

Abstract
TMEM16A is a plasma membrane protein with voltage- and calcium-dependent chloride channel activity. The role of the various TMEM16A domains in expression and function is poorly known. In a previous study, we found that replacing the first ATG of the TMEM16A coding sequence with a nonsense codon (M1X mutation), to force translation from the second ATG localized at position 117, only had minor functional consequences. Therefore, we concluded that this region is dispensable for TMEM16A processing and channel activity. We have now removed the first 116 codons from the TMEM16A coding sequence. Surprisingly, the expression of the resulting mutant, Δ(1–116), resulted in complete loss of activity. We hypothesized that, in the mutant M1X, translation may start at a position before the second ATG, using a non-canonical start codon. Therefore, we placed an HA-epitope at position 89 in the M1X mutant. We found, by western blot analysis, that the HA-epitope can be detected, thus demonstrating that translation starts from an upstream non-ATG codon. We truncated the N-terminus of TMEM16A at different sites while keeping the HA-epitope. We found that stepwise shortening of TMEM16A caused an in parallel stepwise decrease in TMEM16A expression and function. Our results indicate that indeed the N-terminus of TMEM16A is important for its activity. The use of an alternative start codon appears to occur in a naturally-occurring TMEM16A isoform that is particularly expressed in human testis. Future experiments will need to address the role of normal and alternative amino-terminus in TMEM16A structure and function.
Abbreviations: CaCC, Ca2 +-activated Cl− channel; HS-YFP, halide-sensitive yellow fluorescent protein
Keywords: Chloride channel, Structure–function relationship, Protein translation, Non-canonical start codon
Graphical abstract

Highlights
-
•
TMEM16A is a membrane protein with chloride channel activity.
-
•
Two ATG codons are present at positions 1 and 117 of the TMEM16A coding sequence.
-
•
Deletion of the first ATG, in contrast to removal of the first 116 codons, does not abolish TMEM16A function.
-
•
Without the first ATG, TMEM16A translation begins from a non-canonical start codon.
1. Introduction
The TMEM16A protein (also known as anoctamin-1, ANO1) is a main component of Ca2 +-activated Cl− channels, CaCCs [1–3]. The transfection of TMEM16A coding sequence in null systems such as HEK-293 cells generates Ca2 +-dependent Cl− currents [1–7]. Conversely, knockdown of endogenous TMEM16A expression in airway epithelia, smooth muscle, salivary glands and other cell types leads to significant reduction of CaCC function [1,3,5,8–11].
Although the detailed structure of TMEM16A protein is unknown, predictive tools and experimental work suggest the presence of eight transmembrane domains with the N- and C-termini protruding in the cytosolic environment. There is uncertainty about the precise localization of the fifth and sixth transmembrane domains and the size and organization of the connecting region. In one of the studies that described the discovery of TMEM16A, this region was proposed as a reentrant loop facing the extracellular environment and constituting the channel pore [3]. In a more recent study, the putative channel pore region was found to be intracellular [12].
There are multiple isoforms of TMEM16A protein generated by alternative splicing or by the use of an alternative promoter [1,13,14]. A highly basic segment of 22 amino acids, corresponding to exon 6b may be included/skipped in the cytosolic region preceding the first transmembrane domain. Other two alternative segments of 4 and 26 amino acids, coded by exons 13 and 15, respectively, are localized in the first intracellular loop. For simplicity, these three alternative protein segments were labeled as b, c, and d [1]. We also found evidence for a TMEM16A coding sequence (accession no. BC033036) generated by the use of an alternative promoter. In this case, the first two exons, containing the start codon and part of the downstream coding region, were skipped. The expected result is the translation beginning from the first available ATG codon, localized at position 117 of the normal coding sequence. We named the first 116 amino acids of TMEM16A as segment a [1]. The use of the single-letter-labeling of alternative regions allowed a simple coding of TMEM16A isoforms. For example, TMEM16A(ac) and TMEM16A(abc) differ by the absence/presence of the 22 amino acids of segment b (exon 6b). Interestingly, the isoform without segment a (clone BC033036), also lacked the other alternative regions. This isoform, named TMEM16A(0), is a functional channel, although with altered voltage-dependence and ion selectivity [15].
In previous studies, replacement of the first codon of TMEM16A coding sequence with a nonsense (M1X) or an isoleucine codon (M1I) was done to force translation from the second methionine at position 117 [15,16]. The expression of these mutants resulted in no change [15], or a relatively modest change [16], in CaCC properties. Accordingly, we concluded that the first 116 amino acids of TMEM16A protein have a minor role for function [15].
In the present study, we have reconsidered the role of TMEM16A N-terminus. We have found evidence for non-canonical translation start sites. This allows a large part of segment a to be synthesized even in the absence of an upstream methionine codon. Our experiments show that the N-terminus of TMEM16A is essential since complete removal of the first 116 amino acid residues abolishes TMEM16A function.
2. Materials and methods
2.1. Cell culture
HEK-293 cells were cultured in DMEM/Ham's F12(1:1). The medium was supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
2.2. Generation of TMEM16A mutants
The human TMEM16A(abc) coding sequence was cloned into the pcDNA 3.1 plasmid as described previously [1]. We introduced a BspEI restriction enzyme site by mutagenizing a single nucleotide at position 1097 of the TMEM16A(abc) coding sequence. This mutation did not change the amino acid sequence, nor the extent of protein expression and activity as shown by functional measurements. To generate the different truncation mutants carrying the triple HA epitope, we used a strategy based on gene synthesis provided by GenScript (Piscataway, NJ). The different mutants were generated by synthesizing the desired DNA fragments flanked at one end by a KpnI site (corresponding to the one present in the vector polylinker) and at the other end by the BspEI site. These fragments were cut and pasted into the wild type TMEM16A sequence. To generate the BC033036 construct with the triple HA-epitope, a larger fragment was synthesized, flanked by the KpnI site and an EcoRI site localized at position 1836 in the TMEM16A coding sequence. Further details may be provided on request. Single amino acid mutants were generated using the QuikChange XL site-directed mutagenesis kit (Stratagene). All mutants were checked by DNA sequencing.
