

Abstract
Fatalities from schistosome infections arise due to granulomatous, immune-mediated responses to eggs that become trapped in host tissues. Schistosome-specific immune responses are characterized by initial T helper type 1 (Th1) responses and our previous studies demonstrated that myeloid differentiation primary response gene 88 (Myd88)-deficient mice failed to initiate such responses in vivo. Paradoxically, schistosomal antigens fail to stimulate innate cells to release proinflammatory cytokines in vitro. Since Schistosoma mansoni infection is an intestinal disease, we hypothesized that commensal bacteria could act as bystander activators of the intestinal innate immune system to instigate Th1 responses. Using a broad spectrum of orally administered antibiotics and anti-mycotics we analysed schistosome-infected mice that were simultaneously depleted of gut bacteria. After depletion there was significantly less inflammation in the intestine, which was accompanied by decreased intestinal granuloma development. In contrast, liver pathology remained unaltered. In addition, schistosome-specific immune responses were skewed and faecal egg excretion was diminished. This study demonstrates that host microbiota can act as a third partner in instigating helminth-specific immune responses.
Keywords: Host–parasite interaction, gut-microbiota, Th responses, immunopathology, schistosomiasis
Introduction
Schistosoma mansoni is a tropical parasitic helminth and, without treatment, infection evolves into a chronic disease of the liver and bowel. In endemic areas such as Africa and South America, this trematode family currently afflicts 250–300 million people [1–3]. The parasite has a complex life cycle and infection starts with the penetration of the mammalian host by cercariae, the larval life-form released by freshwater snails. Upon morphing into schistosomula, the parasite migrates via the blood and lymphatic system to the lung. Thereafter, juvenile worms travel to the venous system of the liver where they mature, pair up and become fertile. During the first weeks of infection, schistosome-specific T helper type 1 (Th1) [interferon (IFN)-γ)] responses can be observed and it has been shown that both cercariae and schistosomula initiate such reactions [1,4,5]. To complete the life cycle, eggs start to penetrate into the gut lumen and are excreted with the stool after the sixth week of infection [1,2]. During this process, eggs can become trapped within the distal vasculature of the intestine and sinusoids of the liver, and this process triggers an immune-mediated CD4+ T cell-dependent response which leads to granuloma formation [2,6–8]. Murine infections with S. mansoni reflect both the immunological and pathological situations that arise in man, making it an ideal model for studying host–parasite interaction. Antigens secreted by the eggs are further responsible for the induction of Th2 responses [interleukin (IL)-13, IL-5, IL-4], and in contrast to other S. mansoni-infected mouse strains (e.g. CBA) these responses eventually over-ride the initial Th1 reactions in C57BL/6 [2,9–12]. This polarized T cell response is dependent upon IL-4 and, although required by the host for survival during infection, it is also responsible for more serious pathological consequences [13,14]. Infection-associated advanced morbidities that occur in the chronic phase include liver fibrosis, intestinal bleeding and portal hypertension. However, these are only a fraction of schistosome-related symptoms as other chronic complications, such as under-nutrition and infertility, affect the daily lives of sufferers [3].
Interestingly, it remains unclear how eggs penetrate the epithelia and instigate the Th1 response. Dendritic cells (DC) stimulated in vitro with soluble egg antigen (SEA), derived from S. mansoni eggs, elicit little IL-6 and tumour necrosis factor (TNF) production and components thereof are able to dampen Toll-like receptor (TLR) triggering [15–17]. Simultaneously, other components within SEA are able to mediate IL-1β production via the non-obese diabetic-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome demonstrating the complexity of this parasite's immunomodulatory capacity [18]. The ability to modulate the host's ongoing innate responses would indicate that these helminth components could also alter the outcome of T cell responses. Along these lines, we have previously reported that S. mansoni-infected mice lacking the central adaptor protein myeloid differentiation primary response gene 88 (Myd88) fail to mount schistosome-specific Th1 responses, which results in smaller granuloma development [6].
Consequently, we hypothesized that if schistosomal antigens do not initiate strong innate immune responses themselves, additional stimuli must be required to begin specific responses. A possible source of stimuli lies in the host's intestinal flora which could enter the lamina propria after the epithelium is damaged by the penetrating eggs. Alternatively, steady state inflammatory responses could be triggered by gut flora being sampled by lamina propria-residing DC [19,20]. In association, in-vivo studies using Toxoplasma gondii have elucidated the requirement of Gram-negative bacteria such as Escherichia coli in the initiation of intestinal Th1-type immunopathology [21,22]. Murine S. mansoni infections directly parallel the immunopathology that arises in man. Thus, we studied the outcome of S. mansoni-infected mice depleted simultaneously of intestinal flora. The findings presented here point to an important contribution of gut bacteria in modulating schistosome-specific immune responses, granuloma development and egg excretion.
Materials and methods
Mice, S. mansoni infection, egg isolation and SEA preparation
Female specific pathogen-free (spf) C57BL/6 mice were purchased from Harlan Winkelmann GmbH (Borchen, Germany) and received a standard diet and water ad libitum. Mice were infected percutaneously with cercariae from a Brazilian strain of S. mansoni obtained from our in-house cycle of infected Biomphalaria glabrata snails (also of Brazilian origin). All animal experiments were performed under standardized conditions and in accordance with institutional, state and federal guidelines. The study was approved specifically by the ethics committee at the Regierung von Oberbayern, München (55·2-1-54-2531-75-03). The dynamics of schistosome-specific immune responses and stool analyses from S. mansoni-infected mice were performed at weeks 3, 5, 7, 8, 10 and 12 post-infection. For all studies on antibody treatment during infection, mice were killed during the eighth week of infection. Freshly isolated eggs and SEA were prepared according to standard procedure or purchased from BioGlab Ltd, Nottingham, UK, and used for in-vitro culture assays [6–8].
Antibiotic depletion of commensal bacteria
To deplete gut flora, mice were treated with an antibiotic cocktail ad libitum in the drinking water between the sixth and eighth weeks of infection similar to that described in Rakoff-Nahoum et al. [23]. The cocktail contained metronidazole (1 g/l; Fresenius SE, Bad Homburg, Germany), ciprofloxacin (200 mg/l; Bayer AG, Leverkusen, Germany), imipenem (250 mg/l; MSD GmbH, Haar, Germany), ampicillin (1 g/l; Ratiopharm GmbH, Ulm, Germany), vancomycin (500 mg/l; Cell Pharma GmbH, Hannover, Germany) and fluconazol (1 g/l; Pharmacy, Klinikum rechts der Isar, Munich, Germany). The latter was added to avoid overgrowth of Candida. Mice were also treated orally with Diflucan® (20 μg/mouse/day; Pfizer Pharma GmbH, Berlin, Germany) during the first 7 days of antibiotic treatment. Mice were weighed weekly in order to observe their health status. To avoid unnecessary exposure to bacterial contamination, groups of treated mice were handled only by the experimenter during the entire time of investigation.
