

Abstract
The sequestration of carbon and nitrogen by clay-sized particles in soils is well established, and clay content or mineral surface area has been used to estimate the sequestration potential of soils. Here, via incubation of a sieved (<2 mm) topsoil with labelled litter, we find that only some of the clay-sized surfaces bind organic matter (OM). Surprisingly, <19% of the visible mineral areas show an OM attachment. OM is preferentially associated with organo-mineral clusters with rough surfaces. By combining nano-scale secondary ion mass spectrometry and isotopic tracing, we distinguish between new labelled and pre-existing OM and show that new OM is preferentially attached to already present organo-mineral clusters. These results, which provide evidence that only a limited proportion of the clay-sized surfaces contribute to OM sequestration, revolutionize our view of carbon sequestration in soils and the widely used carbon saturation estimates.
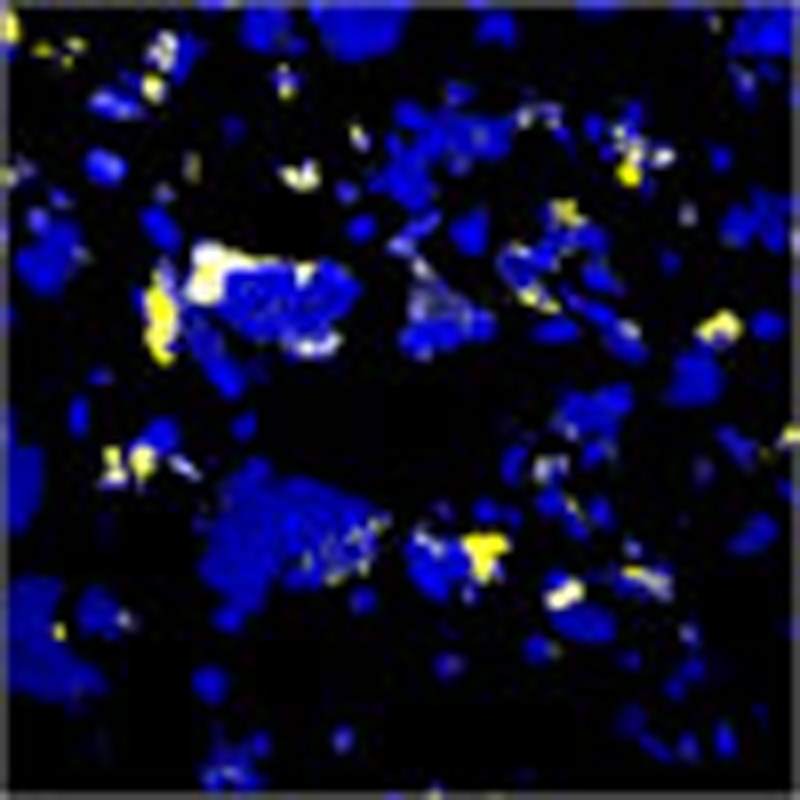 Clay-sized particles bind organic matter and sequester carbon and nitrogen in soils, yet extent and localization of organic matter coverage remain unclear. Using NanoSIMS, Vogel et al. chemically image soils at ultra-high resolution and show that only particles with rough surfaces react with organic matter.
Clay-sized particles bind organic matter and sequester carbon and nitrogen in soils, yet extent and localization of organic matter coverage remain unclear. Using NanoSIMS, Vogel et al. chemically image soils at ultra-high resolution and show that only particles with rough surfaces react with organic matter.
Soils and marine sediments represent one of the largest reservoirs of organic carbon (OC) on Earth1,2,3 and have a crucial role in global trace gas cycles and the associated climate change. In addition to occluding OM in aggregated mineral particles4,5, soils and sediments sequester OC and organic nitrogen (ON) through the formation of strong associations between OM and mineral surfaces in organo-mineral associations6,7. The association between OM and clay-sized particles accounts for a large proportion of sequestered OC in soils8. Clay-sized particles provide a multitude of reactive mineral surfaces for OC and ON binding8. The OC sequestration capacity of a soil is estimated from correlations between OC concentration and the soil’s clay content or specific surface area (SSA)9. Physical fractionation by density and/or particle size8 is widely used to isolate organo-mineral associations. Understanding the dynamics of OM binding within the mineral matrix is essential for understanding the processes controlling the quantity, quality and turnover of OM attached to mineral particles8.
Early research on marine sediments proposed a monolayer coverage of OM on clay mineral surfaces, as indicated by an indirect method (surface area calculations derived from N2 adsorption measurements)10. However, techniques for direct investigation at the appropriate spatial scale are required for a better understanding of the relationship between organic structures and mineral grains8,11 and of the spatiotemporal dynamics of the relationship. Direct analyses by transmission electron microscopy challenged the monolayer coverage concept and suggested that OM exists in patches associated with mineral surfaces12,13, which was confirmed by several other studies14,15,16,17,18. Synchrotron-based analyses showed highly complex forms of OM and reactive mineral materials interacting at the submicron scale19,20. To understand and quantify the OM sequestration potential of soils and sediments, the dynamics of OC and ON accumulation at the micro-scale must be studied. The key technique for tracking the biogeochemical cycles of C and N is stable isotope labelling (13C, 15N). Nano-scale secondary ion mass spectrometry (NanoSIMS) enables the direct visualization of 13C and 15N down to a lateral resolution of 150 nm21,22 and can be used to identify organo-mineral associations by the simultaneous detection of ion species derived from the organic (C, N) and inorganic (Al, Si and Fe) components of the associations18,23,24,25.
