

Abstract
Spontaneous autoimmune polyneuropathy (SAP) in B7-2 knock-out non-obese diabetic (NOD) mice is mediated by myelin protein zero (P0)-reactive T helper type 1 (Th1) cells. In this study, we investigated the role of B cells in SAP, focusing on CD19 as a potential therapeutic target. We found that P0-specific plasmablasts and B cells were increased in spleens of SAP mice compared to wild-type NOD mice. Depletion of B cells and plasmablasts with anti-CD19 monoclonal antibody (mAb) led to attenuation of disease severity when administered at 5 months of age. This was accompanied by decreased serum immunoglobulin (Ig)G and IgM levels, depletion of P0-specific plasmablasts and B cells, down-regulation/internalization of surface CD19 and increased frequency of CD4+ regulatory T cells in spleens. We conclude that B cells are crucial to the pathogenesis of SAP, and that CD19 is a promising B cell target for the development of disease-modifying agents in autoimmune neuropathies.
Keywords: autoimmunity, B cells, CIDP, co-stimulatory molecules, Guillain–Barré syndrome
Introduction
Animal models (induced and spontaneous) are used to study the pathogenetic mechanisms involved in human autoimmune diseases, including Guillain–Barré syndrome (GBS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). A spontaneous autoimmune polyneuropathy (SAP) is triggered in non-obese diabetic (NOD) mice by depletion of regulatory T cells (Tregs) with anti-interleukin (IL)-2 antibody or by elimination of co-stimulatory molecules such as B7-2, intercellular adhesion molecule 1 or programmed death 1 (PD-1) [1–4]. SAP in B7-2 knock-out (KO) NOD mice mimics the chronic progressive form of human CIDP electrophysiologically and histologically, and is ameliorated by immunomodulators such as fingolimod [1,5,6]. We and other investigators have reported that SAP is mediated by interferon (IFN)-γ-producing T helper type 1 (Th1) cells, although a possible contribution of Th17 cells early in the course of disease cannot be excluded [1,7,8]. Furthermore, we have identified myelin protein zero (P0) as a major antigenic target in this model [8,9].
Although Th1 cells are the major effector cells of SAP in the B7-2 KO NOD mouse, the possible contribution of B cells, CD8+ T cells or other cell types in its pathogenesis or disease progression has not been studied. SAP can be induced in NOD severe combined immunodeficient (SCID) animals by CD4+ T cells from affected mice, but not by passive transfer with their sera alone [1,8]. However, humoral responses to P0 have been detected in SAP mice [8]. Furthermore, some CIDP sera that contain immunoglobulin (Ig)G antibodies against P0 caused conduction block and demyelination upon intraneural injection in animals [10,11]. Other autoantibodies such as those reactive against glycolipids are implicated in some forms of GBS, and chronic dysimmune neuropathies [12,13].
The goal of this study was to investigate the role of B cell response in the development or progression of SAP. Aside from production of antibodies by plasmablasts and plasma cells, B cells are involved in antigen presentation, in the production of cytokines and chemokines, as well as in the regulation of T cell subsets [14,15]. One approach to the study of the role of B cells is the use of anti-CD20 or anti-CD19 monoclonal antibodies, which deplete B cells via monocyte-mediated antibody-dependent cellular cytotoxicity [16]. B cell depletion by CD20 monoclonal antibody (mAb) prevents and/or reverses type 1 diabetes in NOD mice [17–19]. Anti-CD20 antibodies primarily deplete mature B cells. CD19, a dominant signalling component of the B cell receptor complex, has a broader expression profile than CD20, encompassing early pre-B cells up to plasmablasts and some plasma cells [20]. Thus, anti-CD19 antibodies may have a more significant impact than anti-CD20 antibodies on the levels of autoantibodies, and could affect other aspects of B cell function due to altered transmembrane signalling.
Whether or not B cell-directed strategy would be effective in preventing or suppressing SAP in B7-2 KO NOD mice has not been investigated. B7:CD28 signalling exerts complex effects on humoral immunity, which includes regulation of germinal centre formation, development of T follicular helper cells and immunoglobulin class-switching in vivo [21–23]. Independent of class-switch recombination, B7-2 regulates the level of IgG1 via a CD19-dependent mechanism. There is also evidence for the intrinsic function of CD28 in the survival of plasma cells [24]. Loss of CD28 or its ligands B7-1 and B7-2 results in altered frequency of plasma cells and antibody levels, although debate remains as to whether CD28 is a positive or negative regulator of plasma cells [25,26]. In this study, we provide evidence that B cells contribute to the pathogenesis of SAP in B7-2 KO NOD mice, irrespective of the complex sequelae of B7-2 elimination on humoral immunity. Furthermore, we posit that targeting of CD19 is a promising strategy for disease intervention in autoimmune diseases affecting the nervous system.
Materials and methods
Clinical and electrophysiological assessment
All animal use procedures were conducted in strict accordance to the National Institutes of Health and University of Chicago institutional guidelines. Female B7-2 KO NOD mice were used in this study unless stated otherwise. For clinical assessment, the following scale was used: 0, normal; 0·5, mild ruffled coat; 1, less active or flaccid tail; 1·5, one leg is curled in when held by tail; 2, mild paraparesis (both legs curled in); 2·5, drags one leg; 3, severe paraparesis (drags both legs); and 3·5, severe tetraparesis; 4, death. Grip strength testing consisted of five separate measurements in each of two trials per session using a grip strength meter (Columbus Instruments, Columbus, OH, USA). Results of two trials were averaged for each mouse per session. After the last grip strength measurement, electrophysiological studies of sciatic nerves were performed as described in our previous publications [1,27]. Latencies, conduction velocities and peak-to-peak amplitudes were measured. Results from stimulation of bilateral sciatic nerves were averaged for each animal, with ‘n’ representing the number of animals in each study group. Animal treatment studies with anti-CD19 mAb versus isotype control mAb were conducted in a randomized and blinded fashion.
Generation and purification of extracellular domain of P0 (P0-ECD)
The P0-ECD construct that contained the 124 amino acid residues (aa 1–124) from the extracellular domain (ECD) of rat P0 was cloned and expressed using the expression vector C5 (GenScript, Piscataway, NJ, USA) or pET23d (+) (EMD Millipore, Billerica, MA, USA). Briefly, His-tagged P0-ECD plasmids were transformed into Escherichia coli strain BL21 (ED3). Protein over-expression was induced in bacterial cells at 30°C with 1 mM isopropylthio-β-D-galactoside (IPTG). Bacterial protein was insolubilized using the BugBuster Protein Extraction Reagent, according to the manufacturer's protocol (EMD Millipore). Recombinant His-tagged protein was obtained from inclusion bodies followed by purification using the His-Bind purification kit (EMD Millipore). The purity of the His-tagged P0-ECD protein was confirmed by Western blot analysis using horseradish peroxidase (HRP)-conjugated goat anti-6-His antibody (1:10 000) (Bethyl Laboratory, Montgomery, TX, USA). To remove endotoxin, we used the ToxinEraser Endotoxin Removal kit (Genscript). The final endotoxin level was <1 EU/μg by the chromogenic LAL method (Genscript). Purified P0-ECD was quantified using the Pierce BCA Protein Assay kit (Thermo Scientific, Rockford, IL, USA), then labelled with Alexa-Fluor 546 using a protein labelling kit (Invitrogen, Eugene, OR, USA), according to the manufacturer's instructions.
