

Abstract
We describe a highly variable clinical presentation of cerebellar ataxia in two sisters. The younger sister demonstrates early onset rapidly progressive cerebellar ataxia accompanied by motor and nonmotor cerebellar features, as well as cognitive decline and psychiatric problems. Mitochondrial respiratory chain enzyme analysis in muscle showed a decrease in complex I + III. Progressive cerebellar atrophy was demonstrated on serial brain MR imaging. Coenzyme Q10 (CoQ10) supplementation, started at the age of 5 years, led to a significant improvement in motor and cognitive abilities with partial amelioration of the cerebellar signs. Discontinuation of this treatment resulted in worsening of the ataxia, cognitive decline, and severe depression.
The older sister, who is 32 years old, has nonprogressive dysarthria and clumsiness from the age of 10 years and MRI reveals cerebellar atrophy.
Exome sequencing identified compound heterozygosity for a known (p. Thr584delACC (c.1750_1752delACC)) and a novel (p.P502R) mutation in the ACDK3 gene.
Conclusions: Patients with primary CoQ10 deficiency due to ADCK3 mutations can demonstrate a wide spectrum of clinical presentations even in the same family. It is difficult to diagnose CoQ10 deficiency based solely on the clinical presentation.
Exome sequencing can provide the molecular diagnosis but since it is expensive and not readily available, we recommend a trial of CoQ10 treatment in patients with ataxia and cerebellar atrophy even before confirmation of the molecular diagnosis.
Introduction
The hereditary ataxias are a group of genetic disorders characterized by slowly progressive incoordination of gait and often associated with poor coordination of hands, speech, and eye movements. Cerebellar atrophy frequently occurs (Bird 2012). A recently published 10-year retrospective study on childhood onset cerebellar ataxia revealed that in the group with a known genetic diagnosis, mitochondrial disease was the most common cause of cerebellar atrophy (Al-Maawali et al. 2012). Other studies on childhood ataxias also confirm that a mitochondrial etiology may be the leading cause of the disease (Ramaekers et al. 1997; Steinlin et al. 1998; Finsterer 2009; Boddaert et al. 2010; Terracciano et al. 2012).
Primary CoQ10 deficiency is a rare, autosomal recessive, clinically heterogeneous disorder caused by defects in proteins involved in the coenzyme Q synthesis pathway (Quinzii and Hirano 2011). Five major phenotypes have been described: encephalomyopathy, cerebellar ataxia, infantile multisystemic form, nephropathy, and isolated myopathy (Emmanuele et al. 2012). Cerebellar ataxia is the most common phenotype. Mutations in the ADCK3 gene have been associated with the ataxic form of CoQ10 deficiency.
We describe two sisters with a highly variable clinical presentation: severe progressive ataxia, cognitive decline, and psychiatric abnormalities versus only mild dysarthria in whom exome sequencing revealed the same heterozygous mutations in the ADCK3 gene.
Clinical Reports
Patient 1
The patient, now 20-year-old, presented to our metabolic-neuro-genetic clinic at the age of 19 years due to a slowly progressive cerebellar disease that had started at the age of 2 years and involved motor, cognitive, and psychiatric functions. Parents are healthy non-consanguineous Ashkenazi Jews. She has an older sister (Patient 2) and two healthy siblings. Perinatal history and early development were normal. She started walking at the age of 1 year and spoke in short sentences at the age of 20 months. At the age of 2 years, her parents noticed that her speech became slurred and her gait less stable. She did not gain any new motor skills. Between 3 and 5 years of age, her balance deteriorated and she sustained frequent falls. Neurologic examination at the age of 5 years demonstrated ataxia, dysarthria, and nystagmus. Cognitive decline and mood changes commenced at the age of 5 years. Her general IQ score deteriorated from 85 to 65 between the ages 10 and 18 years with a performance IQ well below the verbal IQ. She attended a special education school since the age of 12 years. At the age of 14 years, major depression was diagnosed and she was treated with antidepressants. She was hospitalized in a psychiatric hospital at the age of 17 years due to depression with suicidal ideation, faulty social judgment, and unbalanced mood.
