

Abstract
Triple negative breast cancer (TNBC) has increased recurrence and poor survival, despite a high response rate to neoadjuvant chemotherapy. The aim of this study was to determine whether current drug treatment(s) eliminates bulk of tumor cells, but it has a minimal effect on cancer stem cells (CSCs) leading to tumor recurrence. We studied the effects of PARP inhibitors (AZD2281 and BSI-201), paclitaxel, docetaxel, cisplatin and cisplatin plus TRAIL on CSCs derived from CRL-2335 and MDA-MB-468 TNBC cells in vitro. The in vitro data indicate that cisplatin plus TRIAL treatment was most effective in eliminating CSCs compared to PARP inhibitors, cisplatin, paclitaxel and docetaxel. Treatment with cisplatin plus TRAIL also inhibits Wnt-1 signaling and its downstream target, β-catenin, phospho β-catenin, cyclin D1, increased apoptosis, reduced proliferation and mammosphere formation. Inhibition of Wnt-1 by siRNA significantly reduced the ability of CSCs to form mammospheres compared to control. However, maximum effect was seen in cisplatin plus TRAIL-treated cells. Taken together the data suggest that cisplatin plus TRAIL treatment has the potential of providing a new strategy for improving the therapeutic outcome in TNBC patients.
Keywords: triple negative breast tumors, cancer stem cells, apoptosis, cisplatin, TRAIL
Introduction
Breast cancer is a common malignancy diagnosed among women in the United States, and is the second leading cause of mortality. Triple-negative breast cancer (TNBC) is the most aggressive and difficult to treat form of cancer compared to other breast cancers. Approximately, 12-15% of women with breast cancer have TNBC. They are negative for estrogen receptor (ER-) and progesterone receptor (PR-) expression and HER-2/neu amplification (1). They do not benefit from hormonal- or herceptin-based targeted therapies due to loss of target receptors. The disease is diagnosed more frequently in younger and pre-menopausal women (2-6). Current therapeutic approaches such as chemo- and/or radiation therapy is able to shrink the malignant mass and achieve an objective clinical response; however, too often these responses are followed by recurrence of the tumor (7). Unlike most cells within the tumor, cancer stem cells (CSC) were shown to be resistant to conventional chemo- and/or radiation therapy. After treatment, they can regenerate all the cell types in the tumor through their stem cell-like behavior (8,9). There is a critical need for the development of more effective therapies to eliminate both tumor and CSCs in TNBC.
The existence of CSCs has been described in a variety of hematologic and solid tumors, including those of the breast, brain, pancreas, and lung (10). The in vitro and animal models have demonstrated CSCs are relatively resistant to both radiation and chemotherapy (8,9). There is growing evidence to support that cancers originate in tissue stem cells through the dysregulation of self-renewal processes. A number of self-renewal pathways such as Wnt (11,12), Notch (13) and Hedgehog (14) have been shown to be involved in the regulation of stem cells. Furthermore, these pathways are shown to be frequently dysregulated during carcinogenesis (11,12,15). Recent studies have reported a pivotal role of Wnt/β-catenin signaling pathway in the regulation of stem cell self renewal (16). In Wnt/β-catenin pathway secreted proteins of the Wnt family bind to specific Frizzler (FZD) receptors on the surface of target cells to activate intracellular pathways, resulting in the accumulation and nuclear localization of the β-catenin protein. The nuclear β-catenin, binds to T-cell factor 4 (Tcf4) to drive activation of specific target genes, including cyclin D1, c-Myc and survivin, which have been characterized to be critical for cancer development. The strategies aimed at interfering with these interactions represent a rational approach to target breast CSCs. CSCs have distinct properties such as self renewal, potential to proliferate, and eventually leading to tumor recurrence and metastasis (17,18). We investigated the effect of cisplatin plus TRAIL on self renewal pathways such as Wnt/β-catenin signaling, mammosphere formation, cell proliferation and apoptosis in CSCs derived from TNBC cells in vitro.