2.3. Transient transfections
For the functional assay based on the halide-sensitive yellow fluorescent protein (HS-YFP), HEK-293 cells were seeded in 96-well microplates (25,000 cells/well) in 100 μl of antibiotic-free culture medium. After 6 h, cells were co-transfected with plasmids carrying the coding sequence for TMEM16A constructs and the YFP-H148Q/I152L [17]. For each well, 0.2 μg of total plasmid DNA and 0.5 μl of Lipofectamine 2000 (Life Technologies) were first pre-mixed in 50 μl of OPTI-MEM (Life Technologies) to generate transfection complexes (60 min at room temperature), and then added to the cells. After 24 h, the complexes were removed by replacement with fresh culture medium plus antibiotics. The HS-YFP functional assay was performed 48 h after transfection. For patch-clamp experiments, 500,000 HEK-293 cells in 1.5 ml culture medium without antibiotics were mixed with 0.5 ml of OPTIMEM containing 2 μg of plasmid DNA and 5 μl of Lipofectamine 2000 (previously incubated for 60 min at room temperature to allow formation of lipid/DNA complexes). Cell suspension was then plated as 5–6 drops in the center of 35 mm Petri dishes. After 5–6 h, each Petri dish received 2 ml of culture medium without antibiotics. After 24 h, the complexes were removed by replacement with fresh culture medium plus antibiotics. Patch-clamp experiments were carried out 2–3 days after transfection.
2.4. HS-YFP assay
Transfected HEK-293 cells were washed two times with 100 μl of PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 1 mM CaCl2, and 0.5 mM MgCl2; pH = 7.4) and incubated for 30 min with 60 μl of PBS. After incubation cells were transferred to a microplate reader (FluoStar Galaxy; BMG Labtech, Ortenberg, Germany) for CaCC activity determination. The plate reader was equipped with high-quality excitation (ET500/20 ×) and emission (ET535/30 m) filters for YFP (Chroma Technology Corp., Brattleboro, VT, USA). Each assay consisted of a continuous 14 s fluorescence reading with 2 s before and 12 s after injection of 165 μl of modified PBS (Cl− replaced by l− final l− concentration in the well: 100 mM) also containing 1 μM ionomycin. Data were normalized to the initial, background-subtracted, fluorescence. To determine fluorescence quenching rate (QR) associated with l− influx, the final 11 s of the data for each well were fitted with an exponential function to extrapolate the initial slope (dF/dt).
2.5. Western blots
HEK-293 cells were grown to subconfluence on 60 mm-diameter dishes. To express the different TMEM16A protein constructs, cells were transiently transfected using 10 μl of Lipofectamine 2000 and 4 μg of mammalian expression plasmids pcDNA 3.1 carrying the coding sequence for TMEM16A or the HS-YFP. Cells were lysed in RIPA buffer 1 × (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) containing Complete Protease Inhibitor Cocktail (Roche, NJ, USA). The protein concentration in lysates was quantified using the Quantum Protein Assay kit (Euroclone). Total lysates were electrophoresed (30 μg per lane) with NuPAGE Novex Bis-Tris 4–12% gel (Life Technologies) and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA) for Western blotting. The TMEM16A protein was immunodetected with the rabbit SP31 monoclonal antibody (Abcam; 1:1000). TMEM16A proteins tagged with the HA-epitope were also detected with the mouse HA.11 clone 16B12 monoclonal antibody (Covance; 1:1000). Secondary antibodies were anti-rabbit or anti-mouse HRP (Millipore; 1:5000 and 1:10,000, respectively). Membranes were also stripped with the Restore Western Blot Stripping Buffer (Thermo Fisher Scientific Inc.) and incubated with anti-actin goat polyclonal I19 antibody (Santa Cruz Biotechnology) followed by anti-goat HRP-conjugated secondary antibody (Santa Cruz Biotechnology). All antibodies were dissolved in 5% skimmed-milk in Tris-buffered saline–Tween 20. Protein bands were visualized using the ECL Advanced Western Blotting Detection Kit (GE Healthcare Europe, Little Chalfont, UK). Direct recording of the chemiluminescence was performed using the Molecular Imager ChemiDoc XRS System (Bio-Rad). Densitometry analysis of bands was done with the ImageJ software.
2.6. Patch-clamp experiments
Whole-cell membrane currents were recorded in HEK-293 transiently transfected cells. The extracellular solution had the following composition: 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM mannitol, and 10 mM Na-HEPES (pH = 7.4). The pipette (intracellular) solution contained: 130 mM CsCl, 10 mM EGTA, 1 mM MgCl2, 10 mM Cs-HEPES (pH = 7.35), and 1 mM ATP, plus CaCl2 to obtain the desired free Ca2 + concentration: 1 mM for 8 nM, 8 mM for 305 nM, 9 mM for 685 nM, and 9.5 mM for 1.44 μM (calculated with a dissociation constant for the EGTA-calcium complex of 76 nM at pH 7.35). During the experiments, the membrane capacitance and series resistance were analogically compensated using the circuitry provided by the EPC7 patch-clamp amplifier. The stimulation protocol to generate current–voltage relationships consisted in 600 ms-long voltage steps from − 100 to + 100 mV in 20 mV increments starting from a holding potential of − 60 mV. The waiting time between steps was 4 s. Membrane currents were filtered at 1 kHz and digitized at 5 kHz with an ITC-16 (Instrutech) AD/DA converter. Data were analyzed using the Igor software (Wavemetrics) supplemented by custom software kindly provided by Dr. Oscar Moran (Institute of Biophysics, CNR, Genova).
2.7. RT-PCR
One microgram of total RNA derived from 18 different human tissues (Human Total RNA Master panel II, Clontech) was retro-transcribed using IScript RT kit (Biorad) and subsequently amplified by PCR.