Evaluation: Brain MRI at the age of 28 months demonstrated mild cerebellar atrophy that markedly progressed by the next MRI performed at 3.6 years of age. In the following MRI scans at age 4 and 8 years, the atrophy described to be stable (MRI not shown). An MRI done at the age of 18 years demonstrated marked cerebellar atrophy (Fig. 1a,b). Metabolic evaluation was unremarkable. Electroretinogram demonstrated rod and cone dysfunction. Visual-evoked potentials, echocardiography, electrocardiogram, electromyography, and nerve conduction studies were normal. A muscle biopsy revealed normal morphology by light and electron microscopy. A decrease in mitochondrial respiratory chain complexes I + III and IV was demonstrated. Pyruvate dehydrogenase and citrate synthase activities were low. CoQ10 levels were not evaluated.
Fig. 1.
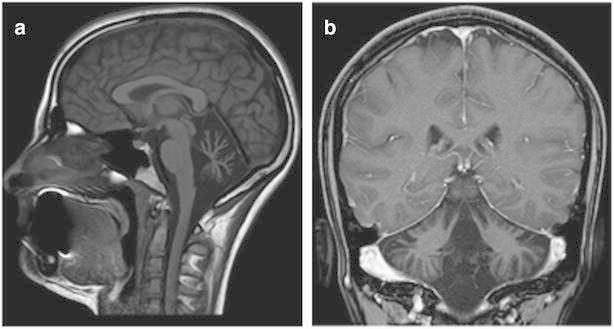
(a, b) Brain MRI of patient 1 at the age of 18 years. Sagittal T1 image and coronal T1 with contrast image demonstrate prominent cerebellar atrophy most prominent in the vermis
Molecular Evaluation: Mitochondrial DNA sequencing, genetic testing for SCA1, SCA 2, SCA 3, and sequencing of the ATM, SURF1, and APTX genes were normal.
At the age of 5 years, CoQ10 20 mg/kg/day treatment was started with partial improvement in motor skills, balance, and strength. After 6 years, she gradually stopped taking the medicine on her own and her condition deteriorated.
On examination at the age of 19 years, she was alert and communicative. She had normal weight, height, and head circumference. Her speech was dysarthric but with a good vocabulary and expressive abilities. She aggressively opposed a physical examination. She demonstrated generalized cerebellar dysfunction: horizontal nystagmus with no opthalmoplegia or oculomotor apraxia, discontinuous pursuit, dysmetria, action tremor, past pointing, abnormal knee heel test, a wide-based ataxic gait, inability to tandem, and difficulties in imitating movements. The BARS (brief ataxia rating scale score) was 8 of 30. She could write a few words in a clear handwriting. The rest of the neurologic exam was normal.
Patient 2
The older sister came to our clinic at the age of 32 years for genetic counseling following her marriage. She described mild dysfluent speech and clumsiness since childhood with no progression. She received a Master of Arts degree and works as a manager. A brain MRI study done at the age of 30 years revealed prominent cerebellar atrophy (Fig. 2a,b). Neurologic exam revealed mild dysarthria but no cerebellar signs.
Fig. 2.
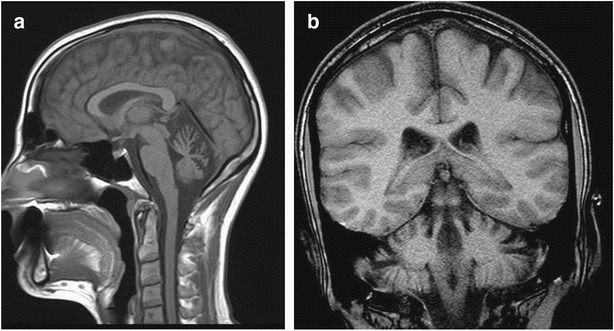
(a, b) Brain MRI of patient 2 at the age of 30 years. Sagittal and coronal T1 images demonstrate prominent cerebellar atrophy
These two sisters showed a diverse clinical picture of cerebellar disorder due to progressive cerebellar atrophy, with partial response to CoQ10 supplement in the younger sister. The clinical picture was suggestive for autosomal recessive CoQ-related disorder. Although the ADCK3 gene was the most appropriate one, there are other genes that are associated with CoQ deficiency syndromes. Thus, for cost-benefit reasons, whole exome sequencing was performed.
