

Abstract
Background
Outcomes for patients with recurrent glioblastoma multiforme (GBM) are poor and may be improved by immunotherapy. We investigated the safety and efficacy of an autologous heat-shock protein peptide complex–96 (HSPPC-96) vaccine for patients with recurrent GBM.
Methods
In this open-label, single-arm, phase II study, adult patients with surgically resectable recurrent GBM were given vaccine after gross total resection. The primary endpoint was overall survival at 6 months. Secondary endpoints included overall survival, progression-free survival, safety, and immune profiling. Outcome analyses were performed in the intention-to-treat and efficacy populations.
Results
Between October 3, 2007 and October 24, 2011, 41 patients underwent gross total resection of recurrent GBM and received a median of 6 doses of HSPPC-96 vaccine. Following treatment, 90.2% of patients were alive at 6 months (95% confidence interval [CI]: 75.9–96.8) and 29.3% were alive at 12 months (95% CI: 16.6–45.7). Median overall survival was 42.6 weeks (95% CI: 34.7–50.5). Twenty-seven (66%) patients were lymphopenic prior to therapy, and patients with lymphocyte counts below the cohort median demonstrated decreased overall survival (hazard ratio: 4.0; 95% CI: 1.4–11.8; P = .012). There were no treatment-related deaths. There were 37 serious (grades 3–5) adverse events reported, with 17 attributable to surgical resection and a single grade 3 constitutional event related to the vaccine.
Conclusion
The HSPPC-96 vaccine is safe and warrants further study of efficacy for the treatment of recurrent GBM. Significant pretreatment lymphopenia may impact the outcomes of immunotherapy and deserves additional investigation.
Keywords: glioblastoma, heat-shock proteins, immunotherapy
Glioblastoma multiforme (GBM), the most common primary brain malignancy, is associated with a universally poor prognosis. Despite standard treatment with maximal safe surgical resection, conformal radiation, and systemic chemotherapy, the median survival from diagnosis remains 15 months.1 All patients eventually recur after initial diagnosis and treatment, at which time the median survival is 3–6 months.2–4 There are limited therapies approved for the treatment of recurrent GBM, including implantable carmustine impregnated wafers (Gliadel) and the humanized monoclonal anti–vascular endothelial growth factor antibody bevacizumab.5 Numerous other targeted therapies have been studied for recurrent GBM with modest results.6–9 Although no individual adjuvant therapy is considered the standard of care, bevacizumab has become the most widely used therapy in the recurrent setting. Studies have demonstrated a median survival from recurrence of between 8 and 11 months with the use of bevacizumab.10–13
Immunotherapy offers the promise of improving outcomes for patients with GBM by evoking specific immune responses that may produce a more sustained and less toxic effect than conventional therapy. Heat-shock proteins (HSPs), which function as intracellular chaperones, can be used to deliver a variety of tumor antigens to antigen presenting cells for immune stimulation.14 Tumor antigenic peptides bound to HSP-96 can be delivered systemically where binding of HSP-96 to the CD91 receptor on dendritic cells results in internalization of the HSP complex and cross-presentation of cleaved tumor peptides on major histocompatibility complex classes I and II.15 By purifying HSP-96 protein complexes from a patient's own tumor, a personalized polyvalent vaccine can be developed and administered for treatment.
We previously studied the safety and efficacy of a heat-shock peptide protein complex–96 (HSPPC-96; Prophage) vaccine in a phase I trial for recurrent GBM.16 Our results demonstrated that the vaccine was well tolerated and resulted in a measurable systemic immune response to the patient's specific tumor antigens. In the present study we evaluate the safety and efficacy of the HSPPC-96 vaccine in patients with recurrent GBM in a phase II, multicenter, clinical trial.
Materials and Methods
Patient Selection
In this single-arm, phase II trial, we enrolled participants from 3 centers in the United States. Individuals over the age of 18 were eligible for inclusion if they had histologically confirmed recurrent GBM after standard initial therapy.1 All participants were required to undergo surgical resection and have a postoperative Karnofsky performance status of at least 70% with a life expectancy >8 weeks. After surgical resection, all participants were screened to ensure an extent of resection >90% of the contrast-enhancing tumor prior to receiving vaccine.