Flow cytometry
Single-cell suspensions from blood, bone marrow and spleen were stained at 4°C using predetermined optimal concentrations of antibodies for 30 min. Cells with the forward- and side-scatter properties of lymphocytes were analysed using the LSR-II flow cytometer (BD Biosciences, San Jose, CA, USA). Background staining was assessed using isotype-matched control antibodies. B cells were identified as B220+ IgM+ cells, plasmablasts as B220+CD44+CD138hi or B220+CD138hi cells, plasma cells as B220loCD138hi cells and Tregs as CD4+CD25+forkhead box protein 3 (FoxP3+) cells. The following antibodies were used: allophycocyanin (APC)-conjugated rat anti-mouse IgM (eBioscience, San Diego, CA, USA), eFluor 450-conjugated anti-mouse CD45R (B220), APC-conjugated rat anti-mouse CD138 (BD Biosciences) and fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD44 (Biolegend, San Diego, CA, USA). Splenic CD1dhiCD5+ B cells were determined using Biolegend's mouse regulatory B cell flow kit, which includes FITC-conjugated anti-mouse CD19, phycoerythrin (PE)-conjugated anti-CD5 and Alexa Fluor 647-conjugated CD1d, with the addition of eFluor450-conjugated B220. For the detection of Tregs, splenocytes were stained with FITC-conjugated anti-mouse CD4 and APC-conjugated anti-mouse CD25 antibodies, fixed, permeabilized and subsequently stained with PE-conjugated anti-mouse FoxP3 antibody (eBioscience).
For B10 cells, splenocytes were suspended in RPMI-1640 media containing 10% fetal calf serum (FCS), 4 mM glutamine, 200 μg/ml penicillin and 200 U/ml of streptomycin, and 5 mM 2-mercaptoethanol. Cells were incubated for 4 h in 96-well plates with lipopolysaccharide (LPS) (10 μg/ml) and a leucocyte activation cocktail containing phorbol mysristate acetate (PMA), ionomycin, brefeldin and BD Golgiplug (BD Pharmingen, San Jose, CA, USA). Cells were then stained with eFluor450-conjugated anti-mouse CD45R (B220) antibody, followed by fixation and permeabilization using the Cytofix Kit prior to staining with PE-conjugated anti-mouse IL-10 antibody (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA) and splenocyte proliferation assays
Serum IgG and IgM concentrations were determined by ELISA using mouse IgG and IgM Ready-SET-Go! Kits (eBioscience). For proliferation assay, splenocytes were cultured at a density of 5 × 105 cells/well in HL-1™ medium plus non-essential amino acids (BioWhittaker, Radnor, PA, USA), L-glutamine (2 mM), sodium pyruvate (1 mM) and β-mercaptopurine (55 μM) in flat-bottomed 96-well plates. Cells were stimulated with P0 (180–199) (20 μg/ml), P0–ECD (20 μg/ml) or ovalbumin (20 μg/ml). On day 3, these cultures were pulsed for 16 h with 1 μCi methyl-[3H]-thymidine, and then harvested on a glass fibre filter. The amount of incorporated methyl-[3H]-thymidine was measured using the Beckman liquid scintillation counter. The stimulation index was defined by counts per minute (cpm) in the presence of antigen divided by cpm in the absence of antigen.
Antibody-dependent cell-mediated cytotoxicity assay
This assay was performed based on the release of lactate dehydrogenase (LDH) with a set effector : target ratio of 2·5:1. Briefly, A20 cells (murine B cell lymphoma) were washed with phosphate-buffered saline (PBS) and resuspended at a density of 0·4 × 106/ml in RPMI-1640 phenol red free media, while effector cells [either J774·2 cells or purified natural killer (NK) cells] were resuspended at a density of 1 × 106/ml. NK cells were isolated using the EasySep mouse NK cell enrichment kit (Stemcell Technologies). Assays were performed in triplicate using 96-well U-bottomed plates, with each well containing 50 μl of target cell suspension, 50 μl of effector cell suspension and 50 μl of the mAb at the appropriate dilution. Plates were spun at 250 g for 4 min to pellet cells, then incubated at 37°C, 5% CO2 for 4 h. LDH release from target cells undergoing cell death was measured using the CytoTox 96 non-radioactive cytotoxicity assay (Promega, Madison, WI, USA), according to the manufacturer's instructions. Absorbance at 490 nm was measured in a plate reader. The percentage cytotoxicity was calculated as (experimental–effector spontaneous – target spontaneous release)/(maximal target–target spontaneous) ×100.
Data analysis
Clinical severity was expressed as mean clinical score ± standard deviation (s.d.) and analysed using analysis of variance (anova) followed by Student's t-test, except in Fig. 3b, where median clinical score was used and analysed using the Mann–Whitney U-test. Results from immunological studies, grip strength measurements and electrophysiology were expressed as mean ± standard error of the mean (s.e.m.). Statistical significance for these data was determined by anova followed by Student's t-test and Bonferroni's method for multiple group experiments. Significance levels were set at P < 0·05.
Fig. 3.
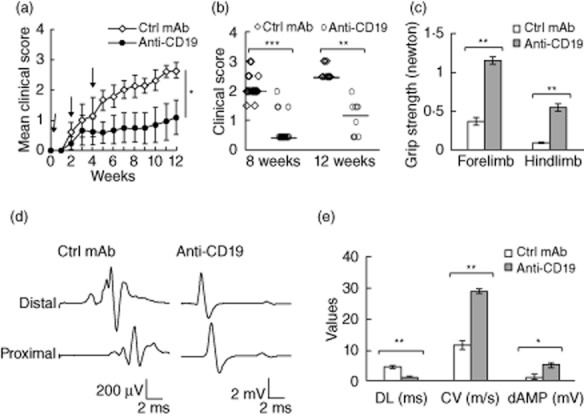
Attenuation of disease severity by B cell depletion with anti-CD19 mAb using the pulse treatment paradigm. (a) Mean clinical score [± standard deviation (s.d.)] over the time–course of SAP starting at 5 months (designated as week 0 here). Arrows: intravenous (i.v.) injection with control (Ctrl) mAb or anti-CD19 mAb (250 μg). For data points up to 8 weeks, n = 21 for Ctrl mAb and n = 18 for anti-CD19 mAb, then n = 10 each for data points beyond 8 weeks post-treatment initiation. *P < 0·000001 between two groups at 8 weeks and beyond. (b) Individual clinical scores at 8 and 12 weeks post-initiation of treatment. Horizontal bars represent the median clinical score (**P < 0·0006 for 12 weeks and ***P < 0·00001 for 8 weeks). (c) Grip strength measurements at the end of study period. Data are shown as ± standard error of the mean (s.e.m.) (n = 8–10 each; **P < 0·00001 for both hindlimb and forelimb). (d) Representative examples of sciatic motor response, demonstrating prolonged distal latency (DL), temporal dispersion, slowed conduction velocity (CV) and decreased distal amplitude (dAMP) in recordings from the Ctrl mAb group, but not in those from the anti-CD19 mAb group. (e) Bar graphs summarizing the electrophysiological data, shown as mean ± s.e.m. (n = 8–10 each). *P < 0·0004 for dAMP, **P < 0·00004 for both DL and CV.