Here, our objective is to identify and quantitatively assess the OM-reactive surfaces in organo-mineral associations that control the C and N sequestration potential of soils. An incubation experiment over 42 days with an arable topsoil of a Luvisol (sieved to <2 mm) with 15N- and 13C-labelled litter is performed to quantify and follow the formation of organo-mineral associations. The soil chosen represents a type of soil common for central Europe, USA and south-east Australia with a mineral composition (illite, chlorite, mixed-layer clay minerals and pedogenic iron oxides) typical for the temperate zone. Microbial biomass-13C, salt-extractable OC-13C and isotopic C and N composition are determined for total bulk soil and selected soil fractions (which are obtained by combined density and particle size fractionation). The spatial distributions of mineral particles, OM and isotopic enrichment are investigated by NanoSIMS. Using this approach, we quantify the associations of OC and ON with mineral surfaces and identify hot spots of OM binding and label accumulation. Our data show that only some of the clay-sized mineral surfaces, namely those with rough surfaces, react with OM and thus contribute to OC and ON sequestration. Furthermore, these rough surfaces are the nuclei for additional OM accumulation and therefore, control the C and N sequestration potential of the soil. Thus, our results indicate that using the amount of clay as a proxy to predict the sequestration potential of soils and sediments is not sufficient. Accordingly, we propose that the contribution of these reactive surfaces must be quantified.
Results
Formation of new organo-mineral associations
During our short-term incubation (42 days), the 13C enrichment in the bulk soils decreased significantly (U-test; P≥0.05), from 0.89 to 0.67 atom-% (Supplementary Table S1, Supplementary Fig. S1a). At 2 h after the start of the incubation, samples with added labelled litter already contained twice as much microbial biomass as the control samples (Supplementary Table S2), pointing to enhanced microbial activity. The progression of litter decomposition was reflected by a linear decrease in C/N ratio (Supplementary Fig. S1b), a decrease in salt-extractable OC (SEOC) (Supplementary Fig. S2), and a strong correlation between microbial biomass and 13C concentration in the bulk soil (r=0.88; P<0.001). Fresh, litter-derived 15N and 13C were found in all the fine mineral fractions shortly after start of the incubation experiment (Fig. 1). Over the course of the incubation period, 13C concentrations in the fine mineral fractions decreased (Supplementary Table S3). In particular, the clay-sized fractions were more enriched in 15N and 13C compared with the other fine mineral fractions (Fig. 1).
Figure 1. Quantitative 15N and 13C enrichment in the fine mineral fractions.

(a) 15N and (b) 13C concentrations (means, error bars indicate SDs, n=3) were calculated from the 15N and 13C excess values (obtained by IRMS) in the fine mineral fractions over the incubation time.
Hot spots of OM sequestration in submicron soil structures
To get insight into the spatial distribution and temporal development of freshly associated OM on clay-sized particles, we used scanning electron microscopy (SEM) and NanoSIMS to analyse the clay-sized fraction at four time points during the incubation period. SEM analysis revealed that some mineral particles appeared as individual particles with mostly plain surfaces, whereas others were aggregated in clusters of several small particles. This clustering causes rough surfaces. The surface roughness of mineral particles due to etch pits, micropores and cracks is generally defined as the ratio of the actual surface area to the surface of a hypothetical smooth surface enveloping the actual surface26. By SEM, we designated the mineral particles as either individual particles with plain surfaces or as clustered particles with rough surfaces (Fig. 2a). Hereafter, we refer to both the surface roughness due to etch pits, micropores and cracks and the observed roughness due to the clustering of mineral particles as ‘rough surface’.
Figure 2. New insights via two-dimensional NanoSIMS imaging.

(a) SEM image and (b) 16O− secondary ion image (linear 0–5191) of a representative clay-sized fraction (time point: 21 days) reveal individual (continuous circle) and clustered (dotted circles) particles. (c) The total OM, as revealed by 12C− and 12C14N− secondary ion images, on the mineral surfaces represents the organo-mineral associations. (d) The new, litter-derived OM is indicated by areas of the mineral surface where the 13C−/12C− and 12C15N−/12C14N− ratios were enriched compared with the ratios for the unlabelled clay-sized fraction. All scale bars represent 5 μm.