Patients were excluded from study entry for known systemic autoimmune diseases, primary or secondary immunodeficiency, concurrent malignancy within the past 5 years (except carcinoma in situ of the uterus or cervix or nonmetastatic nonmelanoma skin cancer), a bleeding diathesis, uncontrolled active infection, or other serious unstable medical condition. Following surgical resection, patients were excluded for histologic diagnoses of pseudoprogression without recurrent tumor, incomplete surgical resection (<90% by volume), documented tumor growth (>10% increase in contrast enhancement) within 4 weeks of surgical resection at the first interval scan, or insufficient tumor to create at least four 25-μg doses of vaccine.
The protocol was submitted to the FDA (IND #12548) and approved by the institutional review board at each participating site. All patients provided written informed consent prior to participation in the study. This study is registered with ClinicalTrials.gov, NCT00293423.
Clinical Procedure
All patients underwent surgical resection and collection of their tumor intraoperatively for production of vaccine. Tumor tissue was freshly frozen and shipped to the manufacturing facility (Agenus) to generate vaccine after histologic confirmation of recurrent disease. Approximately 7 g of tissue was necessary to produce a minimum of four 25-μg vaccine doses. Vaccine quality was confirmed by postproduction testing according to good manufacturing practice guidelines.
All patients underwent a postoperative contrast-enhanced MRI of the brain within 48 h of surgery to assess the extent of resection. Patients were allowed to recover from surgery and wean off perioperative dexamethasone. At 4 weeks following surgery, each patient had another MRI and clinical evaluation. If the repeat MRI demonstrated disease stability, vaccination was initiated. Vaccine was administered in 25-μg doses through intradermal injection every week for 4 weeks, followed by every 2 weeks thereafter. Vaccinations were given until all doses were depleted or the patient's tumor progressed. Surveillance MRI and clinical evaluation were performed every 8 weeks to screen for progression. Progression was defined radiographically using the Macdonald criteria and confirmed in a subset of patients by biopsy or repeat resection.17 Upon progression, further treatment was given at the discretion of the patient's neuro-oncologist. Patients were followed until death to define overall survival (OS).
The primary endpoint was the proportion of patients surviving at 6 months following surgical resection. Secondary endpoints included OS, progression-free survival (PFS), immunologic response, and frequency of adverse events. OS was defined as the time from surgery to death, and PFS was defined as the time from surgery to documented radiographic progression or death if radiographic progression was not documented. We assessed adverse events using the Common Terminology Criteria for Adverse Events, version 4.0, from the National Cancer Institute. Preoperative blood samples drawn from all patients were analyzed by the clinical laboratory at each institution. A complete blood count with differential, including white blood cell count (WBC), absolute lymphocyte count (ALC), and absolute monocyte count (AMC), was obtained to evaluate pretreatment immune status.
Statistical Analysis
We tested the null hypothesis that 60% or fewer patients would survive at 6 months after surgery against the specific alternative hypothesis that 80% or greater would survive at 6 months. With a sample size of 38 patients, the null hypothesis would be rejected in favor of the alternative hypothesis if 27/38 (71%) or more patients were alive at 6 months, with a one-sided type I error of 0.10 and a power of 90%. A total accrual of 50 patients was planned for the phase I (previously reported) and phase II studies together.
Secondary analyses were performed to study OS and PFS for all patients. Survival curves for the intention-to-treat (ITT) and efficacy populations were generated using the Kaplan–Meier product-limit method. Peripheral immune function was assessed by evaluating leukocyte counts from preoperative blood draws. The distribution of each leukocyte count (WBC, ALC, AMC) was plotted and normality was tested using a Shapiro–Wilk test. If the factor was normally distributed, the median value was determined and patients were assigned to cohorts with values greater than/equal to or less than the median. Overall survival was compared between cohorts univariately using the log-rank test. Factors demonstrating significance in univariate analysis were tested in a multivariate Cox proportional hazards model with known predictors of outcome. Tests were accepted as statistically significant for 2-sided Pvalues <.05. All statistical analyses were performed using SPSS (version 20).