Results
Plasmablasts and B cells in SAP mice
B7-2 KO NOD mice begin to exhibit hindlimb weakness at 6–7 months, progressing to generalized paralysis with time [1,5]. To examine B cell responses to P0 in SAP, flow cytometry was used to identify plasmablasts (CD44+CD138+B220+) and B cells (B220+IgM+ and B220+IgG+) reactive to labelled P0-ECD in spleens of 8-month-old female SAP and wild-type (WT) NOD mice. As shown in Fig. 1a, there was no difference in the percentage of splenic plasmablasts between WT NOD and SAP mice. P0-ECD-specific plasmablasts were detected in spleens of SAP mice but not in WT NOD mice (n = 6 each). The specificity of staining was demonstrated by preblocking with unlabelled P0-ECD, which eliminated the strongly positive cells but not the weakly positive cells (non-specific staining). Labelled acetylcholine receptor and ovalbumin were used as irrelevant antigens (n = 6). The percentage of IgM+ B cells in the spleen was similar between WT NOD and SAP mice. IgM+ B cells (%) were 39·1 ± 6·5 in WT and 42·8 ± 4·3 in SAP (n = 3 each). In contrast, there was an increase in the percentage of IgG+ B cells in spleens of SAP mice compared to those from WT NOD mice [33·0 ± 1·2 in WT versus 37·3 ± 1·5 in SAP (n = 5 each), P < 0·03]. Furthermore, P0-ECD-reactive B cells were increased in SAP spleens, particularly in the IgG+ B cell population, but not in WT NOD spleens (Fig. 1b). The purity of His-tagged P0-ECD used in the above studies was demonstrated by Western blot analysis, which showed a band with an expected molecular weight of 16 kDa when probed with HRP-conjugated goat anti-6-His antibody (Fig. 1c).
Fig. 1.
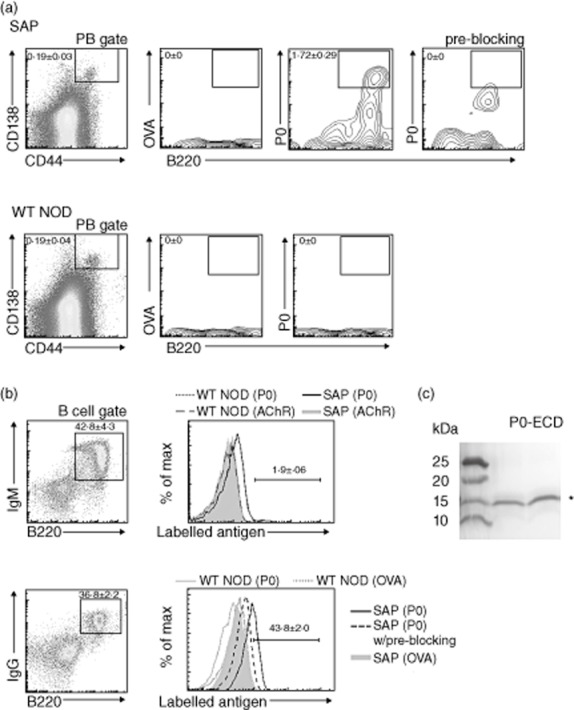
Increased frequency of protein zero (P0)-specific plasmablasts (PB) and B cells in spleens of spontaneous autoimmune polyneuropathy (SAP) mice. (a) Antigen-specific PB. Splenocytes with forward- and side-scatter properties of lymphocytes were analysed for CD138 and CD44 to identify PB, followed by selection based on B220 staining and binding of labelled antigens. Gating on antigen-specific PB was set only on strongly positive cells, whose binding was eliminated by pre-blocking for 1 h with unlabelled P0-extracellular domains (ECD). Mean ± standard error of the mean (s.e.m.) percentages of P0-specific PB are indicated in the scatterplots [n = 6 mice each, P < 0·0005 for SAP versus wild-type non-obese diabetic mice (WT NOD)]. (b) Antigen-specific splenic B cells [immunoglobulin (Ig)M+B220+, IgG+B220+]. Splenic lymphocytes were initially gated based on IgM and B220 or IgG and B220 staining, followed by selection based on the binding of labelled antigens. Scatterplots shown are from SAP mice. The mean (± s.e.m.) percentages of B cells reactive to P0-ECD in spleens of SAP mice are indicated in the histograms (n = 3–5 each, P < 0·00002 for both P0-ECD-reactive IgM+ B cells, and P0-ECD-reactive IgG+ B cells comparing SAP versus WT NOD). Acetylcholine receptor (AChR) and ovalbumin (OVA) were used as irrelevant antigens in (a) and (b). (c) Western blot analysis of purified His-tagged P0-ECD protein detected by horseradish peroxidase (HRP)-conjugated goat anti-6-His antibody (1:10 000).
Effect of B cell depletion with anti-CD19 mAb on SAP
To deplete B cells in female B7-2 KO NOD mice, we utilized the endotoxin-free monoclonal anti-CD19 mAb (clone 1D3), which has been used in another study [28]. The efficacy of anti-CD19 mAb was examined in vitro using the antibody-dependent cellular cytotoxicity (ADCC) assay. Figure 2a depicts the ADCC effector function mediated by anti-CD19 mAb, although it was not as robust as that described for anti-CD20 antibodies [29]. Anti-CD19 mAb did not induce complement-dependent cytotoxicity (Fig. 2b). Aside from the less efficient ADCC effector function of anti-CD19 mAb, there is some evidence that NOD mice exhibit a reduced sensitivity to B cell depletion with other antibodies such as anti-CD20 mAb compared to C57BL/6 mice [18]. Hence, we first investigated the depletion of B cells and plasmablasts in the blood using an acute treatment schedule that consisted of a single cycle of three intravenous (i.v.) injections of anti-CD19 mAb (250 μg) every other day with experiments performed on day 8. Examples of scatterplots of peripheral blood B cells (B220+IgM+) and plasmablasts (B220+CD138hi) from the acute depletion paradigm are shown in Fig. 2c. We found that pulse treatment with anti-CD19 mAb would deplete B cells to the same extent as the acute depletion paradigm. For pulse treatment, anti-CD19 mAb (250 μg) was injected i.v. every other week for three doses. The time–course of B cell depletion in the blood by anti-CD19 mAb over a study period of 12 weeks is shown in Fig. 2d. Total serum IgG and IgM levels at 8 weeks and 12 weeks were measured using ELISA, and results were pooled together. As shown in Fig. 2e, serum IgG and IgM levels were reduced in anti-CD19 mAb-treated animals when compared to mice treated with control mAb.