Materials and methods
Cell lines and reagents
The human breast triple-negative breast cell line CRL2335 and MDA-MB-468 cells were obtained from the American Type Culture Collection (ATCC). Cells were maintained in ATCC-recommended culture media. All cells obtained from ATCC were immediately expanded and frozen down such that all cell lines could be restarted every 3-4 months from a frozen vial of the same batch of cells, and no additional authentication was done in our laboratory. DR4/TRAILR1 monoclonal antibody and DR5/TRAILR2 polyclonal antibody was purchased from Imgenex (San Diego, CA). Monoclonal antibody actin was purchased from BD Biosciences (San Diego, CA). Phospho-β-catenin was purchased from Cell Signaling Technology (Danvers, MA). TRAIL was purchased from R&D systems (Minneapolis, MN). Wnt-1 (H-89), cyclin D1 (CD1.1), β-catenin (10H8) antibodies were purchased from Santa Cruz Biotechnology. PARP inhibitors AZD 2281 (Olaparib) and BSI-201 (Iniparib) were purchased from Chemietek (Indianapolis, IN), docetaxel was purchased from LC Laboratories (Wobum, MA). Cisplatin, paclitaxel was purchased from Sigma (St. Louis, MO). Reagents for protein concentration analysis and protein gel electrophoresis were obtained from Bio-Rad (Hercules, CA). All other chemicals, unless otherwise specified, were purchased from Sigma in the highest suitable purities.
ALDEfluor assay and separation of the ALDH-positive population by fluorescence-activation cell sorter
The ALDEfluor assay was performed as described by the manufacturer using the ALDEfluor kit from StemCell Technologies. Briefly, cells were incubated in an ALDEfluor assay buffer containing ALDH substrate (1 μmol/l per 1×106 cells). In each experiment, a sample of cells was stained under identical conditions with 50 mmol/l of diethylaminobenzaldehyde, a specific ALDH inhibitor, as a negative control. The sorting gates were established using propidium iodide-stained cells for viability. Flow cytometry work was done at Imaging and cytometry resources core at The Karmanos Cancer Institute, WSU.
Mammosphere culture
Mammosphere culture was performed as described by Dontu et al (19) with minor modification. In brief, CRL2335 mammospheres were cultured in suspension (1,000 cells/ml) in serum-free RPMI-1640 media, supplemented with B27 (1:100; Invitrogen). MDA-MB-468 mammosphere were cultured in suspension (1,000 cells/ml) in serum-free DMEM media, supplemented with B27 (1:100; Invitrogen), N2 (1:50; Invitrogen) and 10 ng/ml of EGF. For mammosphere formation assay, cultured mammospheres were enzymatically dissociated by incubation in a trypsin-EDTA solution (Invitrogen) at 37°C. Cells were plated at 4000 cells per well of six-well ultra low-attachment plate (Corning, MA). Mammospheres were counted after 5-7 days. Mammosphere counting; mammospheres were centrifuged and transferred to a 96-well flat bottomed plate in 100 μl of the media and counted using a microscope under low magnification. Experiments were done in triplicates.
MTT assay
In brief, 5×104 cells were added in 96-well ultra low-attachment plate. After 24 h, cells were treated with TRAIL (10 ng/ml), cisplatin (10 μg/ml), or combination of TRAIL plus cisplatin or other drugs for another 24 h. Following treatments, 100 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (1 mg/ml) was added into each sample and incubated for 3 h under 5% CO2 and 37°C. The cell viability was measured by MTT, which is converted by succinate dehydrogenase in mitochondria of viable cells to yield a purple formazan dye. The formazan dye was dissolved in dimethyl sulfoxide (DMSO) and measured by absorption at a wavelength of 550 nm using Benchmark® microplate reader from Bio-Rad.
RNA interference assay
Cultured mammospheres were enzymatically dissociated by treating them with trypsin-EDTA. Cells (viable) were plated in 6-well ultra low-attachment plate in Opti MEM I medium (Invitrogen) at a density of 2×105 per well. After 16 h, cells were transfected with 75 nmol of Wnt-1 siRNA, random siRNA with scrambled sequence was used as control. Lipofectamine 2000 (Invitrogen) transfection reagent was used to transfect sequence according to the manufacturer's instructions. After 72 h of transfection, cells were dissociated and used to determine the mammosphere formation and remaining cells were harvested for Western blot analysis.
Apoptosis assay
Apoptosis was assessed using the cell death detection ELISAPLUS kit (Roche) according to the manufacturer's instructions. This kit quantitatively detected cytosolic histone-associated DNA fragments. In brief, mammospheres were dissociated and 2×104 cells were plated in 96-well ultra low attachment plate. Cells were treated with cisplatin, TRAIL and cisplatin plus TRAIL for 16 h. Apoptosis was quantified by ELISA and normalized to values measured in untreated cells. Data are mean ± SE of triplicate determinations.