Results
Between October 3, 2007 and October 24, 2011, 68 patients with histopathologically proven recurrent GBM were screened and underwent surgical resection. One patient had less than a 90% extent of resection and 4 patients had a postoperative KPS <70% and were excluded. Of the remaining 63 patients, insufficient tumor was obtained to generate vaccine in 13. Of the 50 patients with sufficient tumor resected, 9 demonstrated disease progression at the first follow-up prior to initial vaccination and were therefore excluded according to the study protocol.
Forty-one patients met all pre- and postoperative inclusion criteria and were assigned to receive the HSPPC-96 vaccine for recurrent GBM (Table 1). This group comprised the ITT population. The median time from surgery to first vaccination was 31 days (range, 23–55). Patients received a median of 6 vaccinations (range, 1–15), with 3 patients receiving less than the protocol minimum of 4 vaccinations. These 3 patients were included in the ITT population but excluded from the efficacy population. Patients discontinued vaccination due to depletion of vaccine, tumor progression, withdrawal from the study, or investigator decision (Table 1). All patients were followed until death or closure of the data analysis on January 12, 2013. No patients were lost to follow-up.
Table 1.
Characteristics of patients receiving HSPPC-96 vaccine
| Characteristic | Patients (n = 41) |
|---|---|
| Age, y | |
| Median (range) | 55 (21–75) |
| ≤40 | 3 (7%) |
| 41–50 | 10 (24%) |
| 51–60 | 11 (27%) |
| 61–70 | 15 (37%) |
| >70 | 2 (5%) |
| Gender | |
| Male | 30 (73%) |
| Female | 11 (27%) |
| Ethnicity | |
| White | 39 (95%) |
| Nonwhite | 2 (5%) |
| Karnofsky performance score | |
| 90 | 15 (37%) |
| 80 | 20 (49%) |
| 70 | 6 (14%) |
| Time from diagnosis to progression, wk | |
| Median | 35 |
| Range | 11–321 |
| Time from surgery to first dose of vaccine, days | |
| Median | 31 |
| Range | 23–55 |
| Number of vaccine doses administered | |
| Median | 6 |
| Range | 1–15 |
| Reason for vaccine discontinuation | |
| Progression | 21 (51%) |
| Vaccine depleted | 13 (32%) |
| Patient withdrew | 5 (12%) |
| Investigator decision | 2 (5%) |
At the time of final analysis, 39 patients (95%) had died. Two patients were alive without evidence of progression and were censored in the PFS and OS analysis. In the ITT population, the median PFS was 19.1 weeks (95% confidence interval [CI]: 14.1–24.1) with a 6-month PFS of 29.3% (95% CI: 16.6–45.7; Fig. 1A). The median OS was 42.6 weeks (95% CI: 34.7–50.5) with a 6-month OS of 90.2% (95% CI: 75.9–96.8) and a 12-month OS of 29.3% (95% CI: 16.6–45.7; Fig. 1B). The median PFS and OS were similar for the efficacy population (Supplemental Table S1).
Fig. 1.

(A) Kaplan–Meier estimates of PFS in 41 patients receiving the HSPPC-96 vaccine for recurrent GBM. Vertical lines indicate the timepoints at which patients were censored. (B) Kaplan–Meier estimates of OS in 41 patients receiving the HSPPC-96 vaccine for recurrent GBM. Vertical lines indicate the timepoints at which patients were censored.
In addition to clinical measures, preoperative blood samples were analyzed for leukocyte counts to assess immune function prior to treatment (Supplemental Table S2). The median ALC was 0.90 × 109 cells/L (range, 0.36–2.34), with 27 patients (66%) having an ALC less than the lower limit of normal (1.0 × 109cells/L), according to the standard laboratory reference range. When OS was stratified by ALC, patients with an ALC greater than or equal to the median had significantly improved survival compared with patients with an ALC less than the median (49.1 vs 37.1 wk, P = .039; Fig. 2). The WBC and AMC were not predictive of outcome (Table 2). A proportional hazards model was constructed for OS including previously identified predictors of outcome (age, KPS), the number of vaccine doses received, and the ALC (Table 3). In this model, ALC was found to be an independent predictor of outcome with a hazard ratio of 4.0 (95% CI: 1.4–11.8; P = .012) for patients with an ALC below the median.
Fig. 2.
