 10.1002/hep.21873. [DOI] [PubMed] [Google Scholar]