Western blot analysis
Cells were grown in 6-well plates, to near confluence in the presence or absence of various treatments. Cells were lysed and Western blotting was performed as described previously (20) using a standard protocol. In brief, cell extracts were obtained by lysing the cells in RIPA buffer (20 mM Hepes, 100 mM NaCl, 0.1% SDS, 1% Nonidet P-40, 1% deoxycholate, 1 mM Na3VO4, 1 mM EGTA, 50 mM NaF, 10% glycerol, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1X protease inhibitor mixture). Samples containing 100 μg of total protein were electrophoresed on 8 or 15% SDS-polyacrylamide gels and transferred on to PVDF membrane by electroblotting. Membranes were probed with antibodies as indicated, followed by HRP-conjugated mouse or rabbit secondary antibodies (Amersham). Anti-actin was used for loading controls.
Immunohistochemical analysis (IHC)
Immunohistochemistry was performed on tumor tissue sections placed on glass slides using the standard laboratory protocols as previously described (21,22). Briefly, after deparaffinizing and hydrating with phosphate-buffered saline (PBS) buffer (pH 7.4), tissue sections were pretreated with hydrogen peroxide (3%) for 10 min to remove endogenous peroxidase, followed by antigen retrieval via steam bath for 20 min in EDTA. A primary antibody was applied, followed by washing and incubation with the biotinylated secondary antibody for 30 min at room temperature. Detection was performed with diaminobenzidine (DAB) and counter stained with Mayer hematoxylin followed by dehydration and mounting. Cancer stem cells (CSCs) in human breast tumors were identified by CD44+ and CD24low/- staining. The staining intensity of each specimen was judged relative to the intensity of a control slide containing known positive tissue section. A score of zero indicates no staining relative to background; 1+, weak staining; 2+, moderate staining; and 3+, strong staining. For comparison of staining among tissues, the results were quantified by calculation of a complete H-score which considers both staining intensity and the percentage of cells stained at a specific range of intensities. A complete H-score was calculated by summing the product of the percentage cells stained (0-100%) and staining intensity (0-3) according to Kerfoot et al (23). Statistical analysis of the complete H-scores obtained for the TNBC tissue and ER+, PR+ and HER2- breast tumors were carried out by using the one-tailed Student's t-test with unpaired data of equal variance.
Results
TNBC tumors express relatively higher levels of cancer stem cells compared to ER-positive, PR-positive and HER2-negative breast tumors
The existence of CSCs has been described in a variety of hematologic and solid tumors, including those of the breast (10). Unlike most cells within the tumor, CSC was shown to be resistant to conventional chemo- and/or radiation therapy. After treatment, they can regenerate all the cell types in the tumor through their stem cell-like behavior (8,9). As tumor recurrence is a major problem in TNBC, we postulated that TNBC tumors may have higher CSC levels compared to other breast cancer tumors. We determine the CSC levels in nine TNBC tumors and eight ER+, PR+ and HER2- human breast tumors by immunohistochemical staining. Interpretation of the slides was performed by microscopic examination by a board certified pathologist. Representative staining pattern of the tumor is shown in Fig. 1. The data indicate that TNBC tumors have CD44+/CD24low/- staining in ∼20-70% of the tumor cells compared to 2-10% of the cells in ER+, PR+ and HER2- breast tumors. However, staining intensity and percentage of stained cells varied in different tumor samples. In order to compare the differences between TNBC and ER+, PR+ and HER2-tumors, we calculated the H-score. H-score takes into account the intensity of staining and percentage of stained cells in each tumor (22). Our data indicate a significant high H-score in TNBC ∼1090 compared to an H-score of ∼200 in ER+, PR+ and HER2- breast tumors (p<0.01) (Table I). Consistent with our postulation data indicate that human TNBC cancers express relatively higher levels of CSCs compared to ER+, PR+ and HER2- breast tumors.
Figure 1.
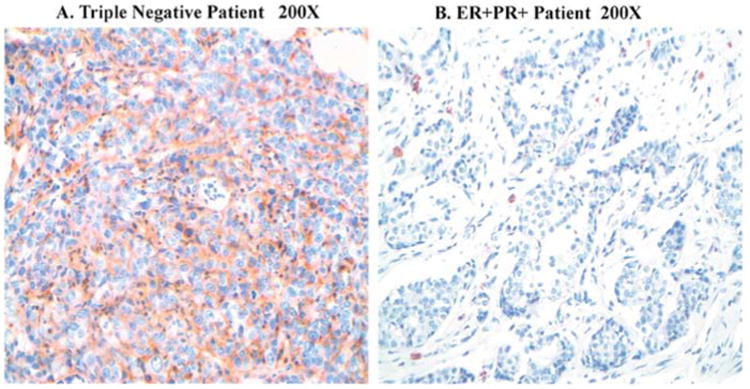
CD44 immunohistochemical staining in human breast tissue specimens (x200). Triple-negative tumor shows strong CD44-positive staining. ER-positive, PR-positive and HER2-negative breast tumor shows mild staining.