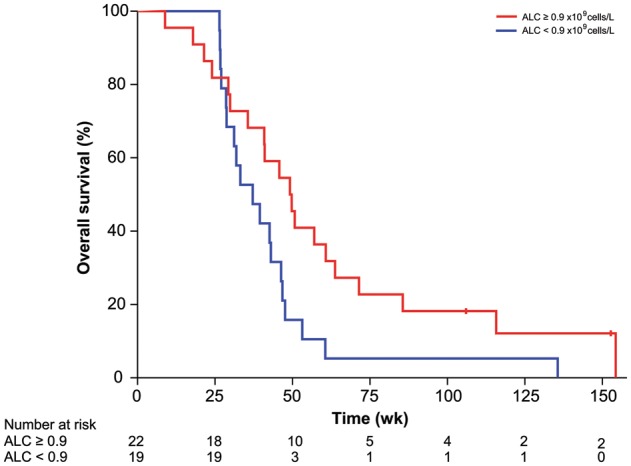
Kaplan–Meier estimates of OS in 41 patients receiving the HSPPC-96 vaccine for recurrent GBM stratified by ALC relative to median expression. Vertical lines indicate the timepoints at which patients were censored.
Table 2.
Overall survival stratified by immune markers in vaccine patients
| Median OS (95% CI) |
P | ||
|---|---|---|---|
| Above Median | Below Median | ||
| White blood cell count | 37.1 (5.8–25.8) | 46.7 (39.6–53.8) | .23 |
| Absolute lymphocyte | 49.1 (38.0–60.3) | 37.1 (26.4–47.9) | .04 |
| Absolute monocyte | 40.9 (29.1–52.6) | 43.0 (36.1–49.9) | .30 |
Table 3.
Proportional hazards for predictors of outcome
| Variable | Hazard Ratio (95% CI) | P |
|---|---|---|
| Age, per 1-y increment | 1.01 (0.97–1.06) | .58 |
| Gender | ||
| Female | 0.76 (0.27–2.10) | .59 |
| Male | 1.0 | |
| KPS | ||
| 70 | 2.28 (0.70–7.42) | .17 |
| 80 | 0.65 (0.30–1.42) | .28 |
| 90 | 1.0 | |
| Vaccine doses (per dose increment) | 0.85 (0.73–0.99) | .04 |
| Absolute lymphoctye count | ||
| Below median | 4.02 (1.37–11.83) | .01 |
| Above/equal to median | 1.0 | |
The number of vaccine doses given was also found to be significantly associated with outcome, with a hazard ratio of 0.85 (95% CI: 0.73–0.99; P = .036) per incremental dose. This association was expected, as patients with longer PFS were eligible to receive more vaccine doses. A post hoc analysis of survival grouped by reason for discontinuation of vaccine was performed to compare outcomes between patients who progressed on vaccine and those who received all eligible doses (Supplemental Table S3). Patients progressing on vaccine expectedly had a shorter PFS (Supplemental Fig. S1), but OS and survival from progression were also significantly shorter compared with patients completing treatment (P < .001; Supplemental Fig. S2).
A summary of all adverse events recorded during the study period is reported in Table 4. The toxicity associated with the vaccine was minimal, related primarily to injection site reactions. A single patient experienced grade 3 fatigue possibly related to vaccine. There were no grade 4 adverse events or deaths attributable to the vaccine. Seventeen serious adverse events (grades 3–4) were associated with surgical resection consistent with the known risks of a craniotomy for GBM.18 There was a single death associated with development of a delayed subdural hematoma in the setting of thrombocytopenia. This occurred 6 months after the tumor resection and was unrelated to vaccine administration.
Table 4.