Fig. 2.
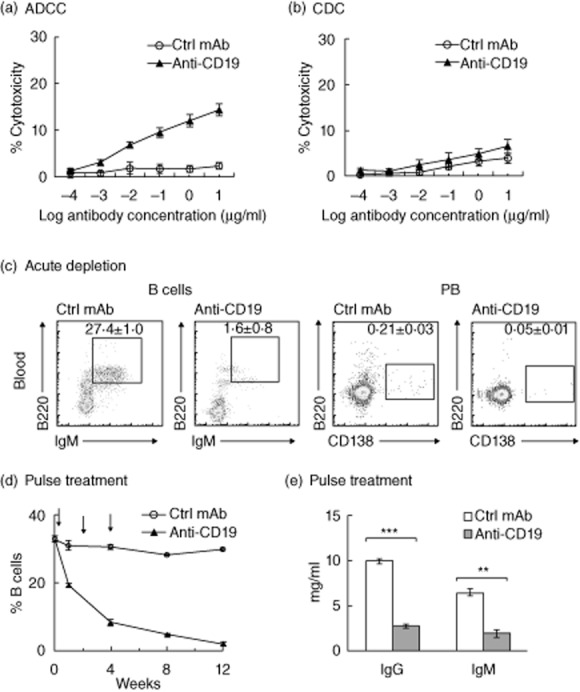
Anti-CD19 monoclonal antibody (mAb)-mediated effector function and B cell depletion in peripheral blood. (a) Antibody-dependent cell-mediated cytotoxicity (ADCC). The lactate dehydrogenase (LDH) release assay was performed using A20 cells (murine B cell lymphoma) as target cells, and J774·2 cell lines (murine monocytic cell line) or purified mouse natural killer (NK) cells as effector cells (n = 4). Effector cell/target cell ratio: 2·5:1. (b) Complement-dependent cytotoxicity (CDC). A20 cells were incubated with anti-CD19 mAb or isotype control at various concentrations in the presence of mouse serum (source of complement) (n = 3). (c) Acute depletion of peripheral blood B cells and plasmablasts (PB) by anti-CD19 mAb (250 μg) administered intravenously (i.v.) thrice every other day. Experiments were performed on day 8. Total lymphocytes were used for gating based on immunoglobulin (Ig)M and B220 for B cells, and CD138 and B220 for PB. Mean ± standard error of the mean (s.e.m.) percentages are indicated in the scatterplots. Comparing control mAb versus anti-CD19, P < 0·00001 for B cells, P < 0·009 for PB (n = 4–6). (d) Time–course of B cell depletion in the blood using the pulse treatment paradigm. Arrows: i.v. injections with control mAb or anti-CD19 mAb (250 μg) thrice every other week (n = 4). Blood samples were obtained by serial retro-orbital bleeding. The first treatment was given the day after the baseline sample was collected. (e) Reduction in serum IgG and IgM levels by pulse treatment with anti-CD19 mAb. Data from 8 weeks and 12 weeks post-treatment initiation were pooled together and shown as mean ± s.e.m. [**P < 0·0014 for IgM and ***P < 0·00001 for IgG (n = 9 each)].
Next, we examined whether B cell depletion would suppress or prevent SAP. When pulse treatment with anti-CD19 mAb was initiated at 7 months (after onset of paraparesis), no reversal of symptoms or suppressive effect could be detected 6 weeks later (n = 4) (data not shown). In contrast, treatment initiated at 5 months of age led to a significant attenuation of disease severity. The control group consisted of 5-month-old animals treated with isotype control mAb (n = 21), 11 of them studied at 8 weeks, and 10 animals studied at 12 weeks post-treatment initiation. The anti-CD19 treatment group consisted of 18 animals, eight of them studied at 8 weeks and 10 animals studied at 12 weeks post-treatment initiation. The beneficial effect of B cell depletion was evident throughout the time–course of our study period, as shown by the mean clinical score (Fig. 3a). Individual clinical scores (with median score indicated) at 8 and 12 weeks are depicted in Fig. 3b. B cell depletion with anti-CD19 mAb also led to an improvement in grip strength and electrophysiological parameters (Fig. 3c–e). Sciatic nerve electrophysiology was performed in only a subset of experiments. Unless otherwise specified, results from immunological studies in subsequent sections are from animals in the pulse treatment paradigm.
Effect of anti-CD19 mAb treatment on immune mechanisms in SAP
Given its dramatic beneficial effect on SAP, we investigated further the immune mechanisms associated with anti-CD19 treatment. Aside from its actions on peripheral blood, treatment with anti-CD19 mAb led to a partial depletion of B cells, plasmablasts and plasma cells (B220loCD138hi) in bone marrow and spleen at 8 and 12 weeks post-treatment initiation, as shown in Fig. 4a,b. The extent of depletion of these cells from bone marrow and spleen was similar to that observed for acute depletion paradigm (data not shown). Further studies revealed that P0-specific plasmablasts and P0-reactive B cells in the spleen were depleted by anti-CD19 mAb (Fig. 5a,b).
Fig. 4.