Table I.
Summary of CD44 immunohistochemical staining intensity in triple negative and ER+PR+ breast tumor tissue.
| Staining intensity | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| Breast tumors | n | 1+ | 2+ | 3+ | Mean complete H-score | P-value |
| Triple negative | 9 | 0 | 2 | 7 | 1090 | |
| ER+PR+Her2- | 8 | 2 | 1 | 1 | 200 | 0.01 |
Cisplatin plus TRAIL had a synergistic effect on cancer stem cells
Clinical trials indicate that PARP-inhibitors AZD2281 (Olaparib) and BSI-201 (Iniparib) have emerged as a promising new class of antineoplastic agents (24,25). However, significant numbers of TNBC tumors reoccur (26,27). We postulated that above drugs eliminates bulk of tumor cells, but they have limited ability to eliminate CSCs and this could lead to tumor recurrence. To test the above concept, we treated the CSCs derived from TNBC cell lines CRL2335 and MDA-MB-468 with cisplatin, TRAIL, cisplatin plus TRAIL, two PARP inhibitors (AZD2281 and BSI-201), paclitaxel and docetaxel. CSCs levels were determined by ALDEfluor kit (StemCell Technologies) using flow cytometric analyses in the presence or absence of the drug(s) after 24 h treatment. The data indicate that untreated CRL2335 cells had ∼36% CSCs (ALDEfluor-positive cells) and treatment with AZD2281 (50 μM) reduced CSCs from 36 to 25%, BSI-201 (50 μM) to ∼20%, docetaxel (10 nM) to ∼24%, paclitaxel (20 nM) had no effect on CSCs, cisplatin (10 μg/ml) to ∼21%, TRAIL (10 ng/ml) to ∼30%, and combination of cisplatin (10 μg/ml) plus TRAIL (10 ng/ml) to ∼1% (Fig. 2A). Similarly, untreated MDA-MB-468 TNBC cells had ∼19% cancer stem cells (ALDEfluor-positive cells), AZD2281 treatment reduced CSCs from ∼19% to 16%, BSI-201 to ∼15%, docetaxel marginally increased CSC to ∼24%, paclitaxel had no effect on CSCs, cisplatin (10 μg/ml) to ∼14%, TRAIL (10 ng/ml) to ∼17%, and combination of cisplatin (10 μg/ml) and TRAIL(10 ng/ml) to ∼2% (Fig. 2B). As cisplatin plus TRAIL had a maximum effect on CSCs compared to other drugs, we also determine the effect of cisplatin plus TRAIL on CSCs and total cells after 24 h treatment. Our data indicate that cisplatin plus TRAIL significantly inhibits both CSCs and total cells compared to cisplatin or TRAIL alone (Fig. 2C). Thus, taken together, the results suggest that cisplatin plus TRAIL treatment increased cell death in both CSCs and total cells in TNBC cells.
Figure 2.
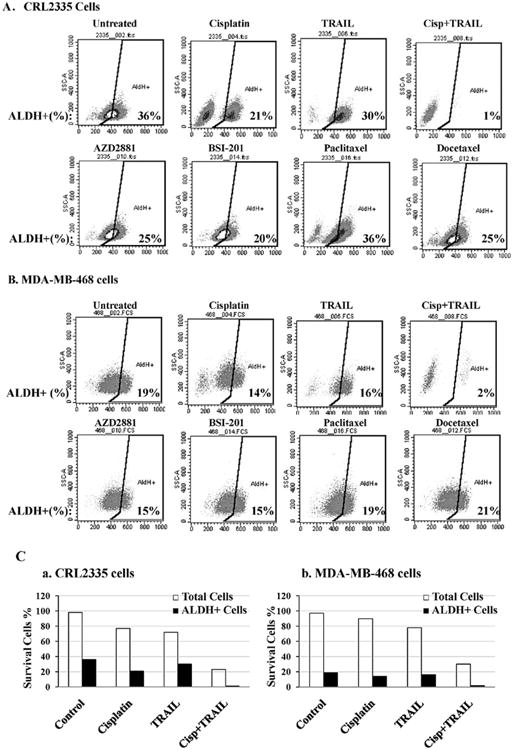
Cisplatin plus TRAIL treatment decreases cancer stem cells. The effect of cisplatin, TRAIL, cisplatin plus TRAIL, PARP inhibitors AZD2881, BSI-20, paclitaxel and docetaxel treatment on ALDEfluor+ (CSCs) population was analyzed after 24 h treatment. The cell percentage numbers for ALDH-positive cells are indicated in the bottom right of each histogram. Maximum decrease in ALDH-positive population was seen in cisplatin plus TRAIL-treated group compared to control in both triple negative cell lines CRL2333 (A) and in MDA-MB-468 cells (B). (C). Cisplatin plus TRAIL-induces cell death in both tumor and cancer stem cells. In general cancer stem cells are resistant to traditional chemotherapy. However, combination of cisplatin plus TRAIL treatment significantly inhibits both tumor cells (white bars) and cancer stem cells (black bars).