We applied for the first time an approach of quantifying the areas visible by NanoSIMS. For each clay-sized fraction and time step, 10–12 spots (40 × 40 μm2 each) were measured. These spots represented a total particle area of 5,815–9,474 μm2 (Table 1), which is more than 10 times the particle area analysed in previous studies of submicron-sized soil particles24. Of the total particle area, 13–26% was assigned to individual particles, and 74–87% was assigned to clustered particles (Table 1). Consequently, in the analysed clay-sized fractions, most of the mineral particles were present in small clusters comprising rough surfaces. The mineral particles are displayed in the NanoSIMS images as 16O− secondary ion images (Fig. 2a). All the OM, which was revealed by the 12C− and 12C14N− secondary ion images, was associated with mineral particles, and the OM was heterogeneously distributed in small patches on the particles (Fig. 2b). By linking the images for OM and for the mineral particles, we found that only 1–2% of the individual particles were covered by OM, whereas 15–21% of the mineral clusters contained OM (Table 1). Determination of the SSA by N2 adsorption showed that, in particular, mineral particles with a large SSA were covered by OM. We calculated that about 77% of the total SSA was covered by OM (Supplementary Table S4).
Table 1. Quantification of exposed two-dimensional areas visible by NanoSIMS.
| Time stepμm2 (%) | Total particle area μm2 (%) | Individual particles μm2 (%) | Clustered particles μm2 (%) | Total area of OM μm2 (%) | Individual particles covered by OM μm2 (%) | Clustered particles covered by OM μm2 (%) | Fresh litter-derived OM μm2 (%) | Fresh OM solely enriched in 13C μm2 (%) | Fresh OM solely enriched in 15N μm2 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 2 h | 7,086 | 959 (14) | 6,120 (86) | 1,343 (19) | 22 (2) | 1,312 (21) | 439 (33) | 8 (2) | 9 (2) |
| 7 d | 5,815 | 1,177 (20) | 4,637 (80) | 918 (16) | 28 (2) | 883 (19) | 403 (44) | 8 (2) | 17 (4) |
| 21 d | 9,016 | 2,330 (26) | 6,684 (74) | 1,164 (13) | 42 (2) | 1,118 (17) | 474 (41) | 3 (1) | 20 (4) |
| 42 d | 9,474 | 1,191 (13) | 8,278 (87) | 1,264 (13) | 12 (1) | 1,247 (15) | 690 (55) | 2 (0) | 35 (5) |
The areas are sums over all images analysed per clay-sized fraction.
Organo-mineral clusters as nuclei for new OM sequestration
We used 13C−/12C− and 12C15N−/12C14N− ratios obtained by means of NanoSIMS to evaluate the spatial distribution of newly incorporated OM in the clay-sized fractions. To follow the changes in the new, litter-derived OM, we quantified as new OM areas where the 13C−/12C−and 12C15N−/12C14N− ratios were enriched compared to the values for the unlabelled clay-sized fraction. Figure 2c shows spots enriched in 13C and 15N, demonstrating distinct micro-scale hot spots of new labelled OM. The 13C−/12C− and 12C15N−/12C14N− ratios measured for numerous spots by NanoSIMS showed a much higher variability in comparison to the isotope ratio mass spectrometry (IRMS) data, indicating a high spatial heterogeneity of the sample (Fig. 3).
Figure 3. Heterogeneity of newly formed organo-mineral associations.

(a) Box plots of 13C−/12C− ratios obtained by NanoSIMS reflecting the number of evaluated regions of interest (ROIs) (medians, error bars indicate data range within 5th and 95th percentiles) and 13C values (dots) obtained by IRMS (medians, error bars indicate SEs, n=3). (b) 12C15N−/12C14N− ratios obtained by NanoSIMS reflecting the number of evaluated ROIs (medians, error bars indicate data range within 5th and 95th percentiles) and 15N values (dots) obtained by IRMS (medians, error bars indicate SEs, n=3). Outliers are marked with asterisks. The ratios for an unlabelled clay-sized fraction were used to prove enrichment and are called natural abundance.
The data obtained by NanoSIMS were generally on the same order of magnitude as the bulk IRMS values (Fig. 3). Most of the isotopically enriched spots (94–96%) showed enrichment in both 13C and 15N isotopes, whereas the rest of the enriched spots showed enrichments in either 13C or 15N (Table 1). We assume that the spots enriched in 13C and 15N (Fig. 2c) represent new OM formed during the incubation experiment, whereas spots that show no enrichment represent pre-existing OM. By overlaying the spots showing total OM (Fig. 2b) with the enriched spots (Fig. 2c), we were able to differentiate spots with old and new labelled OM. The areas enriched in 13C and 15N as a proportion of the total OM area increased from 33–55% during incubation (Table 1), whereas the total OM area changed only marginally over time.
Discussion
Our IRMS data demonstrate the fast formation of new organo-mineral associations in addition to the organo-mineral associations already present from the original soil material. This is because of the rapid decomposition of the added litter. The presence of fresh litter-derived 15N and 13C in the fine mineral fractions point to an immediate incorporation27. This incorporation is not solely caused by sorption processes, but also due to microbial activity, which is indicated by the small C/N ratio in the clay fraction suggesting that the chemical composition is dominated by microbial residues and metabolites8 (Supplementary Table S3).