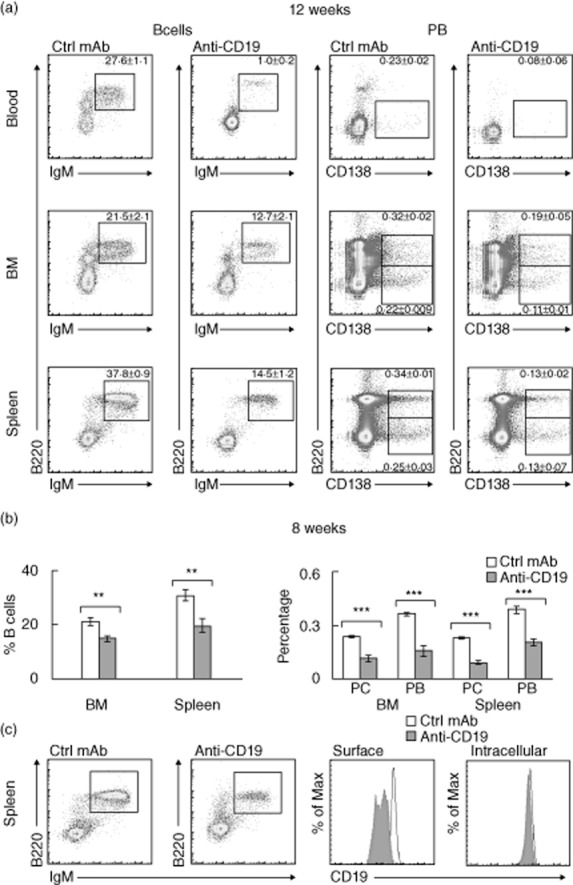
Changes in B cells, plasma cells (PC) and plasmablasts (PB) induced by anti-CD19 mAb using pulse treatment paradigm. (a) The extent of depletion of B cells (IgM+B220+), PB (B220+CD138hi), PC (B220loCD138hi) in the blood, bone marrow (BM) and spleen of spontaneous autoimmune polyneuropathy (SAP) mice studied at 12 weeks. Gating strategy as described in Fig. 1b and Fig 2c. Mean ± standard error of the mean (s.e.m.) percentages are indicated in the scatterplots (n = 4–5 each; P < 0·01 for BM B cells, P < 0·002 for splenic B cells, and P < 0·00002 for blood B cells, P < 0·03 for PB in BM, P < 0·004 for PC in BM and PB in blood, P < 0·0001 for PB and PC in spleen). (b) Data summary from animals studied at 8 weeks, shown as mean ± s.e.m. (n = 5–7). **P < 0·002 for B cells in BM and spleen, ***P < 0·00001 for PB and PC in BM and spleen. (c) Down-regulation/internalization of surface CD19 in splenic B cells at 12 weeks after pulse treatment with anti-CD19 mAb. Lymphocytes were first gated based on B220 and immunoglobulin (Ig)M staining, followed by histograms of CD19 fluorescence. Mean fluorescence intensities (MFI) ± s.e.m. for surface CD19 was 3330·2 ± 50·0 for control mAb treatment, and 1105 ± 30·1 for anti-CD19 treatment (n = 4 each, P < 0·00001). Mean fluorescence intensity (MFI) ± s.e.m. for intracellular CD19 was 3250·5 ± 99·2 for control mAb treatment, and 3116·0 ± 66·3 for anti-CD19 treatment (n = 4 each, P > 0·05).
Fig. 5.
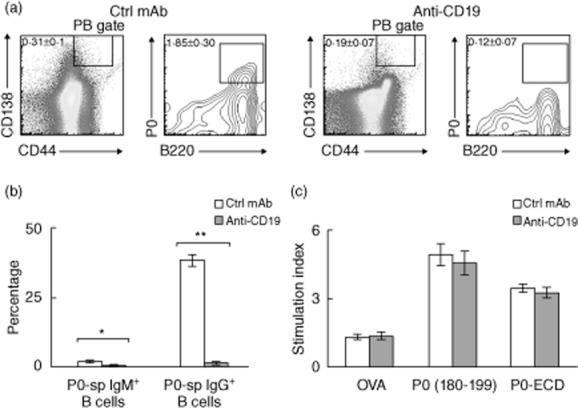
Effect of anti-CD19 mAb (pulse treatment) on B cell and T cell responses to myelin protein zero (P0). (a) Scatterplots showing the depletion of P0-specific plasmablasts (PB) by anti-CD19 mAb (n = 3, P < 0·0008). Gating strategy as described in Fig. 1a. (b) Data summary on the depletion of P0-reactive B cells by anti-CD19 mAb (n = 3). *P < 0·004 for P0-specific IgM+ B cells; **P < 0·0004 for P0-specific IgG+ B cells. (c) Treatment with anti-CD19 mAb did not alter splenocyte proliferation in response to P0 (180–199) and P0-extracellular domains (ECD (ECD) (n = 5 each, P > 0·05). Ovalbumin (OVA) was used as an irrelevant antigen. Data in (a–c) shown as mean ± standard error of the mean (s.e.m.) are from 8 weeks post-treatment initiation.
We also examined the surface staining of CD19 on splenic B cells at 12 weeks post-treatment initiation. Again, B cells were partially depleted by anti-CD19 treatment, but remaining B cells exhibited a significant decrease in surface CD19 mean fluorescence intensities (MFI), but not in intracellular CD19 MFI (post-fixation and permeabilization) (Fig. 4c). This finding suggests that pulse treatment with anti-CD19 mAb led to a sustained down-regulation/internalization of surface CD19 protein.
Because B cells are antigen-presenting cells, we studied the effect of B cell depletion on splenocyte proliferative response to P0 (180–199) and P0-ECD. At 8 weeks post-treatment initiation, there was no difference in the splenocyte proliferative response to 20 μg/ml of P0 (180–199) or P0-ECD between control mAb and anti-CD19 mAb treatment groups (Fig. 5c). To determine whether B cell depletion with anti-CD19 mAb would have an effect on regulatory mechanisms, we examined the frequency of CD4+ Tregs in total splenic lymphocytes. Tregs were identified as CD4+CD25+FoxP3+ cells. The percentage of CD4+ T cells was similar in anti-CD19 mAb and control mAb-treated mice. In contrast, the frequency of Tregs was increased significantly in animals receiving anti-CD19 antibodies when compared to those receiving control mAb, as shown in Fig. 6a. Lastly, we investigated the effect of anti-CD19 treatment on splenic B cell subsets with regulatory activity. B cells that express IL-10 after 4–5 h of ex-vivo phorbol ester and ionomycin are designated as B10 cells, and are found predominantly within the CD1dhiCD5+B220+ subset [30]. Anti-CD19 treatment led to a decrease in the percentage of CD1d+CD5+ cells in B220+ cells and in the percentage of B10 cells (Fig. 6b). The effect of anti-CD19 treatment on Tregs and regulatory B cells can be seen in both acute and pulse treatment paradigms.
Fig. 6.