Cisplatin plus TRAIL inhibited Wnt/β-catenin signaling pathway
Previous studies have shown that Wnt-1 signaling plays a major role in the self-renewal of CSCs (8,28,29). In order to understand the mechanism by which cisplatin plus TRAIL inhibits and/or kills CSCs, we determined whether cisplatin plus TRAIL block self-renewal by inhibiting Wnt/β-catenin signaling in CSCs. The data in Fig. 3 indicated that cisplatin treatment resulted in a significant reduction in β-catenin, phospho β-catenin, and cyclin D1 levels, while similar treatment with TRAIL had a minimal effect on these proteins. In contrast, cisplatin plus TRAIL treatment had maximum reduction on of all the three proteins. TRAIL mediates its anti-apoptotic effects by binding to DR4 and DR5 receptors. Therefore, we determine whether CSCs derived from TNBC cell lines express DR4 and DR5 receptors. Our data show that DR4 and DR5 receptors are expressed by CSCs (Fig. 3B) and cisplatin sensitizes the CSCs to TRAIL-induced apoptosis (Fig. 3C and D). Our data also indicate that there are differences in susceptibility to cisplatin treatment among different cell lines; however combination of cisplatin plus TRAIL had maximum effect in both the TNBC cell lines. Taken together, the data suggest that cisplatin plus TRAIL significantly reduce Wnt1/β-catenin signaling, which could lead to reduced self renewal, and cell proliferation in CSCs.
Figure 3.
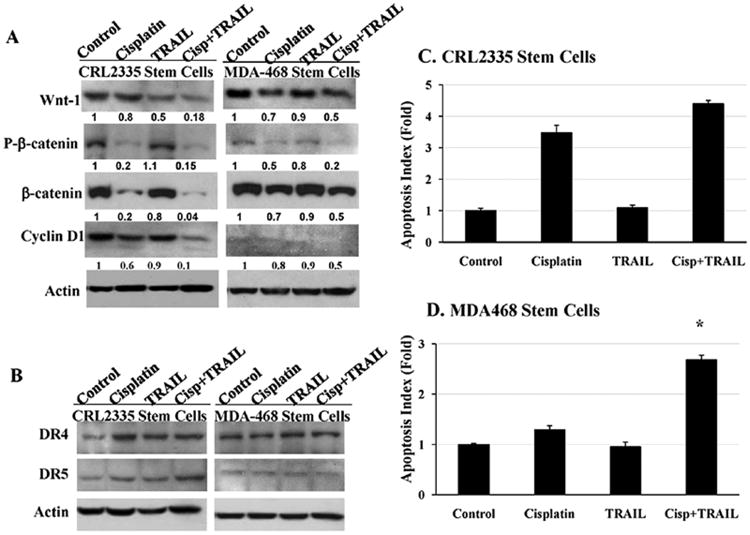
Cisplatin plus TRAIL inhibits Wnt-1 signaling and increases apoptosis in cancer stem cells. Cancer stem cells derived from CRL2335 and MDA-MB-468 TNBC cells were treated with cisplatin, TRAIL or cisplatin plus TRAIL for 24 h. (A). Cancer stem cell extracts were prepared and analyzed for Wnt-1, phospho-β-catenin, β-catenin, cyclin D1, by Western blotting. Equal protein loading was compared with that of actin. (B). Cancer stem cell extracts were prepared and analyzed for DR4 and DR5 receptors by Western blotting. Equal protein loading was compared with that of actin. (C). Apoptosis was quantified by Cell Death ELISA and normalized to values measured in untreated cells. Cancer stem cells showed a significant increase in apoptosis in the presence of cisplatin plus TRAIL in comparison to untreated cells in CRL2335 (C) and MDA-MB-468 (D) derived CSCs (*p<0.001). Data are the mean ± SE of triplicate determinations.