Microbial analysis of stool
During infection, 1 g of excreted stool/cage of infected mice was collected and resuspended in 10 ml 0·9% NaCl (Merck, Darmstadt, Germany). With a starting dilution of 1:200, 50 μl aliquots of progressively smaller serial dilutions were plated onto solid media: Columbia, McConkey, KV and Schaedler (BD, Heidelberg, Germany). Fifty μl of undiluted bacterial stool suspensions was also plated onto Candida-specific growth plates. Bacteria were cultured at 37°C for 48 h under aerobic and anaerobic conditions. Total numbers of bacteria were determined by counting grown colonies on Columbia and Schaedler plates. McConkey and KV were used for quantitative identification of Enterobacteriaceae and Bacteroides spp. The levels of Gram-negative and Gram-positive bacteria were determined by counting the distinctive colony morphotypes on Columbia and Schaedler plates. Bacteria were investigated further by Gram-staining and using the colour strip test API 20E by bioMerieux (Nürtingen, Germany). Results were expressed as colony-forming units (CFU) per gram stool. Upon killing, pieces of small intestine and colon (2 cm) were dissected from individual mice. The luminal content was removed, weighed and resuspended in 500 μl phosphate-buffered saline (PBS). Identification and quantification of bacteria was determined as described above.
Parasite burden
As described previously, egg burden was determined after potassium hydroxide (KOH) digestion of liver and intestinal samples. The number of eggs was then calculated in accordance with the total organ weight and number of adult female worms [6–8]. Worm burden was calculated as adult worm recovery after portal perfusion and microscopic examination of infected livers and intestines. For excreted egg counts, stool samples were gathered over 72 h on week 8 of infection. In brief, 1 g of stool was incubated for 2 h at room temperature in a 30 ml suspension of sodium acetate formalin (SAF). Samples were then homogenized with a glass rod, washed and resuspended in another 25 ml SAF. Ether (5 ml) was then added under vigorous vortex for 30 s, followed by recentrifugation. The total number of eggs was then determined microscopically [6–8].
Histology
Paraffin-embedded sections (4 μm) from the left liver lobe or small bowel of individual mice were stained with Masson's Blue, as described previously [6–8]. Granuloma size was determined microscopically (Axioskop; Zeiss, Göttingen, Germany) after calculating the individual size of 35 liver granulomas/mouse or, where possible, 15 intestinal granulomas per mouse. In a blind fashion, an estimation of inflammation was performed on the intestinal sections of individual mice in a semi-quantitative manner according to the updated Sydney System for the classification and grading of gastritis [24] and scored as ‘0 = absent’, ‘1 = mild’, ‘2 = moderate’ and ‘3 = strong’.
Determination of collagen, immunoglobulin (Ig)E and Th cytokine levels in situ
The collagen content of individually weighed liver samples was determined by first incubating samples with the appropriate concentration of Pepsin (Sigma, Steinheim, Germany) and 0·5 M acetic acid overnight. Collagen levels were then measured colorimetrically using the Sircol™ soluble collagen assay kit (Biocolor Ltd, Newtownabbey, Northern Ireland, UK) [6]. Levels of IgE were measured in the sera (1:500) of individual mice using a commercial enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences, Heidelberg, Germany). Cytokine levels in homogenized liver samples were determined using commercial ELISA kits (R&D Systems GmbH, Wiesbaden, Germany). In brief, small sections of right liver lobes were removed from individual mice, weighed and homogenized (T10 basic Ultra-Turrax® disperser (IKA®-Werke GmbH & Co., Staufen, Germany) in 500 μl of precooled RPMI medium (PAA, Linz, Austria) containing no supplements. Following centrifugation, 16 000 g at 4°C, for 10 min, the resulting supernatant was removed and stored at −20°C. Cytokine concentrations from each mouse were normalized to the number of eggs within the corresponding livers [6,8].
In-vitro immune responses
Schistosome-specific responses
Erythrocyte-depleted mesenteric lymph node (MLN) cells (3 × 105) from individually infected mice, that were treated with or without antibody, were co-cultured with or without SEA (25 μg/ml) or 1 μg/ml α-mouse CD3e antibody (eBiosciences, San Diego, CA, USA) as a polyclonal T cell activator (positive control) in triplicate for 72 h at 37°C. Thereafter, supernatants were removed and tested for cytokine content using commercial ELISA kits (R&D Systems) following the manufacturer's protocol. Developed ELISA plates were read at 450 nm using a 96-well plate ELISA reader and Magellan software (Tecan, Reading, UK).
DC cultures
Bone marrow was obtained by flushing bones with PBS. Cells were depleted of erythrocytes, filtered (100 μm) and centrifuged at 230 g for 10 min (Megafuge 3·0R). Cells were cultured in RPMI-1640 containing 10% fetal calf serum (FCS) (PAA) and 20 ng/ml granulocyte–macrophage colony-stimulating factor (GM-CSF) (PeproTech GmbH, Hamburg, Germany) for 7 days at 37°C. Bone marrow DC (BMDC) (1 × 106 cells/ml) were plated in 96-well plates and then primed with freshly isolated schistosomal eggs for 2 h. Thereafter, cells were stimulated further with TLR stimuli [5 ng/ml Pam3Cys or cytosine–phosphate–guanine (CpG) or 2 ng/ml of LPS or R848; Sigma]. Proinflammatory cytokine secretion was measured after 48 h with commercial ELISA kits.
Transepithelial electrical resistance (TER)
PTK6 cells (protein tyrosine kinase 6; origin: mouse colon epithelium), as described in [25], form polarized monolayers when seeded on high-density Transwell filters [12 mm Transwell-COL collagen-coated 0·4 μm pore polytetrafluoroethylene (PTFE) membrane insert; Corning, New York, NY, USA]. The tightness of the apical junctions can be measured by the flux of ions in the culture medium. Cells were seeded at a density of 1 × 106 cells/Transwell filter. The TER of the epithelial monolayers was measured using a Millicell-ERS volt-ohm meter (Millipore, Eschborn, Germany). The electrodes were soaked in 12 ml of 80% ethanol for 15 min in a 15 ml centrifuge tube, airdried for 30 s and equilibrated with 7 ml of Dulbecco's modified Eagle's medium (DMEM) for another 15 min prior to use. Each Transwell insert was measured three times at different positions to determine basal TER levels before adding increasing amounts of freshly isolated, sterile schistosomal eggs with or without recombinant TNF (10 ng/ml; R&D Systems). Measurements were followed for 72 h.
Statistical analysis
Statistical differences were analysed by either analysis of variance (anova) or Student's t-test using GraphPad Prism software (San Diego, CA, USA). Cytokine concentrations were determined using the standards incorporated into each assay and SigmaPlot (Systat Software GmbH, Erkrath, Germany).