Our quantitative NanoSIMS results reveal for the first time that almost all the OM existing in the clay-sized fraction was bound in mineral clusters. The rough surfaces of clustered particles might provide preferential binding sites for OM13. The results of the SSA determination (Supplementary Table S4) indicate that especially small mineral particles with a high surface roughness got covered by OM, which confirms our findings by NanoSIMS. The OC loading of 1.1±0.1 mg m−2 determined for the clay-sized fractions (Supplementary Table S4) fell into the range proposed for the monolayer coverage9,28,29. However, because the clustered particles contained most of the OM, these organo-mineral clusters exhibited a much higher capacity for OC storage than expected for a monolayer.
We demonstrated for the first time that small mineral particles with rough surfaces in organo-mineral clusters provide the essential reactive interfaces for OM sequestration. Our results clearly indicate that mineral particles with a smooth surface are not suited for substantial OM sequestration, presumably because they do not have etch pits, micropores or cracks. Preferential OM sequestration on mineral clusters with rough surfaces also might be due to better hydration and nutrient conditions at rough surfaces, conditions that offer an advantageous microhabitat for microorganisms30. Using our approach of visualizing and quantifying the exposed surfaces, we discovered that most of the mineral surfaces did not show any OM coverage at all. Clay content is often used as a proxy for the estimation of a soil’s OC sequestration potential9,31. These findings may explain why clay content is sometimes poorly related to OC concentration32.
The heterogeneity of organic patches on mineral surfaces observed by NanoSIMS is generally not detected by bulk IRMS soil analyses25. Synchrotron-based near-edge X-ray spectromicroscopy of mineral assemblages illustrated that OM is present in extremely heterogeneous forms, such as plant or microbial biopolymers, at distinct locations20. The increased variability in the detected 12C15N−/12C14N− ratios might have been caused by fractionation of protein-rich microbial metabolites during the interaction of OM with mineral surfaces33. The organic spots that were enriched in both isotopes represent OM bound to mineral surfaces derived either directly from the litter or from the microbial products originating from litter decomposition. The area enriched solely in 15N increased from 2 to 5% during the incubation period (Table 1). From the accumulation of such OM enriched only in 15N, we infer that this represents material that was more intensively processed by microbes. Thus, NanoSIMS provides detailed insight into the spatial variability of OM accumulation during such an incubation experiment.
At the end of the incubation, the isotopic labels were extensively incorporated over about half of the organo-mineral cluster areas. This high proportion of areas covered with new labelled OM was unexpected on the basis of the enrichments measured for the clay fraction by IRMS, representing only about 44 μg 15N and 310 μg 13C per gram of clay-sized fraction, but is explained by the observation that <19% of the total mineral area is covered by OM. Our findings indicate a preferential attachment of new OM to organo-mineral clusters with rough surfaces containing pre-existing OM. This is demonstrated by an increase of area enriched in 13C and 15N, while the total area covered with OM remains constant (Table 1). Our data are in agreement with a conceptual model of preferential sorption of OM on surfaces already containing sorbed OM34,35. Although we could assign a large proportion of the total area in the clay-sized fractions to individual smooth mineral particles or clusters (80%), they did not contribute to binding of new OM. This result implies that only a limited proportion of the total clay-sized mineral particles were involved in OM sequestration. This finding was unexpected because the clay-sized mineral particles in this soil (illite, mixed-layer clay minerals and pedogenic iron oxides, such as goethite) are all considered to be reactive on all surfaces owing to their permanent and variable charges (Supplementary Fig. S3). These findings have major implications for our understanding of fundamental soil properties that are controlled by the surface reactivity of the fine soil fraction, such as cation exchange capacity, pollutant binding capacity and physical properties. Our data also point to the necessity for careful identification and quantification of the reactive mineral complexes that are responsible for OM sequestration and that control the OM saturation capacity of soils and sediments. Such data could be incorporated into current models for estimating the carbon sequestration capacity of soils and can be expected to considerably improve the predictive power of such models.
Methods
Soil and litter characterisation
For the incubation experiments, we used soil from the uppermost 5 cm of the Ap horizon, a lamellic Luvisol (raptic, arenic and bathygleyic, according to IUSS Working Group WRB soil classification system36). The soil had an organic C content of 13.5±0.9 mg g−1, a N content of 1.4±0.0 mg g−1, a C/N-ratio of 9.6±0.4 and a pH value of 5.5±0.1. The natural abundance for 13C and 15N in the soil were 1.075 and 0.369 atom-%. The clay, silt and sand contents were 18.5%, 18.4% and 63.0% (Supplementary Table S5). The oxalate-extractable iron content was 21.2±0.0 mg g−1, and the dithionite-extractable iron content was 62.8±2.3 mg g−1. The mineral composition of the clay fraction was dominated by chlorite, illite, quartz and mixed-layer minerals, as indicated by X-ray diffraction (Supplementary Fig. S3). The litter we used was derived from a 1:1 mixture of maize (Zea mays) and potato (Solanum tuberosum) dried leaf material. The unlabelled litter had an organic C content of 405.2±1.9 mg g−1, a N content of 23.7±0.1 mg g−1 and a C/N-ratio of 17.1±0.1. The 15N and 13C contents of the unlabelled litter were 0.396 and 1.084 atom-%. A mixture of unlabelled litter and soil was used to determine the natural abundances of the bulk soil and the soil fractions. The labelled litter had an organic C content of 404.2±1.8 mg g−1, a N content of 23.3±0.0 mg g−1 and a C/N-ratio of 17.3± 0.1. The 15N and 13C concentrations were 6.1 and 4.9 atom-%, respectively.