To further determine if inhibition of Wnt-1 is sufficient to achieve similar results as seen in cisplatin plus TRAIL treatment, we inhibited Wnt-1 by Wnt-1 siRNA in CRL2335-derived cancer stem cells. Our data indicate that inhibition of Wnt-1 by siRNA significantly reduced phospho-β-catenin, cyclin D1 levels (Fig. 4A) compared to random siRNA-treated CSCs. However, maximum inhibition was seen in cisplatin plus TRAIL-treated CSCs (Fig. 4A). Consistent with these observations Wnt-1 inhibition by siRNA reduced mammosphere formation by ∼25% compared to random siRNA-treated CRL2335 cells, maximum inhibition ∼95% was seen in cisplatin plus TRAIL-treated CSCs (Fig. 4B).
Figure 4.
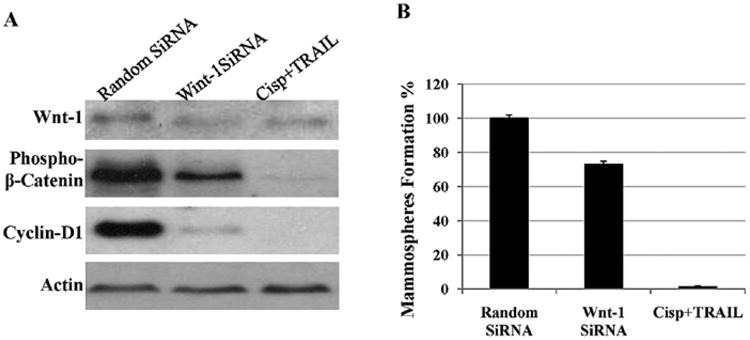
Effects of siRNA knockdown of Wnt-1 on cancer stem cells derived from TNBC cells. Cancer stem cells from CRL2335 were transfected with Wnt-1 siRNA or random siRNA or treated with cisplatin plus TRAIL. Cancer stem cell extracts were prepared and analyzed for Wnt-1, phospho-β-catenin and cyclin D1 by Western blotting (A). Equal protein loading was compared with that of actin. (B) Under similar conditions mammosphere formation ability was assessed using protocols described in methods. Data shows a combination of cisplatin plus TRAIL treatment significantly inhibits Wnt-1 signaling and reduced mammosphere formation ability of CSCs.
Cisplatin plus TRAIL inhibited mammosphere forming ability of CSCs cells
One of the major characteristic features of CSCs is its ability to form mammospheres in suspension culture (19). We investigated the effect of cisplatin plus TRAIL on apoptosis and mammosphere formation in CSCs derived from TNBC cell lines CRL2335 and MDA-MB-468 cells. The results presented in Fig. 5A and B suggested that cisplatin treatment reduced mammosphere formation ability of CSCs by ∼75% compared to an untreated control group, TRAIL treatment reduced mammosphere formation ability by ∼42% and combination of cisplatin plus TRAIL reduced mammosphere formation ability by ∼96% (Fig. 5B). Similarly, in MDA-MB-468 derived CSCs, cisplatin treatment reduced mammosphere formation ability by ∼62%, TRAIL by ∼21% and combination of cisplatin plus TRAIL reduced mammosphere formation ability by ∼87% (Fig. 5B). Thus, taken together, the results suggest that cisplatin plus TRAIL treatment significantly inhibits Wnt-1 signaling, and decreases mammosphere formation in CSCs.
Figure 5.
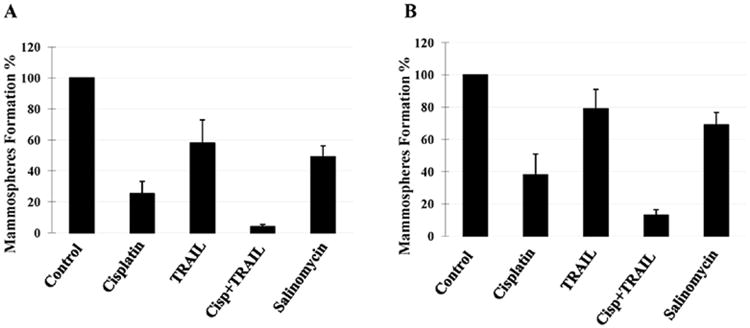
Effects of cisplatin plus TRAIL treatment on the ability of cancer stem cells to form mammospheres. The in vitro mammosphere was quantified by counting the mammospheres after treatment with cisplatin, TRAIL and cisplatin plus TRAIL compared to controls. Cisplatin plus TRAIL treatment had maximum inhibition on the cancer stem cell ability to form mammospheres in CRL2335- (A) and MDA-MB-468- (B) derived CSCs. Salinomycin a well established CSC inhibitor was used as an internal control. Data are the mean ± SE of triplicate determinations.