PCR conditions for the normal 5′-end of TMEM16A and for beta-2 microglobulin were: initial denaturation at 95 °C for 5 min for the initial denaturation; 30 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 40 s; final extension at 72 °C for 7 min. PCR conditions for the alternative 5′-end of TMEM16A were: initial denaturation at 95 °C for 5 min; 30 cycles of 95 °C for 30 s, 63 °C for 30 s, and 72 °C for 40 s; final extension at 72 °C for 7 min.
We used the following forward and reverse primers for the normal TMEM16A 5′-end: 5′-GGCGGTCCCAGCGCACAG and 5′-CCGGTTGCCCGAGGGCC. For the alternative TMEM16A 5′-end the primers were: 5′-GCCGGCACCAAATGCTGACCA and 5′-CCGGTTGCCCGAGGGCC. For beta-2 microglobulin, the primers were: 5′-GCGCTACTCTCTCTTTCTGG and 5′-GATGCCGCATTTGGATTGGA.
The amplified products (260 bp for normal TMEM16A; 286 bp for the alternative TMEM16A, and 371 bp for beta-2 microglobulin) were resolved on 1.5% agarose gels. The identity of bands detected in testis and lung were verified by sequencing.
2.8. Statistics
Data are presented as representative traces/images or as mean ± SEM. Statistical analysis was done with the InStat software (GraphPad). Significant differences between data were calculated with the Student's t test.
3. Results
The region of the TMEM16A protein preceding the first transmembrane domain is composed of nearly 330 amino acids. We previously found evidence for an isoform lacking the first part of this region [1]. Indeed, a cDNA clone (BC033036) contained a different 5′ sequence, as shown in Fig. 1A. Due to the use of an alternative promoter, the transcription of the first two exons is skipped and another sequence is added at the beginning of the transcript. The consequence is the replacement of the first 36 codons with a sequence lacking an ATG (and carrying three non sense codons in frame with the downstream region). The expected result is that the beginning of translation will occur at the next available ATG codon, at position 117 of full length TMEM16A coding region (Fig. 1A). The inspection of the sequences surrounding the two ATGs at position 1 and 117 reveals that the first one may be favored as a translation start site. The general Kozak consensus indicates a purine (A or G) at position − 3 and a G at position + 4 as preferred nucleotides [18]. The first ATG in the TMEM16A coding sequence has the required purine at position − 3, whereas the second ATG has the G at + 4. However, the first ATG may be preferred because it is first encountered by the ribosome during the scanning of the mRNA and because AxxATGA is more probable as a translation start site than CxxATGG [18]. Furthermore, a G at position − 6, present before the first and not the second ATG, seems to be also important. In the absence of the first ATG, as in the BC033036 transcript, translation should start at methionine 117.
Fig. 1.

Analysis of TMEM16A translation start site. A) TMEM16A topology and amino acid sequence of the TMEM16A amino-terminus. The alternative 5′-untranslated region of the cDNA clone BC033036 is shown as an inset. White triangles and the black triangle show sites where the stop codon mutations and the HA epitope were introduced, respectively (for the experiments reported in Fig. 3). B) Electrophoretic mobility of wild type and mutant TMEM16A proteins as detected in western blot experiments (with anti-TMEM16A SP31 antibody). The first ATG was replaced with a nonsense (M1X) or an isoleucine (M1I) codon. A double mutant with methionines 1 and 117 replaced by isoleucine was also studied (M1I-M117I). Where indicated, cell lysates were treated with PNGaseF to remove sugars. Densitometric analysis of TMEM16A expression from four experiments is shown in the bar graph. The intensity of the band in each sample was normalized to the expression of actin and then to the expression of wild type TMEM16A (white bar for glycosylated samples, gray bar for deglycosylated samples). C) Activity of wild type and mutant TMEM16A determined with the HS-YFP assay. Activity, reported as quenching rate (QR), was determined with and without 1 μM ionomycin (***, p < 0.001 vs. mock-transfected cells; n = 5–6).
In our previous study, the isoform corresponding to cDNA clone BC033036 was named TMEM16A(0) because it appeared to lack the 116 amino acids of segment a as well as the other alternative segments b, c, and d [1].
We previously replaced the first ATG with a stop codon (mutant M1X) in order to force the beginning of translation at the second ATG thus skipping segment a [15]. In another study, a similar experiment was carried out by changing the first ATG with ATC, i.e. an isoleucine codon [16]. In the first study, M1X generated Cl− currents very similar to those of the wild type protein [15]. In the second study, M1I, named Δ1,2(5′), also generated Cl− currents but with slightly slower activation at positive membrane potentials [16]. For the present study, we investigated the electrophoretic mobility of proteins lacking the first ATG. Surprisingly, we found that these mutant proteins migrate in the gel with an apparent molecular size (120 kDa) similar, if not identical, to that of the wild type protein (Fig. 1B). In contrast to mobility, the expression level appeared to be reduced by replacement of the first methionine.
The electrophoretic mobility of mature TMEM16A protein as a diffuse band is due to glycosylation. To generate protein bands with a sharper aspect and a more defined molecular weight, we deglycosylated TMEM16A protein with PNGaseF. With this treatment, the relative mobility of M1X and M1I with respect to wild type TMEM16A did not change (Fig. 1B). The three proteins appeared to have essentially the same electrophoretic mobility (~ 100 kDa) although having a calculated molecular weight difference of ~ 12 kDa (mutant vs. wild type protein). Although relatively small, this difference should be visible in the gel. For comparison, we found that a TMEM16A protein lacking the last 44 amino acids at the C-terminus (theoretical loss of ~ 4.5 kDa) ran significantly faster in the gel electrophoresis (not shown). We also studied a third variant which was generated by mutating both M1 and M117 to ATC [16]. In this way, translation should start at methionine 176 thus resulting in the lack of the amino acids coded by exons 1–3 (mutant Δ1,2,3(5′)). Strikingly, the Δ1,2,3(5′) mutant (i.e. M1I-M117I) had the same electrophoretic mobility of the wild type, M1X, and M1I proteins (Fig. 1B). To assess the ion channel function of N-terminal variants, we used the assay based on the halide-sensitive yellow fluorescent proteins (HS-YFP). This assay measures the rate of HS-YFP fluorescence quenching (QR) caused by I− influx through TMEM16A channels. As shown in Fig. 1C, the proteins lacking the ATG codons at the N-terminus were functional with a rate of halide transport smaller than that of the wild type protein but still several folds larger than that of mock-transfected cells. Due to the intriguing results regarding the mobility of TMEM16A proteins lacking the first methionine, we reconsidered the issue of TMEM16A N-terminus.