Results
Assessing schistosome-specific immune responses in vivo and in vitro
During the fifth week of infection, paired S. mansoni worms begin to produce eggs which penetrate into the gut lumen. Trapped eggs initiate Th1 responses which, in C57BL/6 mice, are eventually over-ridden by Th2 responses [2,6,7,11,12]. To verify such schistosome-specific immune profiles, mesenteric lymph node (MLN) cells from 0-, 3-, 5-, 7-, 8-, 10- and 12-week-infected C57BL/6 mice were restimulated ex vivo with SEA for 72 h (Fig. 1). Released Th1 (IFN-γ), Th2 (IL-13, IL-4) or regulatory (IL-10) cytokine production was then determined in the supernatant via ELISA. As shown, the cross-over point from schistosome-specific Th1 to Th2 responses is around the eighth week of infection. Thus, we chose this time-point for further analytical studies. Figure 1 also shows that after 12 weeks of infection the Th2 responses also begin to decrease. Paradoxically, stimulation of DC in vitro with freshly isolated schistosomal eggs fails to elicit the production of proinflammatory cytokines such as TNF and IL-6 (bar 2 in Fig. 2a and b, respectively). Previous reports have shown that SEA can dampen LPS-stimulated innate cells [15–18]. To study whether this phenomenon occurred only with stimulation of TLR-4, we cultured DC with TLR-2 (Pam3Cys), TLR-4 (LPS), TLR-7 (R848) and TLR-9 (CpG) stimuli in the presence or absence of fresh viable schistosomal eggs. As expected, the different TLR stimuli induced strong levels of TNF and IL-6 (Fig. 2a and b, respectively) whereas co-culturing these activated DC with eggs dampened these immune responses, regardless of the activating TLR ligand. Therefore, although eggs fail to stimulate DC to produce proinflammatory cytokines, they possess the capacity to modulate innate immune responses upon TLR triggering which may alter Th responses.
Fig. 1.
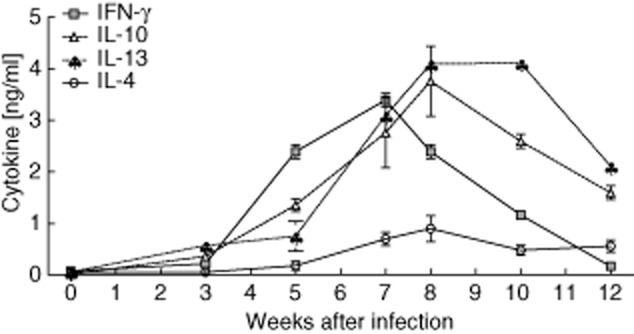
Immune responses to schistosomal antigens in vivo and in vitro. C57BL/6 mice were infected with Schistosoma mansoni for 12 weeks. At time-points 0, 3, 5, 7, 8, 10 and 12 weeks post-infection, draining mesenteric lymph nodes (MLN) were restimulated ex vivo with soluble egg antigen (SEA). After 72 h, the supernatants were removed and analysed for cytokine concentrations via enzyme-linked immunosorbent assay (ELISA). The symbols represent the mean ± standard deviation of six individually assessed mice in two independent infection studies.
Fig. 2.
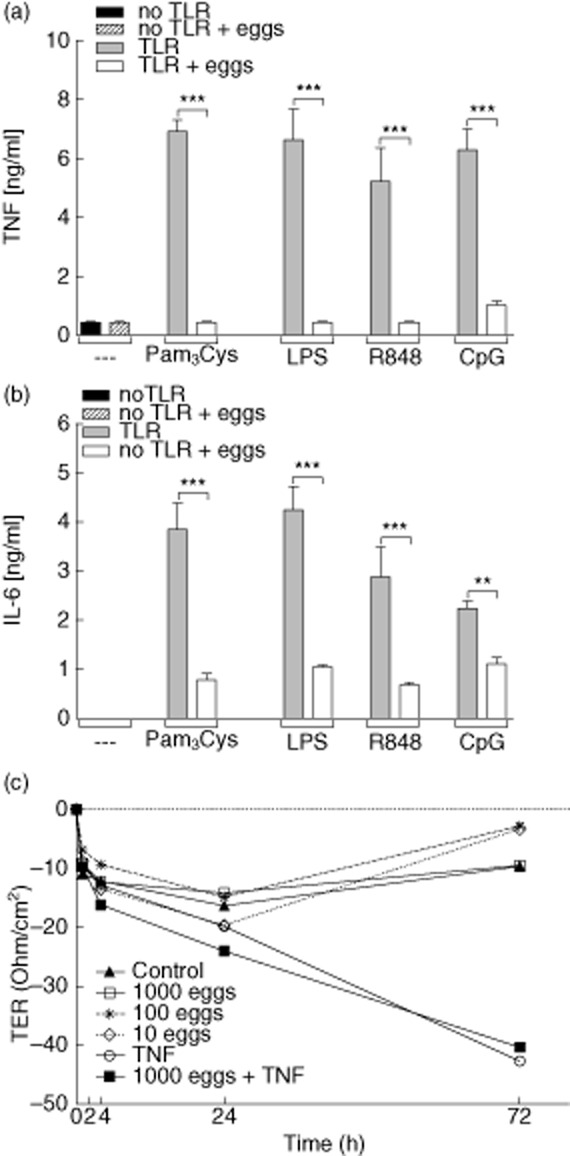
Schistosome eggs suppress innate immune responses and are incapable of disrupting epithelial cell layers in vitro. (a,b) Dendritic cells from C57BL/6 mice were generated from bone marrow cells using granulocyte–macrophage colony-stimulating factor (GM-CSF). After 7 days, dendritic cells (DC) (2 × 105) were co-cultured with 400 viable eggs for 2 h. Thereafter, cells were stimulated with either Pam3Cys, lipopolysaccharide (LPS), R848 or cytosine–phosphate–guanosine (CpG). After an additional 48 h, culture supernatants were removed and analysed for the production of tumour necrosis factor (TNF) (a) and interleukin (IL)-6 (b) via enzyme-linked immunosorbent assay (ELISA). Bars represent mean ± standard deviation (s.d.) of three pooled experiments. (c) Eggs were placed with or without TNF into the upper chamber of an in-vitro system resembling the intestinal epithelium. Epithelial integrity was then monitored at the indicated time-points by measuring the resistance of the epithelial cell layer [transepithelial electrical resistance (TER)]. Symbols represent pooled data from two independent assays. Asterisks indicate significant differences using one-way analysis of variance (anova) (**P < 0·01; ***P < 0·001).