Discussion
TNBC is the most aggressive and difficult to treat form of cancer compared to other forms of breast cancer. TNBC patients do not benefit from hormonal or herceptin-based therapies due to loss of target receptors such as ER, PR and HER-2 (1). Chemotherapy is currently the main stay of systemic treatment, although patients with TNBC, when considered as a group, have a worse outcome after chemotherapy than patients with other subtypes of breast cancer (30,31). One of the reasons may be the inability of the chemotherapeutic drugs to kill CSCs. Some studies suggest that 75% of the tumors arising in women carrying a mutation in BRCA1 gene have a triple-negative phenotype (27,32,33). The PARPs constitute a family of enzymes involved in base-excision repair, a key pathway in the repair of DNA single-strand breaks. Despite exciting developments in the treatment options such as poly(ADP-ribose) polymerase (PARP) inhibitors to target BRCA1-defcient TNBC cancers (24), many women still experience a relapse, and metastatic breast cancer remains a largely incurable disease. Our data show 20-70% of cancer cells from TNBC tumors display cell surface markers for cancer stem cells, such as CD44+/CD24low/- and expression of aldehyde dehydrogenase 1(ALDH1A1). Our data show that PARP inhibitor AZD2281 (olaparib) reduced CSCs from ∼36% to 25% and BSI-201 (iniparib) reduced CSCs from ∼36% to 20% in CRL2335 cells. However, cisplatin plus TRAIL has reduced CSCs from ∼36% to 1% in CRL-2335 cells. Previous studies from preclinical and phase I trials, show a response rate of ∼40% in patients with BRCA1 or BRCA2 mutations (24,25). Our data show that cisplatin plus TRAIL is much more effective in eliminating CSCs compared to cisplatin alone or PARP inhibitors. These observations suggest that cisplatin plus TRAIL is highly effective against TNBC by eliminating both CSCs and bulk of tumor cells. It is also becoming evident that cancer treatment that fails to eliminate CSCs allows relapse of the tumor (34, 35).
One of the defining characteristics of CSCs is their ability to undergo self-renewal. The self-renewal pathways are regulated by Wnt, Notch and Hedgehog signaling. Interestingly, dysregulation of each of these pathways in transgenic mice leads to breast cancer (36-38). Therapies that inhibit a self-renewal pathway(s) could lead to a significant reduction of CSCs in tumor. Previous studies have shown that Wnt activation leads to β-catenin translocation into the nucleus, and cause induction of TCF family of transcription factors leading to increased synthesis of several proteins that are critically involved in cell growth (39). Our data indicate that cisplatin plus TRAIL inhibit Wnt-1 signaling leading to reduced phospho-β-catenin and cyclin D1 levels, which could lead to significant inhibition of self-renewal and proliferation in CSCs. The therapeutic strategies that target CSCs are effective in reducing the risk of relapse and metastasis.
The commonly accepted criteria for clinical efficacy in phase II trials are tumor shrinkage using criteria defined by RECIST (40). The premise is that tumor regression equates with a clinical benefit. Literature suggests that there is only a modest overall survival advantage for pancreatic, prostate and metastatic breast cancer patients even with the tumor regression (41-43). This discrepancy between response and survival may be partially explained by an inability of chemotherapeutic drugs to effectively target the CSCs. Cisplatin plus TRAIL targets both tumor and CSCs in vitro and this could provide new treatment options for TNBC tumors.
We demonstrate for the first time that cisplatin plus TRAIL is highly effective in reducing CSCs compared to PARP inhibitors, docetaxel or paclitaxel in TNBC cells in vitro. Cisplatin plus TRAIL significantly inhibits Wnt-1 signaling pathways, reduces cell proliferation and increases apoptosis in CSCs and tumor cells. Individually, cisplatin and TRIAL are approved by FDA to treat other forms of cancer. Since TNBC patients do not benefit from hormonal or herceptin-based therapies or chemo- and radiation-therapy, cisplatin plus TRAIL treatment is suggested as a potential new cancer treatment strategy in TNBC patients.