Methods
Exome Sequencing
Whole exome sequencing was performed on the patients’ DNA. The sample was enriched with Sureselect Human All Exome v.2 kit which was targeted 50 Mb (Agilent, Santa Clara, CA, USA). Sequencing was carried out on (Illumina, San diego, CA, USA) as 100-bp paired end runs. Image analysis and base calling were performed with the Genome Analyzer Pipeline version 1.5 with default parameters. Reads were mapped to the human reference genome sequence (assembly GRCh37/hg19) using the Burrows-Wheeler Alignment Tool (BWA) version 0.5.8c, and allelic variants were detected using the Genome Analysis Toolkit (GATK). Dataset files including the annotated information were analyzed using ANNOVAR according to the dbSNP database (build 135) and the NHLBI exome variant database with the following filtering steps: autosomal recessive inheritance; variant type including missense, nonsense, and splice-site; not within segmental duplications; minor allele frequency (MAF) less than 0.01; SIFT score < 0.05 when available; PolyPhen2 score > 0.85 when available. Primers for the ADCK3 mutations were designed using the Primer3 software and Sanger sequencing of PCR products was used to verify the mutations in the patients and their parents.
Results
A total of 21,902 coding variants (single-nucleotide variants and small insertions and deletions) were detected in patient 1 and 21,026 in patient 2; after filtering for population frequencies less than 0.01 and synonymous-frameshift-non frameshift changes −1,458 variants were found in patient 1 and 1,500 in patient 2. Homozygous variants-200, none of these in the homozygous region and heterozygous variants-1,200, but only 19 were common in the two patients.
The only candidate gene in the double heterozygous variant list, which segregated with the disease, was ADCK3, previously described in association with cerebellar ataxia. The variants were the novel p.P502R substitution and the previously described p. Thr584delACC (c.1750_1752delACC). The father carries the p.P502R mutation and the mother carries the p. Thr584delACC (c.1750_1752delACC) mutation (Fig. 3). The novel p.P502R mutation is predicted to be deleterious according to Polyphen2, SIFT, and Mutation Taster softwares. The P602 is a conserved amino acid.
Fig. 3.
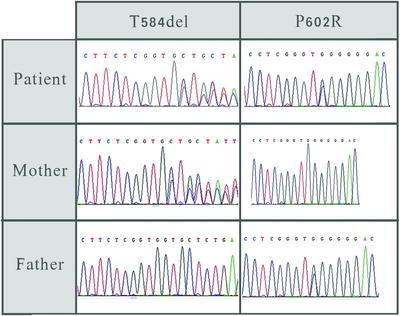
Sequence electropherogram demonstrating the p. Thr584delACC (c.1750_1752delACC) and p.P502R mutations in patient and her parents
Discussion
We describe two sisters with an extremely variable clinical presentation who are compound heterozygotes for two mutations in the ADCK3 gene. The Th584del has been previously described in patients with cerebellar ataxia (Lagier-Tourenne and Tazir 2008), but the P602R mutation is novel (Lagier-Tourenne and Tazir 2008; Mollet et al. 2008; Gerards et al. 2010; Horvath et al. 2012). The pathogenicity of the P602R is supported by a substitution of a neutral amino acid by a basic one, conservation of Proline602 among species and prediction as disease causing by three softwares.
Only 22 patients with ADCK3-associated cerebellar ataxia have been reported since the first publications in 2008 (Lagier-Tourenne and Tazir 2008; Mollet et al. 2008; Gerards et al. 2010; Horvath et al. 2012).
A progressive cerebellar ataxia is described in all reported patients. Early psychomotor development is normal in most children but some of them have been described as clumsy with frequent falls during the first 2–3 years of life. The age of onset varies from 18 to 24 months to 15 years. Adult onset has been described in a single case (Horvath et al. 2012). The first presenting symptom is usually loss of balance with later appearance of other cerebellar signs, including limb incoordination, abnormal eye movements, tremor, and dysarthria. Associated symptoms may include seizures, cognitive decline, depression, exercise intolerance, muscle weakness, dystonia, myoclonus, pyramidal signs, ptosis, migraine, swallowing difficulties, reduced vision, peripheral neuropathy, hearing impairment, and cataracts (Horvath et al. 2012).