Decreased transepithelial resistance is dependent upon proinflammatory cytokines but is not induced by schistosome eggs
The intercellular tight junction is one of the rate-limiting barriers for the permeation of ions and larger solutes in the paracellular pathway [20]. To explore how schistosomal eggs could penetrate the luminal wall, an in-vitro system was established to mimic the integrity of the intestinal epithelium. In this two-chamber system, both luminal and basal components can be used to determine the barrier function of the epithelium by measuring the resistance of the epithelial cell wall. Decreased electrical resistance demonstrates the disruption of the tight junctions and therefore leakage of the epithelium. To analyse if schistosomal eggs were capable of diminishing the resistance of the epithelium they were placed into the upper chamber. Figure 2c clearly shows that after 48 h of incubation with eggs the TER is slightly diminished, but after 72 h the resistance has reconstituted and this process was independent of the amount of eggs. However, applications of TNF or IFN-γ (data not shown) were able to reduce resistance and this ability was not altered upon the addition of eggs. These data suggest that eggs themselves cannot penetrate the intestinal barrier, but would require additional proinflammatory mediators possibly released by macrophages or T cells within the granulomas. Thus, we hypothesized that host responses to intestinal bacteria, leaking through the epithelium, could provide a proinflammatory source, and proceeded to study the role of this third partner during schistosome infection.
Successful depletion of commensal flora during infection
To obtain an impression of the gut microbiota during infection we analysed the bacterial composition of excreted stool samples over the course of infection. In brief, serial dilutions of stool samples were plated onto solid media to identify Gram-negative, Gram-positive, aerobic and anaerobic bacteria. Figure S1a–e (see Supporting information) depicts the bacterial levels in S. mansoni-infected and control groups of mice over 12 weeks. Using this basic bacterial profile we modified an oral bacteria-depletion protocol [23] which included metronidazole, ciprofloxacin, imipenem, ampicillin and vancomycin. To avoid the expansion of Candida in the antibiotic-treated mice, we additionally included fluconazole in our treatment protocol. The latter was given to the groups of mice by oral gavage during the first 7 days of treatment. Since intestinal pathology only starts to develop around the sixth week of infection, we began oral treatment from this time-point. To verify that bacteria had been depleted in both the control and S. mansoni-infected groups (8 weeks post-infection), the stool content of the small and large intestine were plated onto solid agar plates and cultivated for 48 h aerobically and anaerobically. Figure 3 shows the depletion of aerobic and anaerobic bacteria in the small (Fig. 3a) and large (Fig. 3b) intestine from individually analysed mice. As clearly depicted, depletion (open symbols) was successful in both infected and non-infected groups. In addition, the luminal content was cultivated on Candida-specific agar plates and no Candida growth could be observed (data not shown).
Fig. 3.
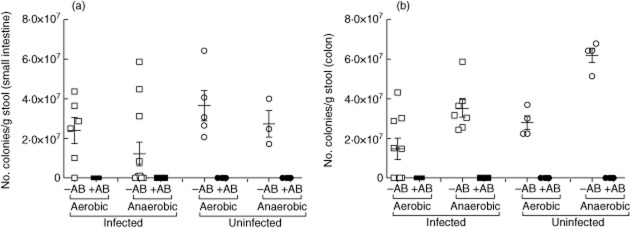
Successful depletion of commensal flora during Schistosoma infection. Groups of S. mansoni-infected or non-infected mice were or were not treated orally 6 weeks post-infection with a broad spectrum antibiotic cocktail (closed and open symbols, respectively). At the eighth week of infection, mice were analysed for the presence of bacteria in the small (a) and large (b) bowel. In brief, the stool content of individual mice was plated on solid agar plates and cultivated for 48 h. Graphs show the number of aerobic and anaerobic colonies per gram stool [mean ± standard deviation (s.d.)] per mouse. Open symbols represent untreated mice (–AB) and closed symbols show antibiotic-treated (+AB) mice. The data shown are representative of one of four separate infection-depleted experiments.
Antibiotic treatment does not affect parasitological aspects of S. mansoni infection
Between the sixth and eighth weeks of S. mansoni infection, groups of mice were left either untreated or given the antibiotic cocktail described in the previous section. At week 8 of infection mice were killed and investigated for alterations in the pathological outcome of S. mansoni infection, such as change in weight (Fig. 4a), levels of total IgE (Fig. 4b), the in-situ cytokine milieu (Fig. 4c) and parasite burden (Fig. 4d–f). Starting from exposure to S. mansoni, groups of mice were weighed every second week. When compared to non-infected control groups, infected mice gained less weight throughout infection. However, no differences in weight were observed between the infected groups of mice upon antibody treatment (Fig. 4a). With regard to total IgE, levels of this antibody in the sera were reduced significantly in infected antibody-treated groups (Fig. 4b). To observe whether or not there were alterations in the cytokine milieu, Th1 and Th2 cytokines were measured in situ from the supernatants of liver homogenates obtained from individual mice. To avoid bias, results were normalized to the number of eggs in the corresponding livers [6]. Figure 4c shows that slight but non-significant differences could be observed between the two groups.
Fig. 4.
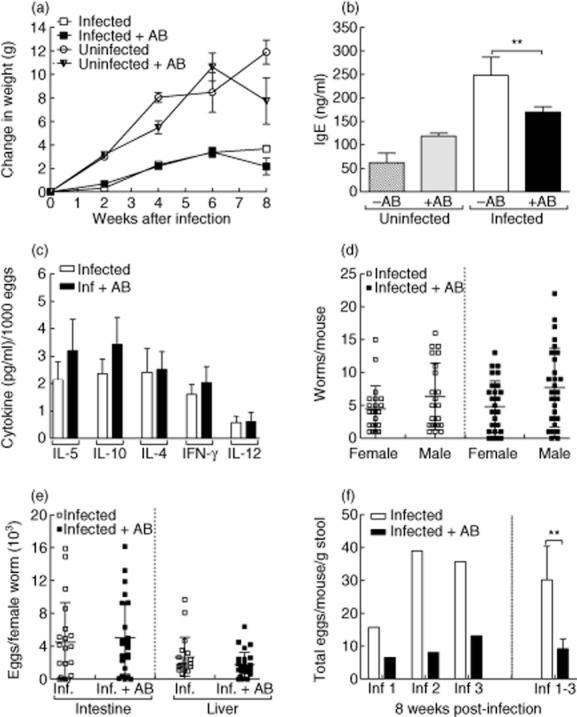
Assessment of infection parameters upon administration of antibiotics. Groups of C57BL/6 mice were infected with Schistosoma mansoni. Between weeks 6–8 post-infection, groups of infected and non-infected control mice were treated with antibiotics. (a) Throughout infection, mice were weighed on a fortnightly basis to observe any alterations in their general health. Symbols represent the change in weight [mean ± standard error of the mean (s.e.m.)] of five mice per group. Data represent one experiment of three. (b) Levels of total immunoglobulin (Ig)E were determined in the sera of individual mice by enzyme-linked immunosorbent assay (ELISA). Bars represent mean ± standard deviation (s.d.) of individual mice from three independent experiments (–AB n = 14 and +AB n = 20). (c) In-situ cytokine levels [interleukin (IL)-5, IL-10, IL-4, interferon (IFN)-γ and IL-12] were measured in homogenized liver samples obtained from 8-week-infected mice by ELISA. Levels were then calculated on individual egg burden in the liver. Bars represent the mean ± s.e.m. of individually assessed mice from four independent experiments (–AB n = 22 and +AB n = 28). (d) Worm burden was calculated following liver perfusion and microscopic examination of infected organs. Symbols represent mean ± s.d. of worm burden per mouse from four independent experiments (–AB n = 22 and +AB n = 28). (e) To determine fecundity, the amount of eggs in the infected tissues was calculated following organ digestion with potassium hydroxide (KOH). Symbols represent the mean ± s.d. of eggs per female worm in individual mice from four independent experiments (–AB n = 22 and +AB n = 28). (f) Eggs secreted in the stool were measured after 8 weeks of infection. The data show egg load per mouse from three independent experiments (Inf 1, 2 and 3). On the right-hand side of the graph, bars represent the mean ± s.d. of the three experiments pooled together. Asterisks indicate a significant difference (** P < 0·01) between the indicated groups following t-test analysis.