The first hypothesis that we formulated was that the translation of wild type TMEM16A protein does not start from the first or the second ATG, as considered before, but from a downstream codon. Therefore, mutagenesis of ATGs at position 1 and 117 should not affect the protein expression and function. This hypothesis was disproved by two types of experiments. We generated a TMEM16A construct with both ATGs mutagenized to stop codons (M1X-M117X). This construct was not functional (Fig. 2A). We also generated a construct, named Δ(1–116)-TMEM16A, entirely lacking the 116 codons of segment a. This construct was also not functional in HS-YFP assays (Fig. 2A). Interestingly, in western blot experiments, the Δ(1–116)-TMEM16A protein showed a markedly smaller size compared to M1X-TMEM16A (80 vs. 100 kDa in deglycosylated samples) as evident from the faster electrophoretic mobility (Fig. 2B). We compared the properties of membrane currents generated by expression of wild type and M1X-TMEM16A (Fig. 2C–E). The amplitude of the currents at different membrane potentials was similar (Fig. 2D), as well as the activation/deactivation kinetics (Fig. 2E). We also calculated the normalized initial current (I0), i.e. the current measured at the beginning of the voltage pulse divided by total current. This value was also similar in the wild type and mutated construct (Fig. 2E). Summarizing, M1X and Δ(1–116) constructs generate completely different results although having in common the TMEM16A coding sequence beginning with codon 117. M1X-dependent channels have wild type characteristics whereas the Δ(1–116) construct is non functional (Fig. 2C,D).
Fig. 2.

Role of segment a in TMEM16A N-terminus. A) Activity of wild type and mutant proteins determined with the HS-YFP assay and reported as quenching rate, QR (***, p < 0.001 vs. mock-transfected cells; n = 5–6). B) Electrophoretic mobility of a TMEM16A protein lacking the first 116 codons, Δ(1–116), compared to the wild type and M1X proteins. The bar graph shows densitometric analysis to estimate TMEM16A protein expression. In each sample, the intensity of the band was normalized to actin expression and then to intensity of the corresponding band of wild type TMEM16A. For experiments with Δ(1–116) and without PNGaseF we considered the top band. White bars: glycosylated samples. Gray bars: deglycosylated samples. Data (mean ± SEM) are from four experiments. C) Representative membrane currents from patch-clamp experiments. The three panels of traces show superimposed currents elicited by voltage steps in the − 100 to + 100 mV range. Cytosolic free Ca2 + concentration was 685 nM. D) Current–voltage relationships from experiments as those shown in C. Currents were measured at the end of voltage pulses. Each point is the mean ± SEM from 6–9 experiments. E) Comparison of wild type and M1X-TMEM16A. The bar graphs report the normalized initial current (I0), the activation time constant (τact), and the deactivation time constant (τdeact) using the voltage pulse to + 100 mV.
To explain these results, we formulated a second hypothesis based on increasing evidence from the literature about the use of non-canonical codons to start translation [19–23]. We postulated that, in the absence of the first ATG (as in M1X and M1I), translation may start from another type of codon present in the first part of segment a. According to the literature, the most probable candidates are CTG and GTG, corresponding to leucine and valine. As shown in Fig. 1A, there are multiples CTG and GTG codons at the beginning of TMEM16A coding sequence. In particular, some of them, like the CTG codons present at positions 31 and 37, have a significant probability of being translation start sites according to the surrounding sequence. For example, both sites show a G and a C at positions -6 and -4, respectively, which are frequently seen in other proteins with noncanonical translation codons [21].
To test the existence of alternative start codons in TMEM16A, we generated several constructs each one carrying a stop codon mutation at different positions within segment a (Fig. 3A). In addition, the sequence upstream of each stop codon was removed. All constructs had also a triple hemagglutinin (HA) epitope between threonine 88 and proline 89 to allow efficient protein detection. The different constructs were analyzed at the functional level with the HS-YFP assay (Fig. 3B). The removal of first ATG in the M1X mutant or removal of the first 16 codons (S16X) caused a similar 40% decrease in activity. Further truncation of the N-terminus (L31X and S33X), resulted in lower but significant levels of activity relative to mock transfected cells (Fig. 3B). The complete loss of halide transport was instead observed for S74X. These constructs were also analyzed in western blot experiments. Importantly, the expression of the M1X construct generated a clear protein band recognized by the anti-HA antibody (Fig. 3C). Since there are no ATGs upstream in the region before the HA sequence, this result demonstrates that translation occurs from a non-canonical start codon. Although the M1X band had a lower intensity with respect to the wild type TMEM16A band, it had similar electrophoretic mobility corresponding to ~ 120 kDa. The constructs S16X, L31X, S33X, and R56X were also detected by the anti-HA antibody as bands of similar size. However, R56X also showed expression of a second band (~ 100 kDa) probably corresponding to a partially glycosylated form of the protein. Indeed, the treatment with PNGase F converted the two bands in a single band of lower molecular weight (Fig. 3C, inset). The expression of the S74X mutant resulted in a weak band of less than 90 kDa (Fig. 3C). Summarizing, progressive truncation of 5′-coding region of TMEM16A results in almost unaltered electrophoretic mobility when the affected region is included between codons 1 and 56. Further downstream truncation results in significant shortening of the protein detected by western blot experiments. Regarding protein expression levels, a first drop occurs for M1X and S16X. A second drop occurs for mutants L31X and S33X. Finally, a more marked loss in expression occurs by truncation at codon 56. We normalized the activity of the different TMEM16A constructs, measured with the HS-YFP assay, to the protein expression level (Fig. 3D). After normalization, the activity of the various truncated versions of TMEM16A (except for S74X) was comparable to that of the wild type protein. This result suggests that the decrease in activity caused by progressive shortening of the N-terminus is due to a decrease in protein expression rather than to an impairment in intrinsic channel activity.