Adverse events in patients receiving HSPPC-96 vaccine
| Adverse Event | Grades 1–2 | Grades 3–5 | Attributable |
|---|---|---|---|
| Cardiac | 5 (12%) | 0 | Unrelated |
| Constitutional | |||
| Anorexia | 4 (10%) | 0 | Unrelated |
| Fatigue | 11 (27%) | 1 (2%) | Vaccine |
| Fever | 4 (10%) | 0 | Unrelated |
| Dermatological | |||
| Injection site reaction | 17 (41%) | 0 | Vaccine |
| Rash | 4 (10%) | 1 (2%) | Unrelated |
| Pruritus | 4 (10%) | 0 | Unrelated |
| Gastrointestinal | 12 (29%) | 2 (5%) | Unrelated |
| Genitourinary/renal | 8 (20%) | 0 | Unrelated |
| Hematological | |||
| Anemia | 5 (12%) | 0 | Unrelated |
| Coagulopathy | 2 (5%) | 1 (2%) | Unrelated |
| Leukopenia | 7 (17%) | 3 (7%) | Unrelated |
| Thrombocytopenia | 0 | 1 (2%) | Unrelated |
| Infection | 7 (17%) | 4 (10%) | Unrelated |
| Lymphatic | 5 (12%) | 0 | Unrelated |
| Metabolic | 13 (32%) | 3 (7%) | Unrelated |
| Musculoskeletal | 8 (20%) | 1 (2%) | Unrelated |
| Neurological | |||
| Cognitive | 17 (41%) | 3 (7%) | Surgery |
| Focal deficit | 23 (56%) | 5 (12%) | Surgery |
| Hydrocephalus | 0 | 4 (10%) | Surgery |
| Mood | 8 (20%) | 0 | Unrelated |
| Seizure | 7 (17%) | 0 | Surgery |
| Pulmonary | 6 (15%) | 0 | Unrelated |
| Surgical site | |||
| Cerebrospinal fluid leak | 0 | 1 (2%) | Surgery |
| Hematoma | 0 | 3 (7%) | Surgery |
| Infection | 1 (2%) | 1 (2%) | Surgery |
| Vascular | 0 | 3 (7%) | Unrelated |
Discussion
There is no currently accepted standard of care for the management of recurrent GBM. Repeat surgical resection is often performed, supported by a number of retrospective studies suggesting that increased cytoreduction through gross total resection at recurrence results in improved survival.19–21 Bevacizumab has gained wide acceptance as the first-line therapy for recurrent disease based on a number of phase II studies demonstrating a median PFS of 11–24 weeks and median OS of 26–44 weeks.10–13,22 Numerous other chemotherapies have been tested in phase II clinical trials for recurrent GBM with a range of median PFS of 9.6–17 weeks and median OS of 21–41 weeks.6–9,23,24 In 2011, Clarke and colleagues25 published a systematic analysis of the results of all North American Brain Tumor Consortium phase II trials for recurrent GBM from 1998 to 2008, demonstrating an aggregate median PFS of 9.3 weeks and median OS of 33.1 weeks. A similar analysis of the European experience between 1999 and 2010 from pooled phase I and II trials from the European Organisation for Research and Treatment of Cancer Brain Tumor Group demonstrated a median PFS of 7.2 weeks and median OS of 24.8 weeks.26
Immunotherapy for GBM promises to improve outcomes for patients by providing a highly specific, nontoxic alternative to conventional chemotherapy. To date, the immunotherapy approaches applied clinically to recurrent GBM have utilized an active vaccination approach with or without an immune-boosting adjuvant. Patients have been vaccinated with tumor-specific peptides conjugated to immunoadjuvants or with autologous dendritic cells cultured and pulsed with tumor peptides ex vivo, with reported median PFS rates of 8–20 weeks and median OS rates of 40–46 weeks.27–29 Here, we report on the use of an HSP-based vaccine for the treatment of recurrent GBM. We previously reported our phase I results, including immune-monitoring data that confirmed a vaccine-specific peripheral immune response in 11/12 patients.16
In the present study we demonstrate a median OS of 42.6 weeks, with 90.2% of patients surviving longer than 6 months. As this study is a single-arm, uncontrolled trial, the outcomes can only be compared with historical controls from similar trials. It is important to note that the patients in this trial represent a highly selected group who may be expected to have better outcomes. As determined by the study design, all patients receiving vaccine underwent a gross total surgical resection and had good functional status prior to vaccination. Therefore, the results of the current trial are most appropriately compared with other surgically based clinical trials for recurrent GBM with similar enrollment criteria. In the original phase III trial supporting the use of carmustine impregnated implantable wafers, Brem and colleagues5 reported a 6-month OS of 56% for patients receiving the carmustine implants versus 36% for patients receiving a placebo implant at resection. They reported >75% extent of resection in over 85% of their patients, with a median OS for the carmustine treatment group of 31 weeks. More recently, Kunwar and colleagues30 studied the efficacy of a chimeric IL-13 to pseudomonas exotoxin fusion protein (IL13-PE38QQR, Cintredekin besudotox) delivered by convection-enhanced delivery following