The amount of worms per individual mouse was determined after performing liver perfusion: worms were expulsed via a cut portal vein. Following perfusion, trapped worms were identified by macroscopic examination of the livers and intestines. Figure 4d shows that between the antibody-treated and non-treated groups, the number of female and male worms per mouse was equal. Thus, antibody treatment did not influence worm development per se. To determine the fecundity of female worms, individual samples of livers and intestines were removed and digested with KOH to release the eggs from the tissues. The number of eggs in each organ were counted and divided through the total amount of female worms per individual mouse. No significant differences between the two groups could be observed (Fig. 4e). Therefore, the fecundity of the worms is not affected by the antibiotic treatment. Eggs which gain access to the gut lumen are visible in the stool from around the sixth week post-infection. To assess egg excretion, we also determined the number of eggs found in the stool just prior to analysis (Fig. 4f). In three independent experiments (left side of Fig. 4f), infected antibody-treated mice excreted significantly fewer eggs than control-infected mice. Combined data are shown on the right side of the figure.
Decreased granuloma development during infection
Histological preparations from the livers and intestines of week 8-infected mice revealed a less pronounced immunopathology in the intestines of antibody-infected mice when compared to control-infected groups. As shown in Fig. 5a (upper panel), there is a marked decrease in inflammation and granuloma development upon comparison of control-infected (far left) with antibody-treated (left) mice. Figure 5b shows the quantitative analysis of the average size of granulomas in each individual mouse [6–8]. Interestingly, although no differences in the size of liver granulomas could be observed between the two infected groups, the size of intestinal granulomas was reduced significantly in the antibody-treated infected mice (Fig. 5b). Because sections were stained with Masson's Blue we were also able to gain an impression of liver fibrosis, as the staining turns collagen fibres blue. In the lower panel of Fig. 5a one can observe that although granuloma development in the liver is not altered, there is a marked increase in collagen fibre intensity in infected, antibody-treated mice (cf. far left with left). We verified this observation by quantitative measurement of soluble collagen in individual livers (Fig. 5c, left). In correlation with the enhanced fibrosis, levels of the major profibrogenetic Th2 cytokine IL-13 were also elevated in liver homogenates of individual, antibody-treated mice (Fig. 5c, right) albeit non-significantly. In addition, an estimation of inflammation activity was performed on individual intestinal sections in a blind fashion. This semi-quantitative assessment was performed in accordance with the updated Sydney System for the classification and grading of gastritis [24]. Scoring ranged from 0 to 3, which corresponds with ‘absent’, ‘mild’, ‘moderate’ and ‘strong’ inflammation, respectively. Figure 5d shows that inflammation in intestines of S. mansoni-infected mice was significantly higher than in infected antibody-treated groups. When apparent, inflammation was observed in the deeper parts of the mucosa adjacent to the crypts within the lamina propria. Superficial villous parts were free of inflammation. There was no architectural alteration of the crypts or villi in specimens from either infected or naive animals. In naive, non-infected animals, there were no apparent histomorphological differences between specimens of animals with or without antibody treatment, especially with regard to the epithelial or the stromal parts of the mucosa.
Fig. 5.

Infected antibody-treated mice present reduced intestinal inflammation and granuloma formation but elevated collagen levels in hepatic granulomas. (a) Histological sections were prepared from the intestines (upper panel) or liver (lower panel) from control and 8-week-infected mice and stained with Masson's Blue. Magnification was ×40 of equally sized sections. Organ sections from infected mice are depicted far left; infected and antibiotic treatment groups, left; naive mice, right; and naive mice treated with antibiotics, far right. (b) From four independent infection studies, the average size of 35 liver granulomas and up to 15 intestinal granulomas were measured in each infected mouse. Symbols represent mean ± standard deviation (s.d.) of individual mice (–AB n = 22 and +AB n = 28). (c) The amount of collagen was determined in individual liver samples using the Biocolor assay kit and then correlated to the number of eggs found in the livers of the corresponding mice. The results shown are the mean ± s.d. obtained from individual mice from two independent infection experiments. In-situ levels of interleukin (IL)-13 were measured in the supernatants of liver homogenates at the time of analysis by enzyme-linked immunosorbent assay (ELISA). Bars represent the mean ± standard deviation (s.d.) of individual mice from three independent infection studies. (d) Semi-quantitative pathological assessment of intestinal inflammation. In a blind fashion, intestinal sections from individual mice were assessed for their level of inflammation (0 = absent; 1 = mild, 2 = moderate inflammation and 3 = strong inflammation). Symbols represent mean ± standard deviation (s.d.) of Inf. n = 10; Inf + AB n = 15; naive n = 5 and naive + AB n = 5). Asterisks indicate a significant difference (*P < 0·05; **P < 0·01; ***P < 0·001) between the indicated groups following analysis of variance (anova) or t-test analysis.
Altered schistosome-specific cytokine responses in antibody-treated mice
To investigate whether depletion of commensal flora influenced schistosome-specific immune responses, lymphocyte preparations from MLN were stimulated in vitro with SEA for 72 h. Thereafter, supernatants were removed and tested for IFN-γ, IL-10 and IL-13 by ELISA (Fig. 6a–c, respectively). Previous research has shown that without priming, neither CD4+ T cells nor bulk lymphocyte populations respond to SEA [6,7] and, as anticipated, cells from the uninfected control groups did not respond here either (data not shown). Interestingly, although SEA-specific IFN-γ and IL-10 responses could be detected in antibody-treated infected mice, they were significantly lower than those observed in control-infected groups. In contrast, although not statistically significant, levels of IL-13 (Fig. 6c) were slightly elevated in the antibody-treated infected mice; this finding correlates with the higher collagen content and histological observations mentioned in Fig. 5a,c.