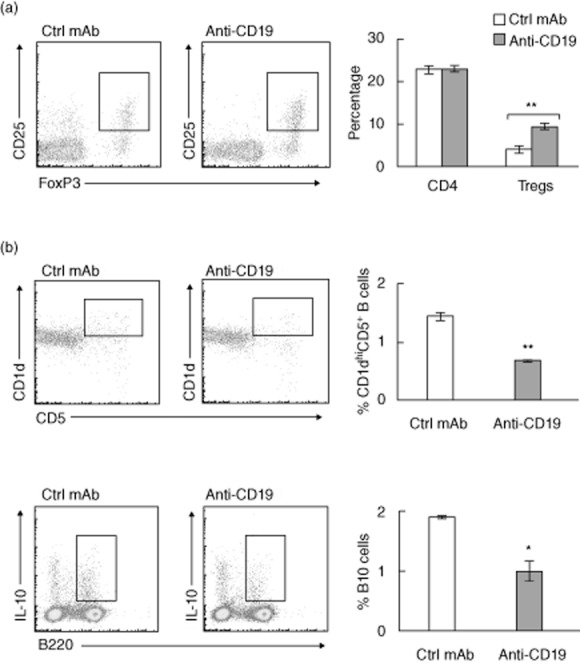
Effect of B cell depletion with anti-CD19 mAb on splenic CD4+ regulatory T cells (Tregs), CD1d+CD5+ B cell subset and B10 cells. (a) Increased frequency of splenic Tregs in B cell-depleted spontaneous autoimmune polyneuropathy (SAP) mice. Tregs were identified as CD4+CD25+forkhead box protein 3 (FoxP3+) cells. Data summary includes two acute depletion and four pulse treatment experiments. **P < 0·0003 for %Tregs. (b) Effect of anti-CD19 mAb treatment on B220+CD1d+CD5+ cells and IL-10+ B cells (B10 cells) in spleens of SAP mice. Summary for B220+CD1d+CD5+ cells includes three acute depletion and three pulse treatments (12 weeks, **P < 0·0002). For B10 cells, splenocytes were stimulated for 4 h ex vivo with lipopolysaccharide (LPS) (10 μg/ml) and leucocyte activation cocktail containing phorbol myristate acetate (PMA), ionomycin, brefeldin and BD Golgiplug prior to staining with anti-B220 and anti-interleukin (IL)-10 antibodies. *P < 0·003, n = 3 each. Bar graphs in (a) and (b) are shown as mean ± standard error of the mean (s.e.m.).
Discussion
GBS and CIDP are autoimmune diseases affecting the peripheral nervous system that involve cell-mediated and humoral immunity. In contrast to some GBS variants and chronic dysimmune neuropathies mediated by antibodies to glycolipids, a single triggering antigen has not yet been found in CIDP. We have reported previously that P0 (180–199)-reactive T cells are pathogenic in B7-2 KO NOD mice, resulting in a spontaneous autoimmune polyneuropathy that mimics CIDP in many aspects [8]. Studies in P0-T cell receptor (TCR) transgenic mice revealed another pathogenic epitope P0 (1–25) in SAP [9]. Central tolerance to P0 is dependent on a transcription factor called the autoimmune regulator (AIRE), which is expressed primarily by medullary thymic epithelial cells. Immune cells in AIRE-deficient mice exhibit increased autoreactivity towards P0 [31]. The role of B cells in SAP has not been elucidated, although passive transfer with SAP sera alone did not induce neuropathy in NOD SCID mice [1]. In this study, we have provided evidence supporting the concept that B cells contribute to the development of SAP.
We found that B cells and plasmablasts from spleens of SAP mice but not WT NOD mice exhibited increased autoreactivity to P0-ECD, consistent with our previous findings that SAP sera contained antibodies against P0 [8]. Previous studies have reported induction of demyelination by passive transfer or intraneural injection of CIDP sera that contain antibodies to P0 [10]. In experimental autoimmune neuritis (EAN), antibody and C5b-9 complex (known as MAC) appear on myelin and Schwann cells before the cellular infiltrate [32]. Other studies in rats have revealed that C5b-9 augments early demyelination, but extensive myelin injury can still occur in the absence of complement in EAN [33].
Although B cell–T cell interactions via CD40–CD40L are involved in the initiation of antigen-specific T cell and humoral responses, early studies in μMT mice suggest that B cells and antibodies to myelin P0 do not perpetuate inflammatory demyelination in EAN [34,35]. In contrast, we found that the severity of SAP is markedly attenuated by B cell depletion with anti-CD19 mAb when administered just prior to the onset of symptoms. Anti-CD19 treatment did not reverse or halt the progression of established disease. These results imply that the effect of B cell depletion is mediated primarily by suppression of the initiation mechanisms in SAP. Once CD4+ T cells infiltrate the nerves, a reduction in B cells, plasmablasts and serum IgG and IgM levels is not sufficient to attenuate myelin damage and axonal loss. This interpretation is consistent with previous studies showing that γ-IFN-producing Th1 cells are the main effector cells in SAP [7–9].
The crucial role of B cells in autoimmunity has been demonstrated in other studies, albeit with variable outcome depending on the timing of B cell depletion. In WT NOD mice, B cell depletion with anti-CD20 mAb inhibits the development of type 1 diabetes, but there is no consensus regarding whether or not it could reverse recent-onset diabetes [17–19]. Interestingly, treatment with anti-CD20 mAb causes exacerbation of EAE when given prior to myelin oligodendrocyte glycoprotein (MOG) immunization, but suppresses progression when given after onset of symptoms (day 14) [36]. Similarly, immunization with P0106–125, led to slightly accelerated onset of disease but ameliorated maximal disease severity in IgHnull (B cell-deficient) mice [37]. These findings demonstrate the critical positive and negative regulatory actions of B cells in disease pathogenesis, with suppressive effect attributed to regulatory B cells (Bregs), while other B cell subsets augment disease progression by promoting activation and expansion of T cells. The effect of B cell depletion also appears to be model-dependent, given that activation status and antigen-specificity of B cells, as well as regulatory mechanisms, may differ in each model. For example, anti-CD20 treatment ameliorates EAE induced by recombinant MOG, but exacerbates EAE induced by MOG p35-55 [38].
The spectrum of B cell depletion varies, depending on the antibody utilized. Anti-CD20 antibodies deplete primarily mature B cells, whereas anti-CD19 antibodies deplete pre-B cells, immature B cells, mature B cells, plasmablasts and some plasma cells [20]. CD19 is important for the development and function of B cells. Changes in CD19 expression correlate with the frequency of CD5+ B cells and with production of autoantibodies [39]. B cells from CD19−/− mice exhibit a decreased proliferative response to mitogens and impaired humoral responses [40,41]. CD19-dependent survival signals are also required for germinal centre formation and maintenance of the follicular and marginal zone B cell compartment [42]. We found that treatment with anti-CD19 mAb not only depletes B cells, but also plasmablasts and plasma cells, and reduces serum IgG and IgM levels in SAP, as reported in other models of autoimmunity [43].