Brain imaging demonstrates cerebellar atrophy in all patients by the time cerebellar signs are already apparent. Other findings include stroke-like lesions (Mollet et al. 2008), pontine atrophy (Gerards et al. 2010), thin corpus callosum, and ventricular enlargement (Horvath et al. 2012).
Nine families have been described with two to four affected siblings. The clinical presentation is described as similar in eight families (Horvath et al. 2012). One family with four affected siblings has been described with clinical variability: all had ataxia and cerebellar atrophy; age of onset varied from 4 to 11 years, only one had mild mental retardation and two had pyramidal signs (Lagier-Tourenne and Tazir 2008). Our patients demonstrate extreme phenotypic variability: The younger sister has early onset progressive ataxia, cognitive decline, and psychiatric involvement while the older one who is 32 years old only has dysarthria with no ataxia. Both sisters, however, demonstrate cerebellar atrophy on MRI (Figs. 1 and 2).
The pathogenesis of the disease is related to energy deficiency due to a defect in CoQ10 metabolism. CoQ10 is an essential electron carrier in the mitochondrial respiratory chain, transferring electrons from complex I and II to complex III and contributing to ATP biosynthesis. It also has a key role as a free radical scavenger, preventing the progression of lipid peroxidation in membranes or regenerating other antioxidants such as vitamin E or ascorbate in other cellular membranes (Artuch et al. 2006; Montero et al. 2007). Studies in both rats and humans have demonstrated that the lowest brain CoQ content is in the cerebellum (Montero et al. 2007), suggesting that antioxidant defenses are very limited in this brain area, and, consequently mild to moderate CoQ deficiencies might lead to cerebellar dysfunction (Montero et al. 2007).
There is no explanation for the phenotypic variations in the reported patients and no correlation has been found between the degree of mitochondrial dysfunction and the disease severity as demonstrated by serum lactate, mitochondrial morphology in muscle biopsy, or respiratory chain activity in muscle, fibroblasts, and lymphocytes (Aure and Benoist 2004; Lagier-Tourenne and Tazir 2008; Mollet et al. 2008; Horvath et al. 2012). There must be other factors, either genetic or environmental, that influence the phenotypic expression.
Clinical improvement following CoQ10 supplementation has been documented in many patients. There is no consensus regarding the oral dose for treatment of CoQ10 deficiency, treatment protocols have not been standardized and results have not been uniform. CoQ10 supplementation has been tried in patients with ADCK3 gene–related ataxia (Lamperti et al. 2003; Aure and Benoist 2004; Mollet et al. 2008; Pineda et al. 2010; Horvath et al. 2012; Emmanuele et al. 2012) in different regimens ranging from 5 to 30 mg/kg/day. Aure et al. reported significant improvement in exercise tolerance and decrease of vomiting episodes under CoQ10 supplementation of 6 mg/kg/day. However, this therapy did not prevent the development of cerebellar atrophy and ataxia. Emmanuele et al. recommend oral supplementation doses of up to 2,400 mg daily in adult patients and up to 30 mg/kg in pediatric patients. Pinedo et al. studied neurologic outcome in patients with CoQ10 deficiency associated cerebellar ataxia after 2 years of treatment with oral CoQ10 supplementation of 30 mg/kg/day regimen. They reported a significant improvement in all patients except in one. The best clinical response among all patients was demonstrated in a patient who showed only mild vermian atrophy (Pineda et al. 2010). All patients with CoQ10 deficiency due to mutations in ADCK3 gene failed to improve and even worsened under treatment with the shorter chain ubiquinone analog, idebenone (Aure and Benoist 2004; Mollet et al. 2008). Our patient showed improvement in motor and academic activity under oral supplementation of 20 mg/kg daily CoQ10 started at 5 years of age but unfortunately she discontinued treatment and a further deterioration occurred including severe psychiatric involvement. Following the establishment of the molecular diagnosis, both sisters started CoQ10 treatment.
Conclusion
Cerebellar ataxia associated with CoQ deficiency due to ADCK3 gene mutation may present with a wide spectrum of clinical phenotypes even in the same family, ranging from early onset progressive cerebellar ataxia with cognitive and psychiatric features to nonprogressive mild cerebellar signs accompanied by cerebellar atrophy. Adult onset of cerebellar symptoms may occur.