Fig. 6.
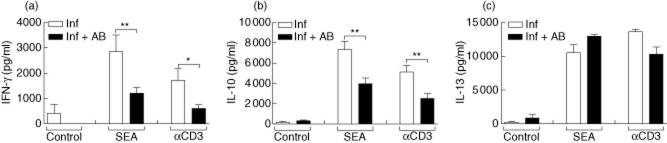
Altered schistosome-specific immune responses in antibody-treated Schistosoma mansoni-infected mice. Mesenteric lymph nodes (MLN) cells from uninfected or S. mansoni-infected mice, with or without antibody treatment, were restimulated in vitro with soluble egg antigen (SEA) (25 μg/ml) or αCD3 (1 μg/ml) for 72 h. Thereafter, the cytokines (a) interferon (IFN)-γ, (b) interleukin (IL)-10 and (c) IL-13 were measured in the supernatant by enzyme-linked immunosorbent assay (ELISA). Bars represent the mean ± standard deviation (s.d.) of individually tested mice from two independent experiments. Asterisks indicate significant differences (*P < 0·05; **P < 0·01).
Discussion
Many helminthic parasites cause chronic inflammation in the gut, and a prominent example is the schistosome family. Egg penetration into the gut lumen is essential for the helminth's survival, but the contributing mechanisms remain poorly understood. During schistosomiasis there is a finely balanced interaction between the host's immune system and the parasite to ensure that both survive, but the involvement of a third partner, the host's commensals, has never been addressed. Moreover, there are only a few studies on alterations in gut flora composition during chronic infectious enteric diseases. For example, during infections with T. gondii a strong increase in Gram-negative bacteria leads to an aggravated intestinal Th1 immunopathology [21], but in experimental Angiostrongylus infection only insignificant increases in these bacteria could be detected [26]. Our studies showed no changes in Gram-positive bacteria but we could detect, albeit insignificantly, variations in Gram-negative bacteria from the fifth week of infection (Supporting information, Fig. S1). This time-point coincides with the onset of fecund females and initial Th1 responses to SEA [7].
As mentioned above, the mechanisms involved in inducing schistosome-specific Th1 responses remain unclear, as does the requirement of egg excretion. Thus, we focused on the primary responses elicited by antigen-presenting cells (APC) upon co-culture with schistosomal eggs and observed that freshly isolated viable eggs failed to stimulate DC in vitro and, like SEA, could even suppress proinflammatory responses towards different TLR ligands (Fig. 2a,b) [15,16]. A similar phenomenon has been reported in studies using the parasite model Fasciola hepatica; here, tegumental antigens were shown to suppress DC maturation and function upon stimulation with TLR ligands [27]. In addition, SEA not only has the capacity to dampen TLR signalling but simultaneously drives NLRP3 inflammasome activation and IL-1β production [18]. Such immune modulation of DC responses would also indicate that these helminth components could alter the outcome of T cell responses, and such deviations have been reported in TLR and inflammasome-deficient mice [6,18]. Interestingly, Benson and colleagues also showed that both mucosal innate and adaptive immune responses to T. gondii rely upon the stimulation of DC by normal gut microflora [28].
As mentioned above, schistosomal eggs have to penetrate the gut epithelium and this critical step is immune-dependent (namely CD4+ T cells), as egg excretion is diminished in immunocompromised patients [29]. In addition, tissue-bound egg numbers in CD4-deficient mice are also reduced and in verification, studies with HIV co-infected patients showed significantly fewer eggs in their faeces [30]. Our studies into the ability of eggs to diminish transepithelial resistance displayed that eggs were incapable of severing tight junctions within the epithelium themselves unless proinflammatory cytokines such as TNF or IFN-γ were present (Fig. 2c). Interestingly, Amiri et al. showed that infected severe compromised immunodeficient (SCID) mice fail to develop granulomas and have no detectable eggs in their stool, a phenotype that could be reverted through the administration of TNF [31]. However, other studies were not able to restore granuloma formation in SCID mice with TNF injection, and egg-laying was delayed but not completely abrogated [32]. Nevertheless, all these data clearly substantiate the hypothesis that schistosomal eggs require local additional factors to gain access to the lumen.
Granuloma formation around the eggs trapped in the liver and the intestine represents the main immunopathology during S. mansoni infection [2,33,34] and, interestingly, there are differences in the development of the hepatic and intestinal granulomas. While liver granulomas shrink, become fibrogenic and possess more regulatory T cells (Treg) in the chronic phase of infection [6], intestinal granulomas do not change in size, indicating the importance of the local milieu in shaping immune responses [34]. Our data show that whereas liver granuloma size remained comparable in both infected groups (Fig. 5b), the granulomas in antibody-treated animals presented enhanced fibrotic markers: more collagen and IL-13 (Fig. 5c). In contrast, in the absence of commensals, intestinal pathology was reduced significantly, with smaller granulomas and less inflammation (Fig. 5b,d). Such deviations in immunopathology have been reported in schistosome-infected IFN-γ receptor-deficient mice [35,36] and, in association, levels of secreted IFN-γ by SEA-stimulated MLN cells were also reduced in infected antibody-treated mice. These observations therefore indicate that granuloma development is influenced by the local effects of the commensals and not simply the result of antibiotics, such as metronidazole, on systemic T cell activity [37]. Our findings also expand upon early studies in S. mansoni-infected germ-free mice [38,39], and indicate that commensals play an important part in helping to perpetuate the local intestinal inflammatory response which appears to aid egg expulsion.
Conversely, Th2 cytokines also play an important role in the intestine during S. mansoni infection, as the absence of IL-4 leads to increased mortality due to hepatocyte damage, impaired intestinal pathology, egg accumulation and a prominent Th1 phenotype [13]. Within this study, schistosome-specific IFN-γ and IL-10 but not IL-13 responses were significantly suppressed in antibody-treated mice (Fig. 6). Interestingly, IL-13 is known to promote collagen synthesis [40], and even though both schistosome-specific IL-13 release from MLN and in-situ IL-13 levels were not significantly elevated, these increases correlated with significantly higher collagen levels (Figs 6c and 5c, respectively). These results indicate that although IgE levels were reduced, the antibody-treated infected group have a stronger Th2 bias at an earlier time-point of infection. Nevertheless, it does not exclude that these cytokine responses are mediated by other helminth-induced cell types such as alternatively activated macrophages, B cells or regulatory T cells. Furthermore, Lukacs and colleagues showed that IFN-γ down-regulates splenic IL-2 and IL-4 production and the corresponding inflammatory granuloma formation [41], indicating that IFN-γ plays a regulatory role in the maintenance of the granulomatous responses [42]. In correlation, studies have also documented cross-regulatory networks in which IFN-γ inhibits Th2 cells and IL-10 inhibits IFN-γ [43]. Thus, as there was a diminished IFN-γ (Fig. 6a) but stronger IL-13 (Fig. 6c) production in antibody-treated mice, it suggests the need of gut bacteria for the production of Th1 cytokines. These data substantiate that Th1 and Th2 responses within the intestine have to be finely regulated, as excessive inflammation induced by Th1 cytokines damages the intestinal tissue, impairs egg excretion and therefore endangers the survival of the host and the parasite.