resection of recurrent GBM, compared with patients receiving carmustine impregnated wafer implantation at resection (PRECISE Trial).30 Patients enrolled in this trial had a median age of 55 years, KPS ≥ 70%, and gross total surgical resection. The authors reported no significant difference in outcomes between treatment groups, with a median OS of 36.4 weeks in the cytotoxin group compared with 35.3 weeks in the carmustine wafer group.30 Relative to the outcomes of these comparable, large, surgically based trials for recurrent GBM, the outcomes from HSPPC-96 vaccination appear promising. The survival results of the current study are also comparable to the best outcomes reported with bevacizumab. Admittedly, many patients receiving bevacizumab in these trials did not undergo prior repeat surgical resection. A proper comparison of the efficacy of HSPPC-96 vaccination with bevacizumab would require equivalent surgical resection in both groups. We believe the findings in the current study support the value of a comparison between the HSPPC-96 vaccine and bevacizumab in surgically accessible recurrent tumors. A 3-arm, randomized phase II clinical trial comparing vaccine to vaccine in combination with bevacizumab to bevacizumab alone following surgical resection is now open and has begun enrolling patients (NCT01814813).
In addition to evaluating standard clinical endpoints, we studied the impact of immunologic status on patient outcomes. Systemic immunosuppression is a well-recognized finding in patients with GBM.31–33 A number of mechanisms have been identified to account for this immunosuppression, including tumor-induced lymphopenia, expansion of regulatory T cells, and expansion of immunosuppressive monocytes/macrophages.34–37 To study the impact of immune status on the efficacy of the HSPPC-96 vaccine, we measured preoperative leukocyte fractions including WBC, ALC, and AMC. The majority of patients were found to be lymphopenic, with those patients having less than the median ALC demonstrating significantly decreased OS in univariate and multivariate analysis. Although it is not surprising that lymphopenia can decrease the efficacy of immunotherapy meant to induce a cytotoxic lymphocytic response, there are few reports correlating pretherapeutic immune status with outcome in phase II or III clinical trials of immunotherapy for GBM. Our findings suggest that patients may benefit from adjuvants to address tumor-induced lymphopenia, and the implications of pretreatment lymphopenia warrant consideration when selecting patients in future tumor vaccine clinical trials.
Supplementary Material
Funding
This work was supported by the National Cancer Institute Special Program of Research Excellence (SPORE); American Brain Tumor Association; National Brain Tumor Society; and the Accelerated Brain Cancer Cure, Inc. Dr. Bloch is partially supported by the Reza and Georgianna Khatib Endowed Professorship in Neurological Surgery. Dr. Parsa is partially supported by the Michael J. Marchese Endowed Chair in Neurological Surgery.
Conflict of interest statement. None declared.
Supplementary Material
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Ballman KV, Buckner JC, Brown PD, et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007;9:29–38. doi: 10.1215/15228517-2006-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamborn KR, Yung WK, Chang SM, et al. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. 2008;10:162–170. doi: 10.1215/15228517-2007-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17:2572–2578. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 5.Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345:1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 6.Kesari S, Schiff D, Doherty L, et al. Phase II study of metronomic chemotherapy for recurrent malignant gliomas in adults. Neuro Oncol. 2007;9:354–363. doi: 10.1215/15228517-2007-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prados MD, Lamborn K, Yung WK, et al. A phase 2 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: a North American Brain Tumor Consortium study. Neuro Oncol. 2006;8:189–193. doi: 10.1215/15228517-2005-010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puduvalli VK, Giglio P, Groves MD, et al. Phase II trial of irinotecan and thalidomide in adults with recurrent glioblastoma multiforme. Neuro Oncol. 2008;10:216–222. doi: 10.1215/15228517-2007-060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC Brain Tumor Group study 26034. J Clin Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 11.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 13.Sathornsumetee S, Desjardins A, Vredenburgh JJ, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010;12:1300–1310. doi: 10.1093/neuonc/noq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava PK, Callahan MK, Mauri MM. Treating human cancers with heat shock protein-peptide complexes: the road ahead. Expert Opin Biol Ther. 2009;9:179–186. doi: 10.1517/14712590802633918. [DOI] [PubMed] [Google Scholar]