Anti-CD19 treatment had no effect on splenocyte proliferative response to myelin P0 in our model, which is counterintuitive but may be due to incomplete B cell depletion in SAP spleens, or due to compensatory changes in other antigen-presenting cells. Other studies have shown that B cell depletion leads to impaired antigen-driven CD4+ T cell expansion [44,45]. CD19-deficient NOD mice exhibit diminished expansion of CD8+ T cells reactive to the islet-specific glucose 5-phosphatase catalytic subunit-related protein (IGRP), but not those reactive to insulin [46]. Conversely, transient enhancement of proliferation of BDC2·5 diabetogenic T cells during the depletion phase of anti-CD20 treatment has been reported [47]. The latter study also revealed that newly generated B cells are the ones that modify the function of T effector (Teff) and Treg cells, and that the protective effect on diabetes is abrogated by ablation of Tregs with anti-CD25 mAb in NOD mice [47]. Indeed, we found that B cell depletion with anti-CD19 mAb led to an increased frequency of CD4+ Tregs in SAP, similar to that reported in other experimental autoimmune diseases [17,48]. It is plausible that B cell depletion-induced changes in the cytokine milieu would shift the balance in favour of generation of Tregs rather than Teff. B cells are a major source of IL-6, which is known to inhibit the differentiation of naive T cells into Tregs [49]. In a study by Barr and co-workers, a decrease in IL-6-producing B cells was thought to underlie the amelioration of EAE by anti-CD20 treatment, although the effect on Tregs was not investigated [50]. Conversely, there is some evidence that B cells promote expansion of Tregs via glucocorticoid-induced TNF receptor ligand, and that B cell deficiency (e.g. μMT mice) or anti-CD20 treatment leads to reduced frequency of Tregs in some disease models [38,51,52].
A specific subset of B cells with a unique CD1dhiCD5+ phenotype was found recently to inhibit autoimmunity via the production of IL-10, but these cells also maintain the capacity for plasma cell differentiation [30,53]. We found that the frequency of B220+CD1dhiCD5+ cells and B10 cells was decreased by anti-CD19 treatment, although not sufficient to cause worsening of SAP. It is plausible that the potential detrimental effects of decreased frequency of B10 cells in SAP is counterbalanced or over-ridden by an increase in Tregs. Possible effects of anti-CD19 mAb on splenic dendritic cell subsets expressing CD19 were not examined in this study.
In summary, this study highlights the role of B cells in the pathogenesis of SAP in B7-2 KO NOD mice. Although SAP is a predominantly Th1-mediated disease, SAP mice exhibit increased B cell autoreactivity to P0 and aberrant expansion of P0-specific plasmablasts when compared to WT NOD mice. Depletion of B cells and plasmablasts with anti-CD19 mAb leads to the prevention/attenuation of SAP when administered just prior to symptom onset. Treatment with anti-CD19 mAb is also associated with increased frequency of CD4+ Tregs in the spleen. Overall, our data also suggest that CD19-based immunotherapeutics have the potential to become valuable disease-modifying agents for autoimmune diseases.
Acknowledgments
B7-2 KO NOD mice were generously provided by Dr J. A. Bluestone (UCSF). We would like to thank our colleagues Drs P. Wilson, M. Jensen and D. White for their helpful discussions during early stages of this project. This work was supported by the National Institute of Health Grant R01 NS064059 to B. Soliven and by a gift from the Miller Group Charitable Trust Fund (M. P. Miller III).
Author contributions
P. M. A. and S. Q. performed the clinical assessment and immunological studies; D. D. performed the electrophysiological studies; P. M. A. and B. S. designed the study and co-wrote the manuscript.
Disclosure
The authors do not have any conflicts of interest to declare.
References
- 1.Salomon B, Rhee L, Bour-Jordan H, et al. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J Exp Med. 2001;194:677–684. doi: 10.1084/jem.194.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida T, Jiang F, Honjo T, Okazaki T. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc Natl Acad Sci USA. 2008;105:3533–3538. doi: 10.1073/pnas.0710951105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horste GM, El-Haddad H, Mausberg AK, Martin S, Hartung HP, Kieseier B. Spontaneous demyelinating autoimmune neuropathy in ICAM-1 deficient NOD mice. Neurology. 2010;74:A490. [Abstract] [Google Scholar]
- 5.Kim HJ, Jung CG, Dukala D, et al. Fingolimod and related compounds in a spontaneous autoimmune polyneuropathy. J Neuroimmunol. 2009;214:93–100. doi: 10.1016/j.jneuroim.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ubogu EE, Yosef N, Xia RH, Sheikh KA. Behavioral, electrophysiological, and histopathological characterization of a severe murine chronic demyelinating polyneuritis model. J Peripher Nerv Syst. 2012;17:53–61. doi: 10.1111/j.1529-8027.2012.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bour-Jordan H, Thompson HL, Bluestone JA. Distinct effector mechanisms in the development of autoimmune neuropathy versus diabetes in nonobese diabetic mice. J Immunol. 2005;175:5649–5655. doi: 10.4049/jimmunol.175.9.5649. [DOI] [PubMed] [Google Scholar]
- 8.Kim HJ, Jung CG, Jensen MA, Dukala D, Soliven B. Targeting of myelin protein zero in a spontaneous autoimmune polyneuropathy. J Immunol. 2008;181:8753–8760. doi: 10.4049/jimmunol.181.12.8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louvet C, Kabre BG, Davini DW, et al. A novel myelin P0-specific T cell receptor transgenic mouse develops a fulminant autoimmune peripheral neuropathy. J Exp Med. 2009;206:507–514. doi: 10.1084/jem.20082113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan WX, Taylor J, Andrias-Kauba S, Pollard JD. Passive transfer of demyelination by serum or IgG from chronic inflammatory demyelinating polyneuropathy patients. Ann Neurol. 2000;47:765–775. [PubMed] [Google Scholar]
- 11.Yan WX, Archelos JJ, Hartung HP, Pollard JD. P0 protein is a target antigen in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50:286–292. doi: 10.1002/ana.1129. [DOI] [PubMed] [Google Scholar]
- 12.Pestronk A, Choksi R. Multifocal motor neuropathy. Serum IgM anti-GM1 ganglioside antibodies in most patients detected using covalent linkage of GM1 to ELISA plates. Neurology. 1997;49:1289–1292. doi: 10.1212/wnl.49.5.1289. [DOI] [PubMed] [Google Scholar]
- 13.Rinaldi S, Willison HJ. Ganglioside antibodies and neuropathies. Curr Opin Neurol. 2008;21:540–546. doi: 10.1097/WCO.0b013e32830b84b7. [DOI] [PubMed] [Google Scholar]