Experimental design
We thoroughly mixed 50 g of sieved soil (<2 mm) with 0.5 g of litter (<200 μm). Three treatments were incubated: (1) labelled litter and soil (three independent replicates), (2) unlabelled litter and soil (three independent replicates) and (3) an additional treatment per time point without litter, as a control. After litter addition, the soils were incubated for 42 days under defined conditions at 14 °C and at 60% maximal water holding capacity. To maintain the water content, we moistened the soil columns at 2 day intervals. Samples were taken at five time points during incubation: after 2 h and after 1, 7, 21 and 42 days.
Bulk analyses
For the determination of SEOC, a 5 g aliquot of fresh soil was shaken with 20 ml of 0.01 M CaCl2 for 45 min, and then the suspension was filtered (Whatman, grade 595 ½, Bruchsal, Germany). SEOC in the filtrate was determined on a total carbon analyser (DIMATOC 2000, Dimatec Analysen GmbH, Essen, Germany) by catalytic high-temperature oxidation. Microbial biomass (Cmic) was determined according to the fumigation–extraction method37. A 5 g aliquot of fresh soil was extracted with 0.01 M CaCl2, as described for SEOC measurements. Cmic was calculated as the difference between Ctotal in fumigated (fum) and non-fumigated (n-fum) samples using a kEC value of 0.45 (ref. 38). Atom-% 13C in the extracts was measured via online liquid chromatography coupled with stable IRMS (MAT 253, Thermo Fisher Scientific, Dreieich, Germany)39. The atom-% 13C in microbial biomass (atom-% 13Cmic) was computed as
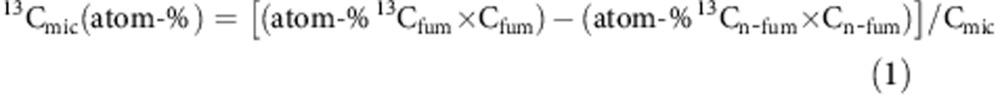 |
where 13Cfum and 13Cn-fum are atom-% 13C values of the fumigated and non-fumigated extracts, respectively, and Cfum and Cn-fum are the respective carbon concentrations40.
For further analyses, the bulk soil was dried at 40 °C. Aliquots were ground and homogenized with a vibrating ball mill with zircon-grinding tools (Pulverisette 23, Fritsch, Germany). OC, 13C, N and 15N were determined in duplicate by means of an isotope ratio mass spectrometer (Delta V Advantage, Thermo Fisher Scientific, Dreieich, Germany) coupled to an elemental analyser (Euro EA, Eurovector, Milan, Italy). Appropriate standards were included for normalization correction, instrument linearity and precision41. Values of 15N and 13C atom-% excess were calculated by subtracting 13C and 15N enrichment of the respective soil with unlabelled litter from the corresponding enrichments obtained for the treatments with labelled litter. All calculated contents were determined from the atom-% excess values.
Physical soil fractionation
To remove all free labelled litter and to extract the organo-mineral associations, we conducted a combined density and particle size fractionation. Dry soil material (30 g, <2 mm) was saturated with 300 ml of sodium polytungstate solution (density 1.8 g cm−3) and allowed to settle overnight. The floating free particulate organic matter fraction (fPOM) was removed by means of a water jet pump. For disruption of the soil aggregates, the remaining slurry was ultrasonically dispersed (Sonopuls HD 2200, Bandelin, Germany; VS 70 T Sonotrode Ø 13 mm with a liquid coverage of 1.5 cm) with an energy input of 450 J ml−1. Subsequently, the dispersed sample was centrifuged (15 min at 3019 × g) to separate the occluded particulate OM from the mineral residue. To remove the sodium polytungstate from the fPOM and occluded particulate OM, the fractions were washed with deionized water on a pressure filtration unit (Durapore Membrane Filters, pore size 0.22 μm, Millipore, Switzerland). The remaining organo-mineral fraction was centrifuged several times with deionized water until the electrical conductivity was <50 μS cm−3. The mineral residues were wet-sieved at 63 μm and 20 μm mesh sizes to separate the sand and coarse silt fractions, respectively. The soil material fraction of <20 μm was separated into medium and fine silt (20–2 μm) and clay (<2 μm) via sedimentation. All fractions were dried (40 °C) and weighed to obtain the mass proportion of each fraction relative to the bulk soil. We analysed fPOM and the smaller mineral fractions (coarse, medium and fine silt, and clay) for OC, 13C, N and 15N as described above for the bulk soil. Values of 13C atom-% and 15N atom-% excess were calculated by subtracting the values for the respective fractions of unlabelled soil (fractions of soil with unlabelled litter) from the enrichments obtained from the labelled fractions for each time point.