Although host commensals are definitely not the only factor, this study provides evidence that during S. mansoni infection they play an important role in the outcome of intestinal granuloma formation and schistosome-specific immune responses and could thereby influence egg excretion. Thus, we hypothesize that intestinal flora act as a symbiotic partner for both host and parasite, as without commensal-induced responses (e.g. TNF), penetration of eggs into the bowel lumen is hampered. However, to avoid potentially harmful reactions elicited by the continuous exposure to commensals, helminth-derived factors dampen innate cell responses and continue to modulate the host's responses to their own advantage. Further studies are required, however, to investigate these processes at the intestinal barrier. Since a recent report has also demonstrated the essential requirement of gut flora in helminth survival [44], the interplay of these microbes may offer a new route of anti-helminthic therapies.
Acknowledgments
We thank M. Herdlika, S. Paul and S. Schmidt for excellent technical assistance. This work was supported by the Else Kröner-Fresenius-Stiftung (EKFS) project A94/06 and E. L.-V. was funded through project A47/2010, also by the EKFS.
Disclosures
There are no financial or commercial conflicts of interest.
Author contributions
M. H., L. E. L., K. M., R. V., R. L. and C. P. de C. performed the experiments and analysis, H. W. provided expertise and financial support, M. H., L. E. L. and C. P. de C. wrote the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's website:
Fig. S1. Microbiological analysis of commensal flora during Schistosoma infection. C57BL/6 mice were infected with S. mansoni for 12 weeks. At the indicated time-points post-infection, serial dilutions of stool samples were analysed for microbial content. After 48 h of culture, bacterial species were identified by morphotypes, colour strip tests and Gram staining. Graphs show changes in Gram-positive (a) Lactobacilli, (b) Enterococci, (c) Peptostreptococci and Gram-negative bacteria (d) Escherichia coli and (e) Bacteroides species. The data represent results from one infection study (naive n = 4 and Inf + AB n = 5).
References
- 1.Pearce EJ, MacDonald AS. The immunobiology of schistosomiasis. Nat Rev Immunol. 2002;2:499–511. doi: 10.1038/nri843. [DOI] [PubMed] [Google Scholar]
- 2.Wilson MS, Mentink-Kane MM, Pesce JT, et al. Immunopathology of schistosomiasis. Immunol Cell Biol. 2007;85:148–154. doi: 10.1038/sj.icb.7100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson S, Vennervald BJ, Kadzo H, et al. Health implications of chronic hepatosplenomegaly in Kenyan school-aged children chronically exposed to malarial infections and Schistosoma mansoni. Trans R Soc Trop Med Hyg. 2010;104:110–111. doi: 10.1016/j.trstmh.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mountford AP, Coulson PS, Pemberton RM, et al. The generation of interferon-gamma-producing T lymphocytes in skin-draining lymph nodes, and their recruitment to the lungs, is associated with protective immunity to Schistosoma mansoni. Immunology. 1992;75:250–256. [PMC free article] [PubMed] [Google Scholar]
- 5.Wynn TA, Oswald IP, Eltoum IA, et al. Elevated expression of Th1 cytokines and nitric oxide synthase in the lungs of vaccinated mice after challenge infection with Schistosoma mansoni. J Immunol. 1994;153:5200–5209. [PubMed] [Google Scholar]
- 6.Layland LE, Wagner H, da Costa CU. Lack of antigen-specific Th1 response alters granuloma formation and composition in Schistosoma mansoni-infected MyD88−/− mice. Eur J Immunol. 2005;35:3248–3257. doi: 10.1002/eji.200526273. [DOI] [PubMed] [Google Scholar]
- 7.Layland LE, Rad R, Wagner H, et al. Immunopathology in schistosomiasis is controlled by antigen-specific regulatory T cells primed in the presence of TLR2. Eur J Immunol. 2007;37:2174–2184. doi: 10.1002/eji.200737063. [DOI] [PubMed] [Google Scholar]
- 8.Layland LE, Mages J, Loddenkemper C, et al. Pronounced phenotype in activated regulatory T cells during a chronic helminth infection. J Immunol. 2010;184:713–724. doi: 10.4049/jimmunol.0901435. [DOI] [PubMed] [Google Scholar]
- 9.Pearce EJ, Kane CM, Sun J, et al. Th2 polarisation during infection with the helminth parasite Schistosoma mansoni. Immunol Rev. 2004;201:117–126. doi: 10.1111/j.0105-2896.2004.00187.x. [DOI] [PubMed] [Google Scholar]
- 10.Pearce EJ. Priming of the immune response by schistosome eggs. Parasite Immunol. 2005;27:265–270. doi: 10.1111/j.1365-3024.2005.00765.x. [DOI] [PubMed] [Google Scholar]
- 11.Cheever AW, Duvall RH, Hallack TA, Jr, et al. Variation of hepatic fibrosis and granuloma size among mouse strains infected with Schistosoma mansoni. Am J Trop Med Hyg. 1987;37:85–97. doi: 10.4269/ajtmh.1987.37.85. [DOI] [PubMed] [Google Scholar]
- 12.Rutitzky LI, Hernandez HJ, Stadecker MJ. Th1-polarizing immunization with egg antigens correlates with severe exacerbation of immunopathology and death in schistosome infection. Proc Natl Acad Sci USA. 2001;98:13243–13248. doi: 10.1073/pnas.231258498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fallon PG, Richardson EJ, McKenzie GJ, et al. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol. 2000;164:2585–2591. doi: 10.4049/jimmunol.164.5.2585. [DOI] [PubMed] [Google Scholar]
- 14.King CH, Sturrock RF, Kariuki HC. Transmission control for schistosomiasis – why it matters now. Trends Parasitol. 2006;22:575–582. doi: 10.1016/j.pt.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Kane CM, Cervi L, Sun J, et al. Helminth antigens modulate TLR-initiated dendritic cell activation. J Immunol. 2004;173:7454–7461. doi: 10.4049/jimmunol.173.12.7454. [DOI] [PubMed] [Google Scholar]
- 16.van Liempt E, van Vliet SJ, Engering A, et al. Schistosoma mansoni soluble egg antigens are internalized by human dendritic cells through multiple C-type lectins and suppress TLR-induced dendritic cell activation. Mol Immunol. 2007;44:2605–2615. doi: 10.1016/j.molimm.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Correale J, Farez M. Helminth antigens modulate immune responses in cells from multiple sclerosis patients through TLR2-dependent mechanisms. J Immunol. 2009;183:5999–6012. doi: 10.4049/jimmunol.0900897. [DOI] [PubMed] [Google Scholar]