- 14.Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–337. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalakas MC. B cells in the pathophysiology of autoimmune neurological disorders: a credible therapeutic target. Pharmacol Ther. 2006;112:57–70. doi: 10.1016/j.pharmthera.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Uchida J, Hamaguchi Y, Oliver JA, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CY, Rodriguez-Pinto D, Du W, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiu Y, Wong CP, Bouaziz JD, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008;180:2863–2875. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 19.Serreze DV, Chapman HD, Niens M, et al. Loss of intra-islet CD20 expression may complicate efficacy of B-cell-directed type 1 diabetes therapies. Diabetes. 2011;60:2914–2921. doi: 10.2337/db11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:572–577. doi: 10.1038/nrrheum.2009.184. [DOI] [PubMed] [Google Scholar]
- 21.Borriello F, Sethna MP, Boyd SD, et al. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- 22.Salek-Ardakani S, Choi YS, Rafii-El-Idrissi Benhnia M, et al. B cell-specific expression of B7-2 is required for follicular Th cell function in response to vaccinia virus. J Immunol. 2011;186:5294–5303. doi: 10.4049/jimmunol.1100406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Good-Jacobson KL, Song E, Anderson S, Sharpe AH, Shlomchik MJ. CD80 expression on B cells regulates murine T follicular helper development, germinal center B cell survival, and plasma cell generation. J Immunol. 2012;188:4217–4225. doi: 10.4049/jimmunol.1102885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kin NW, Sanders VM. CD86 regulates IgG1 production via a CD19-dependent mechanism. J Immunol. 2007;179:1516–1523. doi: 10.4049/jimmunol.179.3.1516. [DOI] [PubMed] [Google Scholar]
- 25.Rozanski CH, Arens R, Carlson LM, et al. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med. 2011;208:1435–1446. doi: 10.1084/jem.20110040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Njau MN, Kim JH, Chappell CP, et al. CD28–B7 interaction modulates short- and long-lived plasma cell function. J Immunol. 2012;189:2758–2767. doi: 10.4049/jimmunol.1102728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DH, Muthyala S, Soliven B, Wiegmann K, Wollmann R, Chelmicka-Schorr E. The beta 2-adrenergic agonist terbutaline suppresses experimental allergic neuritis in Lewis rats. J Neuroimmunol. 1994;51:177–183. doi: 10.1016/0165-5728(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 28.Keren Z, Naor S, Nussbaum S, et al. B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging. Blood. 2010;117:3104–3112. doi: 10.1182/blood-2010-09-307983. [DOI] [PubMed] [Google Scholar]
- 29.Herbst R, Wang Y, Gallagher S, et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther. 2010;335:213–222. doi: 10.1124/jpet.110.168062. [DOI] [PubMed] [Google Scholar]
- 30.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Su MA, Davini D, Cheng P, et al. Defective autoimmune regulator-dependent central tolerance to myelin protein zero is linked to autoimmune peripheral neuropathy. J Immunol. 2012;188:4906–4912. doi: 10.4049/jimmunol.1200493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoll G, Schmidt B, Jander S, Toyka KV, Hartung HP. Presence of the terminal complement complex (C5b-9) precedes myelin degradation in immune-mediated demyelination of the rat peripheral nervous system. Ann Neurol. 1991;30:147–155. doi: 10.1002/ana.410300205. [DOI] [PubMed] [Google Scholar]
- 33.Tran GT, Hodgkinson SJ, Carter NM, et al. Membrane attack complex of complement is not essential for immune mediated demyelination in experimental autoimmune neuritis. J Neuroimmunol. 2010;229:98–106. doi: 10.1016/j.jneuroim.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Y, Bao L, Zhu S, et al. CD4 and CD8 T cells, but not B cells, are critical to the control of murine experimental autoimmune neuritis. Exp Neurol. 2002;177:314–320. doi: 10.1006/exnr.2002.7944. [DOI] [PubMed] [Google Scholar]
- 35.Zhu W, Mix E, Jin T, Adem A, Zhu J. B cells play a cooperative role via CD40L–CD40 interaction in T cell-mediated experimental autoimmune neuritis in Lewis rats. Neurobiol Dis. 2007;25:642–648. doi: 10.1016/j.nbd.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunn A, Utermohlen O, Sanchez-Ruiz M, et al. Dual role of B cells with accelerated onset but reduced disease activity in P0(1)(0)(6)(–)(1)(2)(5)-induced experimental autoimmune neuritis of IgH (0)(/)(0) mice. Acta Neuropathol. 2010;120:667–681. doi: 10.1007/s00401-010-0724-8. [DOI] [PubMed] [Google Scholar]
- 38.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68:369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J Immunol. 1996;157:4371–4378. [PubMed] [Google Scholar]
- 40.Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 41.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 42.Otero DC, Anzelon AN, Rickert RC. CD19 function in early and late B cell development: I. Maintenance of follicular and marginal zone B cells requires CD19-dependent survival signals. J Immunol. 2003;170:73–83. doi: 10.4049/jimmunol.170.1.73. [DOI] [PubMed] [Google Scholar]
- 43.DiLillo DJ, Griffiths R, Seshan SV, et al. B lymphocytes differentially influence acute and chronic allograft rejection in mice. J Immunol. 2011;186:2643–2654. doi: 10.4049/jimmunol.1002983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouaziz JD, Yanaba K, Venturi GM, et al. Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci USA. 2007;104:20878–20883. doi: 10.1073/pnas.0709205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greeley SA, Moore DJ, Noorchashm H, et al. Impaired activation of islet-reactive CD4 T cells in pancreatic lymph nodes of B cell-deficient nonobese diabetic mice. J Immunol. 2001;167:4351–4357. doi: 10.4049/jimmunol.167.8.4351. [DOI] [PubMed] [Google Scholar]
- 46.Ziegler AI, Le Page MA, Maxwell MJ, et al. The CD19 signalling molecule is elevated in NOD mice and controls type 1 diabetes development. Diabetologia. 2013 doi: 10.1007/s00125-013-3038-2. PMID 24013782. [DOI] [PubMed] [Google Scholar]
- 47.Xiang Y, Peng J, Tai N, et al. The dual effects of B cell depletion on antigen-specific T cells in BDC2.5NOD mice. J Immunol. 2012;188:4747–4758. doi: 10.4049/jimmunol.1103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamel KM, Cao Y, Ashaye S, et al. B cell depletion enhances T regulatory cell activity essential in the suppression of arthritis. J Immunol. 2011;187:4900–4906. doi: 10.4049/jimmunol.1101844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 50.Barr TA, Shen P, Brown S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun JB, Flach CF, Czerkinsky C, Holmgren J. B lymphocytes promote expansion of regulatory T cells in oral tolerance: powerful induction by antigen coupled to cholera toxin B subunit. J Immunol. 2008;181:8278–8287. doi: 10.4049/jimmunol.181.12.8278. [DOI] [PubMed] [Google Scholar]
- 52.Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–3198. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maseda D, Smith SH, DiLillo DJ, et al. Regulatory B10 cells differentiate into antibody-secreting cells after transient IL-10 production in vivo. J Immunol. 2012;188:1036–1048. doi: 10.4049/jimmunol.1102500. [DOI] [PMC free article] [PubMed] [Google Scholar]