Specific surface area
The SSA of the clay-sized fraction was determined using a standard method for measuring surface area in soils based on the physisorption of N2 gas on a sample at 77 K using the Brunauer–Emmett–Teller equation42. Nitrogen adsorption was measured at 11 points in the partial pressure range of 0.05–0.3 with an Autosorb-1 analyser (Quantachrome, Syosset, NY, USA). Before measurement, the samples were outgassed for 14–20 h in vacuum under helium flow at 40 °C. The SSA was measured for the clay-sized fraction before and after OM removal to assess the coverage of SSA by OM15. OM was removed by oxidation with a 10% H2O2 solution at room temperature.
NanoSIMS analysis
Clay-sized fractions at the time points of 2 h and 7, 21 and 42 days, as well as a clay-sized fraction with unlabelled litter as natural abundance, were analysed by means of NanoSIMS (Cameca NanoSIMS 50l, France). A 1 mg sample of dried clay-sized fraction was dispersed in 20 ml of deionised water, and 100 μl of the dispersion was dropped on a silica wafer, which was dried overnight in a desiccator. The homogeneous distribution of the particles was checked with a reflected-light microscope (Axio Imager Z2m, Zeiss, Germany). Reflected-light microscopic images of the whole sample were taken for documentation and later orientation on the sample using the CCD camera of the NanoSIMS. SEM images (Jeol JSM-5900LV, Eching, Germany) were taken to support interpretation of the NanoSIMS results. Prior to SEM measurements, the samples were coated with gold (~5 nm; SCD 005 sputter coater, Bal-tec GmbH, Germany) to avoid charging.
For NanoSIMS analysis, the Cs+ primary ion probe was used with a primary ion impact energy of 16 keV. Prior to analysis, contaminants and an additional gold coating layer (~30 nm) were sputtered away by a high primary beam current (pre-sputtering). During the pre-sputtering, the reactive Cs+ ions were implanted into the sample to enhance the secondary ion yields. The primary beam (~1.2 pA) focused at a lateral resolution of 100–200 nm was scanned over the sample (40 × 40 μm), and 12C−, 13C−, 12C14N−, 12C15N−, 16O− and 27Al16O− secondary ions were collected on electron multipliers with an electronic dead time fixed at 44 ns. The estimated depth resolution with 16 keV Cs+ ions as primary ion beam is about 15 nm. The measurements were performed at high mass resolving power to circumvent the occurrence of mass interferences (for example, 12C1H− versus 13C−, 12C14N− versus 12C21H2− and 12C15N− versus 13C14N−). We compensated for the charging due to the non-conductive mineral particles by using the electron flood gun of the NanoSIMS instrument. All measurements were done in imaging mode. The ion images were acquired using a dwell time of 1–3 ms/pixel, 512 × 512 pixels and one cycle. For every sample, 10–12 spots were analysed to obtain a reliable data basis for the calculation of the fate of 13C and 15N.
NanoSIMS images were evaluated using the Open MIMS Image plugin for the ImageJ software. All images were corrected for electron multiplier dead time (44 ns). ROIs were chosen according to the distribution of the 12C− and 12C14N− secondary ions as an OM indicator. The threshold option of the ImageJ software was used to automatically generate the ROIs for these images. For this, the triangle algorithm was used43. The ROIs of the 12C− and 12C14N− images were combined afterward to obtain the ROIs for the OM distribution. Areas and sums of secondary ion counts for each ROI were extracted from all images. All ROIs with an area greater than 10 pixels were used for further calculations. Mineral particles were selected as ROIs on the basis of 16O− and 27Al16O− images. For structural analyses, the visible mineral surface areas were divided into clustered particles and individual particles on the basis of the corresponding SEM images.
Data presentation and statistical analysis
Mean values and standard deviations were calculated using Microsoft Excel (2007). IRMS data were statistically analysed with IBM SPSS Statistics (ver. 19.0, IBM, Armonk, NY, USA). For this analysis, the means of three independent replicates were tested using a non-parametric test (U-test) with a significance level of 95%. Figures were prepared and regressions were performed with SigmaPlot software (ver. 11.0, Systat Software, Erkrath, Germany).