- 18.Ritter M, Gross O, Kays S, et al. Schistosoma mansoni triggers Dectin-2 which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci USA. 2010;107:20459–20464. doi: 10.1073/pnas.1010337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niess JH, Adler G. Enteric flora expands gut lamina propria CX3CR1+ dendritic cells supporting inflammatory immune responses under normal and inflammatory conditions. J Immunol. 2010;184:2026–2237. doi: 10.4049/jimmunol.0901936. [DOI] [PubMed] [Google Scholar]
- 20.Marchiando AM, Graham WV, Turner JR. Epithelial barriers in homeostasis and disease. Annu Rev Pathol. 2010;5:119–144. doi: 10.1146/annurev.pathol.4.110807.092135. [DOI] [PubMed] [Google Scholar]
- 21.Heimesaat MM, Bereswill S, Fischer A, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 22.Heimesaat MM, Fischer A, Siegmund B, et al. Shift towards pro-inflammatory intestinal bacteria aggravates acute murine colitis via Toll-like receptors 2 and 4. PLOS ONE. 2007;2:662. doi: 10.1371/journal.pone.0000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Dixon MF, Genta RM, Yardley JH, et al. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;10:1161–1181. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Whitehead RH, Robinson PS, Williams JA, et al. Conditionally immortalized colonic epithelial cell line from a Ptk6 null mouse that polarizes and differentiates in vitro. J Gastroenterol Hepatol. 2008;23:1119–1124. doi: 10.1111/j.1440-1746.2008.05308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nobre V, Serufo JC, Carvalho Odos S, et al. Alteration in the endogenous intestinal flora of Swiss Webster mice by experimental Angiostrongylus costaricensis infection. Mem Inst Oswaldo Cruz. 2004;99:717–720. doi: 10.1590/s0074-02762004000700009. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton CM, Dowling DJ, Loscher CE, et al. The Fasciola hepatica tegumental antigen suppresses dendritic cell maturation and function. Infect Immun. 2009;6:2488–2498. doi: 10.1128/IAI.00919-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benson A, Pifer R, Behrendt CL, et al. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karanja DM, Colley DG, Nahlen BL, et al. Studies on schistosomiasis in western Kenya: I. Evidence for immune-facilitated excretion of schistosome eggs from patients with Schistosoma mansoni and human immunodeficiency virus coinfections. Am J Trop Med Hyg. 1997;56:515–521. doi: 10.4269/ajtmh.1997.56.515. [DOI] [PubMed] [Google Scholar]
- 30.Doenhoff M, Musallam R, Bain J, et al. Studies on the host–parasite relationship in Schistosoma mansoni-infected mice: the immunological dependence of parasite egg excretion. Immunology. 1978;35:771–778. [PMC free article] [PubMed] [Google Scholar]
- 31.Amiri P, Locksley RM, Parslow TG, et al. Tumour necrosis factor alpha restores granulomas and induces parasite egg-laying in schistosome-infected SCID mice. Nature. 1992;356:604–607. doi: 10.1038/356604a0. [DOI] [PubMed] [Google Scholar]
- 32.Cheever AW, Poindexter RW, Wynn TA. Egg laying is delayed but worm fecundity is normal in SCID mice infected with Schistosoma japonicum and S. mansoni with or without recombinant tumor necrosis factor alpha treatment. Infect Immun. 1999;67:2201–2208. doi: 10.1128/iai.67.5.2201-2208.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boros DL. Immunopathology of Schistosoma mansoni infection. Clin Microbiol Rev. 1989;2:250–269. doi: 10.1128/cmr.2.3.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva LM, Fernandes AL, Barbosa A, et al. Significance of schistosomal granuloma modulation. Mem Inst Oswaldo Cruz. 2000;95:353–361. doi: 10.1590/s0074-02762000000300010. [DOI] [PubMed] [Google Scholar]
- 35.Oliveira VR, El-Cheikh MC, Aguiar AM, et al. Schistosoma mansoni egg-induced hepatic granulomas in mice deficient for the interferon-gamma receptor have altered populations of macrophages, lymphocytes and connective tissue cells. Microbes Infect. 2000;2:1817–1826. doi: 10.1016/s1286-4579(00)01341-1. [DOI] [PubMed] [Google Scholar]
- 36.Rezende SA, Oliveira VR, Silva AM, et al. Mice lacking the gamma interferon receptor have an impaired granulomatous reaction to Schistosoma mansoni infection. Infect Immun. 1997;65:3457–3461. doi: 10.1128/iai.65.8.3457-3461.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grove DI, Mahmound AA, Warren KS. Suppression of cell-mediated immunity by metronidazole. Int Arch Allergy Appl Immunol. 1977;54:422–427. doi: 10.1159/000231857. [DOI] [PubMed] [Google Scholar]
- 38.Viera LQ, Moraes-Santos T. Schistosomiasis mansoni: evidence for a milder response in germfree mice. Rev Inst Med Trop Sao Paulo. 1987;29:37–42. doi: 10.1590/s0036-46651987000100006. [DOI] [PubMed] [Google Scholar]
- 39.Vieira LQ, Moraes-Santos T, Vieira EC. Alterations in cell number, size and collagen content in livers of conventional and germfree mice bearing Schistosoma mansoni. Prog Clin Biol Res. 1985;181:235–238. [PubMed] [Google Scholar]
- 40.Wynn TA, Thompson RW, Cheever AW, et al. Immunopathogenesis of schistosomiasis. Immunol Rev. 2004;201:156–167. doi: 10.1111/j.0105-2896.2004.00176.x. [DOI] [PubMed] [Google Scholar]
- 41.Lukacs NW, Boros DL. Lymphokine regulation of granuloma formation in murine Schistosomiasis mansoni. Clin Immunol Immunopathol. 1993;68:57–63. doi: 10.1006/clin.1993.1095. [DOI] [PubMed] [Google Scholar]
- 42.Boros DL. The role of cytokines in the formation of the schistosome egg granuloma. Immunobiology. 1994;191:441–450. doi: 10.1016/S0171-2985(11)80450-X. [DOI] [PubMed] [Google Scholar]
- 43.Chensue SW, Warmington KS, Ruth J, et al. Cross-regulatory role of interferon-gamma (IFN-gamma), interleukin (IL)-4 and IL-10 in schistosome egg granuloma formation: in-vivo regulation of T helper activity and inflammation. Clin Exp Immunol. 1994;98:395–400. doi: 10.1111/j.1365-2249.1994.tb05503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayes KS, Bancroft AJ, Goldrick M, et al. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science. 2010;328:1391–1394. doi: 10.1126/science.1187703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.