Acknowledgments
This work was supported in part by Department of Pathology, WSU and the National Institute of Health Grant RO1 CA 118089 (to K.B.R). The microscopy, imaging and cytometry resources core is supported, in part, by NIH center grant P30CA22453 to The Karmanos Cancer Institute, WSU and Perinatology research branch of the National Institutes of Child health and development, Wayne State University.
Abbreviations
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- DR4
death receptor 4
- DR5
death receptor 5
- PARP
poly(ADP-ribose) polymerase
- EGFR
epidermal growth factor receptor
- ER
estrogen receptor
- PR
progesterone receptor
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 3.Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 4.Haffty BG, Yang Q, Reiss M, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24:5652–5657. doi: 10.1200/JCO.2006.06.5664. [DOI] [PubMed] [Google Scholar]
- 5.Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109:25–32. doi: 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- 6.Tischkowitz M, Brunet JS, Begin LR, et al. Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC Cancer. 2007;7:134. doi: 10.1186/1471-2407-7-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol. 2010;4:404–419. doi: 10.1016/j.molonc.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 9.Yu F, Yao H, Zhu P, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 10.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 13.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–R615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dievart A, Beaulieu N, Jolicoeur P. Involvement of Notch1 in the development of mouse mammary tumors. Oncogene. 1999;18:5973–5981. doi: 10.1038/sj.onc.1202991. [DOI] [PubMed] [Google Scholar]
- 16.Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 17.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–1115. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. 2010;28:4006–4012. doi: 10.1200/JCO.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nabha SM, Glaros S, Hong M, et al. Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene. 2005;24:3166–3176. doi: 10.1038/sj.onc.1208502. [DOI] [PubMed] [Google Scholar]
- 21.Visscher DW, Sarkar FH, Kasunic TC, Reddy KB. Clinico pathologic analysis of amphiregulin and heregulin immunostaining in breast neoplasia. Breast Cancer Res Treat. 1997;45:75–80. doi: 10.1023/a:1005845512804. [DOI] [PubMed] [Google Scholar]
- 22.Yin S, Sethi S, Reddy KB. Protein kinase Cdelta and caspase-3 modulate TRAIL-induced apoptosis in breast tumor cells. J Cell Biochem. 2010;111:979–987. doi: 10.1002/jcb.22786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerfoot C, Huang W, Rotenberg SA. Immunohistochemical analysis of advanced human breast carcinomas reveals down-regulation of protein kinase C alpha. J Histochem Cytochem. 2004;52:419–422. doi: 10.1177/002215540405200314. [DOI] [PubMed] [Google Scholar]
- 24.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 25.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 26.Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8:235–244. doi: 10.1016/S1470-2045(07)70074-8. [DOI] [PubMed] [Google Scholar]
- 27.Reddy KB. Triple-negative breast cancer: an updated review on treatment options. Curr Oncol. doi: 10.3747/co.v18i4.738. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Wakeman TP, Lathia JD, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 31.Tan DS, Marchio C, Jones RL, et al. Triple negative breast cancer: molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res Treat. 2008;111:27–44. doi: 10.1007/s10549-007-9756-8. [DOI] [PubMed] [Google Scholar]
- 32.Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- 33.Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52:108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 34.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 35.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 36.Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148:567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly OG, Pinson KI, Skarnes WC. The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development. 2004;131:2803–2815. doi: 10.1242/dev.01137. [DOI] [PubMed] [Google Scholar]
- 38.Soriano JV, Uyttendaele H, Kitajewski J, Montesano R. Expression of an activated Notch4(int-3) oncoprotein disrupts morphogenesis and induces an invasive phenotype in mammary epithelial cells in vitro. Int J Cancer. 2000;86:652–659. doi: 10.1002/(sici)1097-0215(20000601)86:5<652::aid-ijc8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 39.Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. J Mol Med. 2009;87:1097–1104. doi: 10.1007/s00109-009-0518-4. [DOI] [PubMed] [Google Scholar]
- 40.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 41.Abratt RP, Brune D, Dimopoulos MA, et al. Randomised phase III study of intravenous vinorelbine plus hormone therapy versus hormone therapy alone in hormone-refractory prostate cancer. Ann Oncol. 2004;15:1613–1621. doi: 10.1093/annonc/mdh429. [DOI] [PubMed] [Google Scholar]
- 42.Huff CA, Matsui W, Smith BD, Jones RJ. The paradox of response and survival in cancer therapeutics. Blood. 2006;107:431–434. doi: 10.1182/blood-2005-06-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rocha Lima CM, Green MR, Rotche R, et al. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol. 2004;22:3776–3783. doi: 10.1200/JCO.2004.12.082. [DOI] [PubMed] [Google Scholar]