Fig. 3.

Effect of progressive N-terminus truncation. A) Schematic representation of the various truncation mutants used in the study. The position of the two methionines (1 and 117), the HA epitope, and possible (alternative) translation start sites is shown. Additional information about the constructs is also depicted in Fig. 1A. The dashed line reported for the BC033036 construct indicates the presence of the alternative 5′-end which contains three stop codons in frame with the downstream coding sequence. B) Ca2 +-activated halide transport measured for the various constructs using the HS-YFP assay (** and ***, p < 0.01 and p < 0.001, respectively, vs. mock-transfected cells; n = 6). C) Electrophoretic mobility for wild type, BC033036 (TMEM16A(0)), and truncation mutants. All proteins were tagged and detected using the HA epitope. The bar graph shows the densitometric analysis of TMEM16A protein normalized to actin expression (n = 6). For this analysis, we considered the intensity of the band with the highest molecular weight. The inset on the right shows results from an experiment in which lysates from R56X-transfected cells were treated with/without PNGaseF. D) Normalized TMEM16A activity. Anion transport activity (QR) from HS-YFP experiments was normalized to TMEM16A protein expression.
We carried out a series of control experiments to check the validity of our conclusions. First, we verified that the stop codon introduced at the beginning of our constructs was indeed preventing the translation from upstream sites. To do so, we placed the 3XHA epitope right before the M1X mutation, without other codons in between (Fig. 4A). The expression of this construct (HA-M1X) generated a protein that, as expected, was detected by the anti-TMEM16A (SP31) antibody but not by the anti-HA antibody (Fig. 4A). This finding demonstrates that the results shown in Fig. 3 are not affected by a combination of events that involve: i) start of translation from a codon localized further upstream in the plasmid; ii) readthrough of the stop codon introduced at the beginning of TMEM16A coding sequence. We also checked that the anti-HA antibody was unable to detect the untagged TMEM16A protein (Fig. 4B).
Fig. 4.
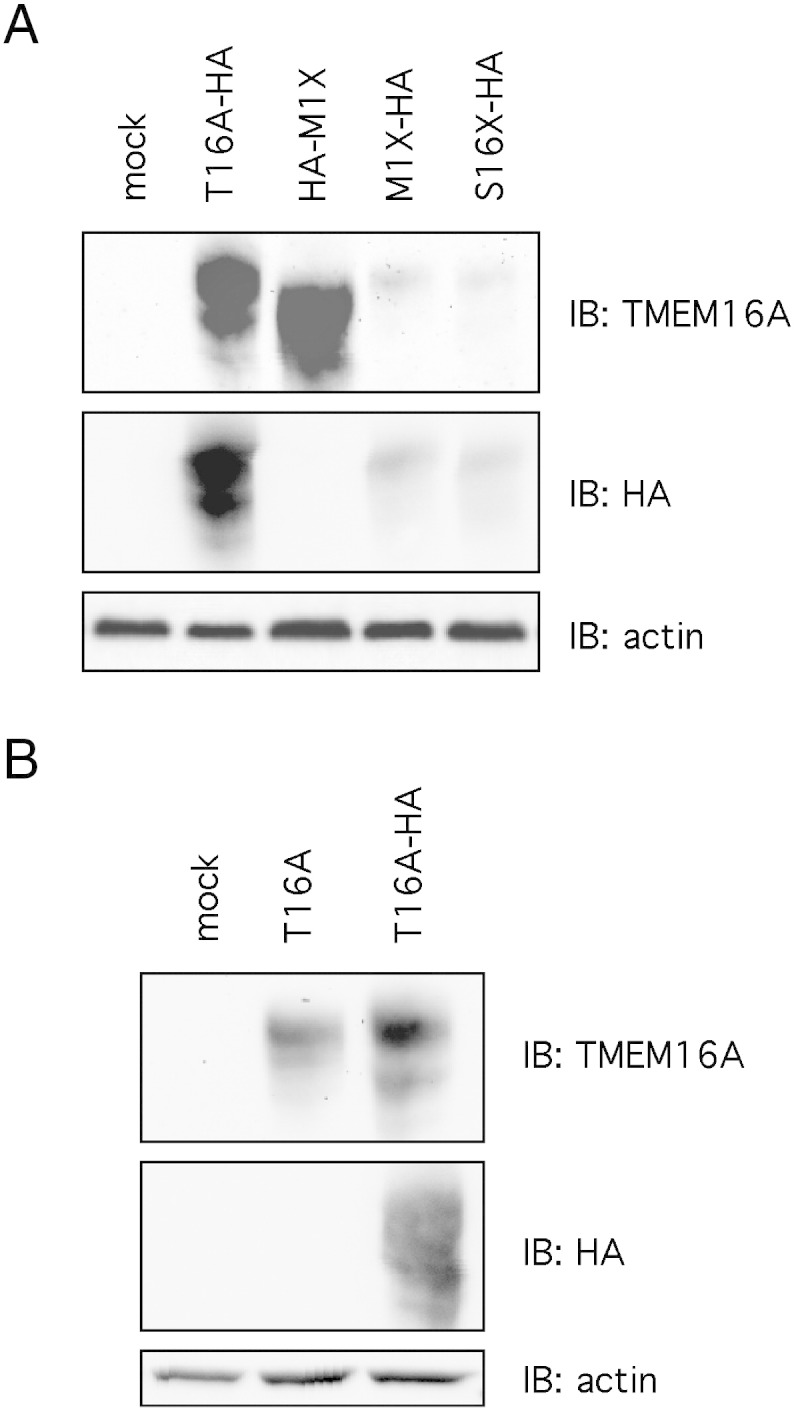
Western blot analysis of TMEM16A protein constructs with anti-TMEM16A and anti-HA antibodies. A) Results show that anti-HA detects the protein when the HA epitope is placed after (M1X-HA, S16X-HA) and not before (HA-M1X) the stop codon. B) The anti-HA antibody does not react with the wild type TMEM16A protein.