Author contributions
C.V. performed experiments, collected and analysed data, developed analytical approaches for NanoSIMS output data and wrote the manuscript; C.W.M. supervised the analysis, developed analytical approaches for NanoSIMS output data and wrote the manuscript; K.H. performed experiments, supervised the analysis and wrote the manuscript; C.H. conducted NanoSIMS analyses, gave technical support and conceptual advice and edited the manuscript; F.B. conducted IRMS analyses, gave technical support and edited the manuscript; S.S. designed and performed the experiment, collected and analysed data and edited the manuscript; M.S. performed experiments, supervised the analysis and edited the manuscript; I.K.-K. performed experiments, supervised the analysis and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Additional information
How to cite this article: Vogel, C. et al. Submicron structures provide preferential spots for carbon and nitrogen sequestration in soils. Nat. Commun. 5:2947 doi: 10.1038/ncomms3947 (2014).
Supplementary Material
Supplementary Figures S1-S3 and Supplementary Tables S1-S5
Acknowledgments
We are grateful to Dr Marianne Hanzlik (Institute of Electron Microscopy, Technische Universität München, Garching) for assistance with the SEM measurements. We thank Uta Lindstedt, Maria Greiner, Bärbel Angres and Johann Lugmeier for their laboratory assistance. This project was funded within the framework of the priority program 1315 ‘Biogeochemical Interfaces in Soil’, which is funded by the German Research Foundation. Further, we acknowledge financial support from the Distributed Infrastructure for Experimentation in Ecosystem Research (EXPEER) under FP7 -262060 and we are grateful for the financial support by the International Graduate School of Science and Engeneering of the TU München within the Focus Area Water.
References
- Lal R. Soil carbon sequestration impacts on global climate change and food security. Science 304, 1623–1627 (2004). [DOI] [PubMed] [Google Scholar]
- Schmidt M. W. I. et al. Persistence of soil organic matter as an ecosystem property. Nature 478, 49–56 (2011). [DOI] [PubMed] [Google Scholar]
- Hedges J. I. & Keil R. G. Sedimentary organic matter preservation: an assessment and speculative synthesis. Mar. Chem. 49, 81–115 (1995). [Google Scholar]
- Lützow M. V. et al. Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions—a review. Eur. J. Soil Sci. 57, 426–445 (2006). [Google Scholar]
- Six J., Elliott E. T. & Paustian K. Soil macroaggregate turnover and microaggregate formation: a mechanism for C sequestration under no-tillage agriculture. Soil Biol. Biochem. 32, 2099–2103 (2000). [Google Scholar]
- Lalonde K., Mucci A, Ouellet A & Gelinas Y. Preservation of organic matter in sediments promoted by iron. Nature 483, 198–200 (2012). [DOI] [PubMed] [Google Scholar]
- Torn M. S., Trumbore S. E., Chadwick O. A., Vitousek P. M. & Hendricks D. M. Mineral control of soil organic carbon storage and turnover. Nature 389, 170–173 (1997). [Google Scholar]
- Kögel-Knabner I. et al. Organo-mineral associations in temperate soils: integrating biology, mineralogy, and organic matter chemistry. J. Plant Nutr. Soil Sci. 171, 61–82 (2008). [Google Scholar]
- Feng W., Plante A. & Six J. Improving estimates of maximal organic carbon stabilization by fine soil particles. Biogeochemistry 112, 81–93 (2013). [Google Scholar]
- Mayer L. M. Surface area control of organic carbon accumulation in continental shelf sediments. Geochim. Cosmochim. Acta 58, 1271–1284 (1994). [Google Scholar]
- Watling L. Small-scale features of marine sediments and their importance to the study of deposit-feeding. Mar. Ecol.-Prog. Ser. 47, 135–144 (1988). [Google Scholar]
- Ransom B., Bennett R. H., Baerwald R. & Shea K. TEM study of in situ organic matter on continental margins: occurrence and the ‘monolayer’ hypothesis. Mar. Geol. 138, 1–9 (1997). [Google Scholar]
- Chenu C. & Plante A. F. Clay-sized organo-mineral complexes in a cultivation chronosequence: revisiting the concept of the ‘primary organo-mineral complex’. Eur. J. Soil Sci. 57, 596–607 (2006). [Google Scholar]
- Arnarson T. S. & Keil R. G. Organic-mineral interactions in marine sediments studied using density fractionation and X-ray photoelectron spectroscopy. Org. Geochem. 32, 1401–1415 (2001). [Google Scholar]
- Mayer L. M. & Xing B. Organic matter-surface area relationships in acid soils. Soil Sci. Soc. Am. J. 65, 250–258 (2001). [Google Scholar]
- Kinyangi J. et al. Nanoscale biogeocomplexity of the organomineral assemblage in soil. Soil Sci. Soc. Am. J. 70, 1708–1718 (2006). [Google Scholar]
- Lehmann J., Kinyangi J. & Solomon D. Organic matter stabilization in soil microaggregates: implications from spatial heterogeneity of organic carbon contents and carbon forms. Biogeochemistry 85, 45–57 (2007). [Google Scholar]