- 15.Binder RJ, Srivastava PK. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc Natl Acad Sci U S A. 2004;101:6128–6133. doi: 10.1073/pnas.0308180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crane CA, Han SJ, Ahn B, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. 2013;19:205–214. doi: 10.1158/1078-0432.CCR-11-3358. [DOI] [PubMed] [Google Scholar]
- 17.Macdonald DR, Cascino TL, Schold SC, Jr., Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 18.Chang SM, Parney IF, McDermott M, et al. Perioperative complications and neurological outcomes of first and second craniotomies among patients enrolled in the Glioma Outcome Project. J Neurosurg. 2003;98:1175–1181. doi: 10.3171/jns.2003.98.6.1175. [DOI] [PubMed] [Google Scholar]
- 19.Bloch O, Han SJ, Cha S, et al. Impact of extent of resection for recurrent glioblastoma on overall survival: clinical article. J Neurosurg. 2012;117:1032–1038. doi: 10.3171/2012.9.JNS12504. [DOI] [PubMed] [Google Scholar]
- 20.Lacroix M, Abi-Said D, Fourney DR, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95:190–198. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]
- 21.McGirt MJ, Chaichana KL, Gathinji M, et al. Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J Neurosurg. 2009;110:156–162. doi: 10.3171/2008.4.17536. [DOI] [PubMed] [Google Scholar]
- 22.Raizer JJ, Grimm S, Chamberlain MC, et al. A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer. 2010;116:5297–5305. doi: 10.1002/cncr.25462. [DOI] [PubMed] [Google Scholar]
- 23.Kong DS, Lee JI, Kim JH, et al. Phase II trial of low-dose continuous (metronomic) treatment of temozolomide for recurrent glioblastoma. Neuro Oncol. 2010;12:289–296. doi: 10.1093/neuonc/nop030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry JR, Belanger K, Mason WP, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010;28:2051–2057. doi: 10.1200/JCO.2009.26.5520. [DOI] [PubMed] [Google Scholar]
- 25.Clarke JL, Ennis MM, Yung WK, et al. Is surgery at progression a prognostic marker for improved 6-month progression-free survival or overall survival for patients with recurrent glioblastoma? Neuro Oncol. 2011;13:1118–1124. doi: 10.1093/neuonc/nor110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorlia T, Stupp R, Brandes AA, et al. New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: a pooled analysis of EORTC Brain Tumour Group phase I and II clinical trials. Eur J Cancer. 2012;48:1176–1184. doi: 10.1016/j.ejca.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Izumoto S, Tsuboi A, Oka Y, et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg. 2008;108:963–971. doi: 10.3171/JNS/2008/108/5/0963. [DOI] [PubMed] [Google Scholar]
- 28.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terasaki M, Shibui S, Narita Y, et al. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen—A24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol. 2011;29:337–344. doi: 10.1200/JCO.2010.29.7499. [DOI] [PubMed] [Google Scholar]
- 30.Kunwar S, Chang S, Westphal M, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12:871–881. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brooks WH, Netsky MG, Normansell DE, Horwitz DA. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J Exp Med. 1972;136:1631–1647. doi: 10.1084/jem.136.6.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elliott LH, Brooks WH, Roszman TL. Cytokinetic basis for the impaired activation of lymphocytes from patients with primary intracranial tumors. J Immunol. 1984;132:1208–1215. [PubMed] [Google Scholar]
- 33.Roszman TL, Brooks WH. Immunobiology of primary intracranial tumours. III. Demonstration of a qualitative lymphocyte abnormality in patients with primary brain tumours. Clin Exp Immunol. 1980;39:395–402. [PMC free article] [PubMed] [Google Scholar]
- 34.Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 35.Fong B, Jin R, Wang X, et al. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One. 2012;7:e32614. doi: 10.1371/journal.pone.0032614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Learn CA, Fecci PE, Schmittling RJ, et al. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clin Cancer Res. 2006;12:7306–7315. doi: 10.1158/1078-0432.CCR-06-1727. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues JC, Gonzalez GC, Zhang L, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010;12:351–365. doi: 10.1093/neuonc/nop023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.