Patch-clamp experiments were also carried out to analyze the functional properties of truncated TMEM16A constructs (Fig. 5). The removal of the first ATG with the M1X mutation did not cause a significant decrease in activity. Actually, cells expressing M1X-TMEM16A displayed large CaCC currents indistinguishable from those elicited by wild type TMEM16A (Fig. 5). In particular, with an intracellular Ca2 + concentration of 685 nM, the currents elicited by expression of the wild type and mutant protein were similarly activated when the membrane potential was turned from holding (− 60 mV) to positive values. Large tail currents were observed at the end of the voltage step. Membrane currents with similar aspect, although of smaller amplitude, were also observed with the expression of S16X, L31X, and S33X mutants. We compared the normalized initial current (I0) among the different constructs. This value was significantly higher in L31X, S33X, and R56X relative to wild type protein (Fig. 5B). This result indicates that the fraction of the total current that is activated by membrane depolarization is decreased by progressive truncation of the N-terminus. Regarding kinetics, the time constants of current activation were all similar to each other, as already shown for wild type and M1X (Fig. 2E). Instead, a significant increase (p < 0.05) in the time constant of current deactivation was seen for S16X (139 ± 4 ms), L31X (108 ± 9 ms), and S33X (115 ± 16 ms) compared to wild type TMEM16A (75 ± 6 ms) (not shown). Cells transfected with S74X-TMEM16A were instead devoid of significant CaCC channel activity (Fig. 5A), a result consistent with the results from the HS-YFP assay (Fig. 3B).
Fig. 5.

Functional analysis of TMEM16A truncation mutants. A) Representative membrane currents (left) and current–voltage relationships (right). measured in HEK-293 cells transfected with the indicated constructs. Cytosolic free Ca2 + concentration was 685 nM. B) Normalized initial current (I0) measured at + 100 mV for the various constructs (* and **, p < 0.05 and p < 0.01, respectively, vs. wild type; n = 3–9). C) Ca2 +-dependence of wild type and R56X-TMEM16A. The membrane conductance in each experiment was calculated from the tail current elicited at − 60 mV following the pulse at + 100 mV. The normalized conductance (G/Gmax) was then plotted vs. the cytosolic free Ca2 + concentration. Each point is the mean ± SEM of 5 experiments. Data were fitted with the Hill equation.
We asked whether the decrease in the amplitude of membrane currents found for the mutants with the largest truncation, such as R56X, is due to an alteration in Ca2 + sensitivity. Therefore, we measured currents at various cytosolic Ca2 + concentrations. These experiments showed a modest shift in the Ca2 +-dependence for R56X-TMEM16A compared to the wild type protein. Indeed, the half effective concentrations were 490 and 340 nM, respectively (Fig. 5C). This result indicates that the marked decrease in anion transport caused by truncation of the N-terminus is mainly due to a decrease in protein expression.
We also studied the isoform corresponding to the BC033036 cDNA clone, that we previously named TMEM16A(0). Our previous interpretation was that the protein corresponding to this cDNA clone is devoid of the first 116 amino acids because of the lack of the first ATG codon [1]. However, because of our findings with the Δ(1–116) mutant, we performed experiments in which the BC033036 sequence was tagged with the 3XHA epitope in the same position of the other constructs (between codons 88 and 89; Fig. 3A). Interestingly, in immunoblotting experiments, we detected a band using the anti-HA antibody (Fig. 3C). This clearly indicates that the translation starts from a non-ATG codon also in this construct. At the functional level, we found that the HA-tagged BC033036 is significantly functional although less than the wild type protein. In the HS-YFP assay, the rate of halide transport was 7-fold larger than that of mock-transfected cells (Fig. 3B). In patch-clamp recordings, the expression of BC033036 was associated with significant membrane currents compared to mock-transfected cells (Fig. 5A). Similarly to other mutants with comparable level of truncation, such as L31X and S33X, BC033036 had reduced activation at positive membrane potentials compared to currents elicited by the expression of wild type protein (Fig. 5B). Interestingly, the currents of BC033036 had a more marked voltage-dependent component than previously observed [15].
We evaluated various human tissues looking for the expression of the alternative 5′-end. For this purpose, we used two separate RT-PCRs: one reaction to detect the regular TMEM16A 5′-end and the other reaction for the alternative end. Regular TMEM16A was expressed in lung, prostate, salivary gland, placenta, and, to a lower level, in testis, kidney, liver, skeletal muscle, heart, and thyroid (Fig. 6). In contrast, the alternative 5′-end was expressed only in testis and, more weakly, in lung. The amplification products were sequenced to confirm their identity. Sequencing demonstrated that the BC033036 5′-end was indeed expressed in testis.
Fig. 6.

Tissue expression of TMEM16A with normal and alternative 5′-end. RT-PCR was done on RNA extracted for indicated human organs. The expression of β2-microglobulin (β2m) was also determined for control. Data are representative of three similar experiments.
4. Discussion
TMEM16A is a member of a novel family of membrane proteins which play a variety of physiological functions linked to ion transport. TMEM16 proteins probably share a series of structural features including the overall topology, consisting of eight transmembrane domains. The region between the sixth and the seventh transmembrane domain may be particularly important for the regulation of protein activity by cytosolic Ca2 +. In particular, two glutamic acid residues are probably involved in direct Ca2 + binding, at least in TMEM16A and TMEM16F [12,24]. The first intracellular loop is also involved in the regulation of TMEM16A channel activity by Ca2 + and membrane potential [7,13]. The role of the other protein domains is unclear.