Clinical improvement under CoQ10 supplementation may be remarkable, but the dosage regimen should be appropriate and the treatment should be started early. Due to the diagnostic difficulties of CoQ deficiency–related cerebellar ataxia, a therapeutic trial of CoQ10 should be offered to patients with cerebellar ataxia and cerebellar atrophy of unknown origin even before establishing the molecular diagnosis or enzymatic diagnosis.
Footnotes
Competing interests: None declared
Contributor Information
Dorit Lev, Email: dorlev@post.tau.ac.il.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Al-Maawali A, Blaser S, Yoon G. Diagnostic approach to childhood-onset cerebellar atrophy: a 10-year retrospective study of 300 patients. J Child Neurol. 2012;27:1121–1132. doi: 10.1177/0883073812448680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artuch R, Brea-Calvo G, Briones P, Aracil A, Galva´n A, Espinos C, Corral J, Volpini V, Ribes A, Andreu AL, Palau F, Sa´nchez-Alca´zar JA, Navas P, Pineda M. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme Q10 supplementation. J Neurol Sci. 2006;246:153–158. doi: 10.1016/j.jns.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombe‘s A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–729. doi: 10.1212/01.WNL.0000134607.76780.B2. [DOI] [PubMed] [Google Scholar]
- Bird TD (2012) Hereditary ataxia overview. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) GeneReviews™ [Internet]. University of Washington, Seattle, 28 Oct 1993–1998 [updated 2012 Nov 01] [PubMed]
- Boddaert N, Desguerre I, Bahi-Buisson N, et al. Posterior fossa imaging in 158 children with ataxia. J Neuroradiol. 2010;37(4):220–230. doi: 10.1016/j.neurad.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Emmanuele V, López LC, Berardo A, Naini A, Tadesse S, Wen B, D’Agostino E, Solomon M, DiMauro S, Quinzii C, Hirano M. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012;69(8):978–983. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J. Mitochondrial ataxias. Can J Neurol Sci. 2009;36:543–553. doi: 10.1017/s0317167100008027. [DOI] [PubMed] [Google Scholar]
- Gerards M, van den Bosch B, Calis C, et al. Nonsense mutations in ADCK3cause progressive cerebellar ataxia and atrophy. Mitochondrion. 2010;10:510–515. doi: 10.1016/j.mito.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Horvath R, Czermin B, Gulati S, et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry. 2012;83:174–178. doi: 10.1136/jnnp-2011-301258. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Tazir M. Lo’pez LC, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamperti C, Naini A, Hirano M, et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206–1208. doi: 10.1212/01.WNL.0000055089.39373.FC. [DOI] [PubMed] [Google Scholar]
- Mollet J, Delahodde A, Serre V, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82:623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero R, Pineda M, Aracil A, Vilaseca MA, Briones P, Sánchez-Alcázar JA, Navas P, Artuch R. Clinical, biochemical and molecular aspects of cerebellar ataxia and Coenzyme Q10 deficiency. Cerebellum. 2007;6(2):118–122. doi: 10.1080/14734220601021700. [DOI] [PubMed] [Google Scholar]
- Pineda M, Montero R, Aracil A, et al. Coenzyme Q(10)-responsive ataxia: 2-yeartreatment follow-up. Mov Disord. 2010;25(9):1262–1268. doi: 10.1002/mds.23129. [DOI] [PubMed] [Google Scholar]
- Quinzii CM, Hirano M. Primary and secondary CoQ10 deficiencies in humans. Biofactors. 2011;37:361–365. doi: 10.1002/biof.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaekers VT, Heimann G, Reul J, et al. Genetic disorders and cerebellar structural abnormalities in childhood. Brain. 1997;120(Pt 10):1739–1751. doi: 10.1093/brain/120.10.1739. [DOI] [PubMed] [Google Scholar]
- Steinlin M, Blaser S, Boltshauser E. Cerebellar involvement in metabolic disorders: a pattern-recognition approach. Neuroradiology. 1998;40(6):347–354. doi: 10.1007/s002340050597. [DOI] [PubMed] [Google Scholar]
- Terracciano A, Renaldo F, Zanni G, et al. The use of muscle biopsy in the diagnosis of undefined ataxia with cerebellar atrophy in children. Eur J Paediatr Neurol. 2012;16(3):248–256. doi: 10.1016/j.ejpn.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]