- Remusat L. et al. NanoSIMS study of organic matter associated with soil aggregates: Advantages, limitations, and combination with STXM. Environ. Sci. Technol. 46, 3943–3949 (2012). [DOI] [PubMed] [Google Scholar]
- Solomon D., Lehmann J., Kinyangi J., Liang B. & Schäfer T. Carbon K-Edge NEXAFS and FTIR-ATR spectroscopic investigation of organic carbon speciation in soils. Soil Sci. Soc. Am. J 69, 107–119 (2005). [Google Scholar]
- Lehmann J. et al. Spatial complexity of soil organic matter forms at nanometre scales. Nature Geosci. 1, 238–242 (2008). [Google Scholar]
- Herrmann A. M. et al. Nano-scale secondary ion mass spectrometry – a new analytical tool in biogeochemistry and soil ecology: a review article. Soil Biol. Biochem. 39, 1835–1850 (2007). [Google Scholar]
- Mueller C. W. et al. Advances in the analysis of biogeochemical interfaces: NanoSIMS to investigate soil microenvironments. Adv. Agron. 121, 1–46 (2013). [Google Scholar]
- Heister K., Höschen C., Pronk G., Mueller C. & Kögel-Knabner I. NanoSIMS as a tool for characterizing soil model compounds and organomineral associations in artificial soils. J. Soils Sediments 12, 35–47 (2012). [Google Scholar]
- Hatton P.-J., Remusat L., Zeller B. & Derrien D. A multi-scale approach to determine accurate elemental and isotopic ratios by nano-scale secondary ion mass spectrometry imaging. Rapid Commun. Mass Spectrom. 26, 1363–1371 (2012). [DOI] [PubMed] [Google Scholar]
- Keiluweit M. et al. Nano-scale investigation of the association of microbial nitrogen residues with iron (hydr)oxides in a forest soil O-horizon. Geochim. Cosmochim. Acta 95, 213–226 (2012). [Google Scholar]
- Anbeek C. Surface roughness of minerals and implications for dissolution studies. Geochim. Cosmochim. Acta 56, 1461–1469 (1992). [Google Scholar]
- Davidson E. A., Hart S. C., Shanks C. A. & Firestone M. K. Measuring gross nitrogen mineralization, and nitrification by 15N isotopic pool dilution in intact soil cores. J. Soil Sci. 42, 335–349 (1991). [Google Scholar]
- Mayer L. M. Relationships between mineral surfaces and organic carbon concentrations in soils and sediments. Chem. Geol. 114, 347–363 (1994). [Google Scholar]
- Keil R. G., Montlucon D. B., Prahl F. G. & Hedges J. I. Sorptive preservation of labile organic matter in marine sediments. Nature 370, 549–552 (1994). [Google Scholar]
- Wang G. & Or D. Hydration dynamics promote bacterial coexistence on rough surfaces. ISME J. 7, 395–404 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassink J. The capacity of soils to preserve organic C and N by their association with clay and silt particles. Plant Soil 191, 77–87 (1997). [Google Scholar]
- Percival H. J., Parfitt R. L. & Scott N. A. Factors controlling soil carbon levels in New Zealand grasslands: is clay content important? Soil Sci. Soc. Am. J. 64, 1623–1630 (2000). [Google Scholar]
- Liu X. et al. STXM and NanoSIMS investigations on EPS fractions before and after adsorption to goethite. Environ. Sci. Technol. 47, 3158–3166 (2013). [DOI] [PubMed] [Google Scholar]
- Sollins P. et al. Organic C and N stabilization in a forest soil: evidence from sequential density fractionation. Soil Biol. Biochem. 38, 3313–3324 (2006). [Google Scholar]
- Kleber M., Sollins P. & Sutton R. A conceptual model of organo-mineral interactions in soils: self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 85, 9–24 (2007). [Google Scholar]
- WRB, I.W.G. World Reference Base for Soil Resources 2006 FAO Rome, first update (2007). [Google Scholar]
- Vance E. D., Brookes P. C. & Jenkinson D. S. An extraction method for measuring soil microbial biomass C. Soil Biol. Biochem. 19, 703–707 (1987). [Google Scholar]
- Joergensen R. G. The fumigation-extraction method to estimate soil microbial biomass: Calibration of the kEC value. Soil Biol. Biochem. 28, 25–31 (1996). [Google Scholar]
- Krummen M. et al. A new concept for isotope ratio monitoring liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 18, 2260–2266 (2004). [DOI] [PubMed] [Google Scholar]
- Marx M. et al. Determination of the fate of 13C labelled maize and wheat rhizodeposit-C in two agricultural soils in a greenhouse experiment under 13C-CO2-enriched atmosphere. Soil Biol. Biochem. 39, 3043–3055 (2007). [Google Scholar]
- Werner R. A. & Brand W. A. Referencing strategies and techniques in stable isotope ratio analysis. Rapid Commun. Mass Spectrom. 15, 501–519 (2001). [DOI] [PubMed] [Google Scholar]
- Brunauer S., Emmett P. H. & Teller E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 60, 309–319 (1938). [Google Scholar]
- Zack G. W., Rogers W. E. & Latt S. A. Automatic measurement of sister chromatid exchange frequency. J. Histochem. Cytochem. 25, 741–753 (1977). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1-S3 and Supplementary Tables S1-S5