In previous studies, we introduced mutations in the first ATG of the coding sequence in order to force the translation from the second ATG thus skipping the first 116 amino acid residues [15,16]. This intervention resulted in no change or in a relatively minor modification of TMEM16A channel kinetics. Such results indicated that the first part of the N-terminus may be removed without a dramatic loss in channel function. However, as demonstrated in the present study, the previous experiments were affected by the unexpected involvement of non-canonical translation start codons. Indeed, by analyzing the electrophoretic mobility, we have now found that the mutants lacking the first methionine codon have an apparent size comparable to that of the wild protein despite a theoretical loss of nearly 12 kDa. To explain this finding, we initially considered the possibility that the translation of TMEM16A protein actually starts always from the second ATG, at position 117. This would explain the similarity in molecular size and function between wild type and mutant proteins. However, this hypothesis was disproved by removing the entire coding sequence corresponding to the first 116 codons. In this case, the activity was totally lost. This result also indicated that translation does not start from a third downstream methionine codon. Therefore, we considered the possibility of a translation start from a non-ATG codon. This hypothesis was supported by the increasing evidence that translation of proteins may actually start from other codons, in particular from CTG and GTG [19–23]. In a recent study, global analysis of protein translation by ribosome profiling revealed an unexpected large use in cells of alternative codons, particularly of CTG [20]. Therefore, we generated a series of constructs in which we removed the first methionine and progressively shortened the downstream coding sequence. The translation start from a noncanonical codon was demonstrated by using an HA epitope placed before the second methionine. By using this approach, we found that M1X and the other mutant proteins lacking the first methionine were detected by the anti-HA antibody. Furthermore, most of these proteins had a size close to that of the wild protein. Regarding protein expression level, a first drop was observed for mutants M1X and S16X. A second was instead found for L31X and S33X. Therefore, progressive truncation of the N-terminus resulted in a stepwise decrease and not in a sharp loss of expression and function at a single site of protein sequence. These results suggest that more than one alternative start codon may be used. The precise identity of these codons is not certain. However, we postulate two main CTG codons at position 31 and 37. Leucine 31 could be used in the case of mutants M1X and S16X. Leucine 37 could be instead the start codon for mutants L31X and S33X. Other downstream non canonical codons must be used for mutants R56X and S74X. In particular, S74X is still recognized by the anti-HA antibody but the size of the resulting TMEM16A protein appears significantly shortened.
Our findings are relevant to the amino acid composition of the TMEM16A(0) isoform (i.e. BC033036 clone). We previously concluded that, by lacking the first methionine codon, this protein was also devoid of the first 116 amino acids [1]. Instead, our present results indicate that the TMEM16A(0) protein has additional amino acids at the N-terminus. This has been demonstrated by finding that the HA epitope, placed in a site that was considered part of the 5′-untranslated region, was actually included in the protein. As shown in Fig. 1A, the start codon for the TMEM16A(0) isoform could be leucine 37. If so, this isoform would have 80 additional amino acids at the N-terminus, a feature that could be important for the protein function. Indeed, without this region, as in the case of the Δ(1–116) mutant, the protein is totally inactive. Using RT-PCR, we have determined the expression among different human organs of the RNA sequence corresponding to the alternative N-terminus . The skipping of the first two exons appears as an infrequent event, identified only in testis and lung. The particular expression of this alternative form in testis is not surprising for various reasons. Actually, the BC033036 clone, that was initially identified as the one coding for TMEM16A(0) isoform, was derived from a testis cDNA library [1]. In addition, a search using BLAST alignment reveals that many ESTs from testis have the alternative end. This finding is consistent with various reports showing that in testis, for reasons that are still unknown, the expression of many genes is affected by the use of alternative transcription and translation start sites [19,25–28]. This phenomenon may be related to the particular processes associated with sperm cell development and function [29]. Interestingly, human spermatozoa have been shown to possess a Ca2 +-dependent Cl− channel, a kind of activity that could be attributed to TMEM16A expression. Indeed, the use of a TMEM16A inhibitor significantly reduced acrosome reaction in spermatozoa [30].
Our results are also relevant to the findings obtained in patients with diabetic gastroparesis [16]. In these patients, an abnormal TMEM16A transcript was found, characterized by the loss of the first three exons, i.e. Δ1,2,3(5′). The conclusion in the previous study was that the resulting protein would have altered channel gating properties. However, as discussed in the introduction, this conclusion derived from the mutagenesis of the methionines in position 1 and 117 [16]. We now know that the results from these experiments were affected by the presence of alternative translation start sites and that the TMEM16A isoform generated by the skipping of the first three exons is actually totally devoid of activity. This indicates that the function of interstitial cells of Cajal (ICCs) in diabetic patients would be more severely impaired than previously expected. A drastic decrease in TMEM16A function, causing the loss of chloride conductance and excitability of ICCs, would explain the gastroparesis.
Summarizing, our study have several important implications. First, in contrast to previous conclusions, the initial part of the TMEM16A N-terminus has an important role in protein expression and function. Second, TMEM16A is another of the many proteins that are affected by translation from noncanonical start sites. The physiological meaning of this phenomenon is unclear but may be important when alternative promoters are activated as in testis.
Acknowledgements
This work was supported by the Ministero della Salute (Ricerca Finalizzata: GR-2009-1596824; Ricerca Corrente: Cinque per mille), the Telethon Foundation (GGP10026), the Fondazione Italiana Fibrosi Cistica (FFC#2/2012), and the NIH (grant DK57061).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L.J.V. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 2.Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y.D., Cho H., Koo J.Y., Tak M.H., Cho Y., Shim W.S., Park S.P., Lee J., Lee B., Kim B.M., Raouf R., Shin Y.K., Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 4.Davis A.J., Forrest A.S., Jepps T.A., Valencik M.L., Wiwchar M., Singer C.A., Sones W.R., Greenwood A., Leblanc N. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am. J. Physiol. 2010;299:C948–C959. doi: 10.1152/ajpcell.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romanenko V.G., Catalán M.A., Brown D.A., Putzier I., Hartzell H.C., Marmorstein A.D., Gonzalez-Begne M., Rock J.R., Harfe B.D., Melvin J.E. Tmem16A encodes the Ca2 +-activated Cl− channel in mouse submandibular salivary gland acinar cells. J. Biol. Chem. 2010;285:12990–13001. doi: 10.1074/jbc.M109.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian Y., Kongsuphol P., Hug M., Ousingsawat J., Witzgall R., Schreiber R., Kunzelmann K. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J. 2011;25:1058–1068. doi: 10.1096/fj.10-166884. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Q., Yu K., Perez-Cornejo P., Cui Y., Arreol J., Hartzell H.C. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc. Natl. Acad. Sci. U. S. A. 2011;108:8891–8896. doi: 10.1073/pnas.1102147108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta A.K., Khimji A.K., Kresge C., Bugde A., Dougherty M., Esser V., Ueno Y., Glaser S.S., Alpini G., Rockey D.C., Feranchak A.P. Identification and functional characterization of TMEM16A, a Ca2 +-activated Cl− channel activated by extracellular nucleotides, in biliary epithelium. J. Biol. Chem. 2011;286:766–776. doi: 10.1074/jbc.M110.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manoury B., Tamuleviciute A., Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J. Physiol. 2010;588:2305–2314. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ousingsawat J., Martins J.R., Schreiber R., Rock J.R., Harfe B.D., Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2 +-dependent chloride transport. J. Biol. Chem. 2009;284:28698–28703. doi: 10.1074/jbc.M109.012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rock J.R., O'Neal W.K., Gabriel S.E., Randell S.H., Harfe B.D., Boucher R.C., Grubb B.R. Transmembrane protein 16A (TMEM16A) is a Ca2 +-regulated Cl− secretory channel in mouse airways. J. Biol. Chem. 2009;284:14875–14880. doi: 10.1074/jbc.C109.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu K., Duran C., Qu Z., Cui Y.Y., Hartzell H.C. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ. Res. 2012;110:990–999. doi: 10.1161/CIRCRESAHA.112.264440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrera L., Caputo A., Ubby I., Bussani E., Zegarra-Moran O., Ravazzolo R., Pagani F., Galietta L.J.V. Regulation of TMEM16A chloride channel properties by alternative splicing. J. Biol. Chem. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Driscoll K.E., Pipe R.A., Britton F.C. Increased complexity of Tmem16a/Anoctamin 1 transcript alternative splicing. BMC Mol. Biol. 2011;12:35. doi: 10.1186/1471-2199-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrera L., Scudieri P., Sondo E., Caputo A., Caci E., Zegarra-Moran O., Ravazzolo R., Galietta L.J.V. A minimal isoform of the TMEM16A protein associated with chloride channel activity. Biochim. Biophys. Acta. 2011;1808:2214–2223. doi: 10.1016/j.bbamem.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazzone A., Bernard C.E., Strege P.R., Beyder A., Galietta L.J.V., Pasricha P.J., Rae J.L., Parkman H.P., Linden D.R., Szurszewski J.H., Ördög T., Gibbons S.J., Farrugia G. Altered expression of Ano1 variants in human diabetic gastroparesis. J. Biol. Chem. 2011;286:13393–13403. doi: 10.1074/jbc.M110.196089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galietta L.J.V., Haggie P.M., Verkman A.S. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499:220–224. doi: 10.1016/s0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- 18.Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerashchenko M.V., Su D., Gladyshev V.N. CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J. Biol. Chem. 2010;285:4595–4602. doi: 10.1074/jbc.M109.070532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov I.P., Firth A.E., Michel A.M., Atkins J.F., Baranov P.V. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 2011;39:4220–4234. doi: 10.1093/nar/gkr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kochetov A.V. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays. 2008;30:683–691. doi: 10.1002/bies.20771. [DOI] [PubMed] [Google Scholar]
- 23.Starck S.R., Jiang V., Pavon-Eternod M., Prasad S., McCarthy B., Pan T., Shastri N. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science. 2012;336:1719–1723. doi: 10.1126/science.1220270. [DOI] [PubMed] [Google Scholar]
- 24.Yang H., Kim A., David T., Palmer D., Jin T., Tien J., Huang F., Cheng T., Coughlin S.R., Jan Y.N., Jan L.Y. TMEM16F forms a Ca2 +-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151:111–122. doi: 10.1016/j.cell.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleene K.C. Patterns, mechanisms, and functions of translation regulation in mammalian spermatogenic cells. Cytogenet. Genome Res. 2003;103:217–224. doi: 10.1159/000076807. [DOI] [PubMed] [Google Scholar]
- 26.Kleene K.C., Cataldo L., Mastrangelo M.A., Tagne J.B. Alternative patterns of transcription and translation of the ribosomal protein L32 mRNA in somatic and spermatogenic cells in mice. Exp. Cell Res. 2003;291:101–110. doi: 10.1016/s0014-4827(03)00339-2. [DOI] [PubMed] [Google Scholar]
- 27.Rauschendorf M.A., Zimmer J., Hanstein R., Dickemann C., Vogt P.H. Complex transcriptional control of the AZFa gene DDX3Y in human testis. Int. J. Androl. 2011;34:84–96. doi: 10.1111/j.1365-2605.2010.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasta V., Sonnenburg W.K., Yan C., Soderling S.H., Shimizu-Albergine M., Beavo J.A. Identification of a new variant of PDE1A calmodulin-stimulated cyclic nucleotide phosphodiesterase expressed in mouse sperm. Biol. Reprod. 2005;73:598–609. doi: 10.1095/biolreprod.104.039180. [DOI] [PubMed] [Google Scholar]
- 29.Eddy E.M. Male germ cell gene expression. Recent Prog. Horm. Res. 2002;57:103–128. doi: 10.1210/rp.57.1.103. [DOI] [PubMed] [Google Scholar]
- 30.Orta G., Ferreira G., José O., Treviño C.L., Beltrán C., Darszon A. Human spermatozoa possess a calcium-dependent chloride channel that may participate in the acrosomal reaction. J. Physiol. 2012;590:2659–2675. doi: 10.1113/jphysiol.2011.224485. [DOI] [PMC free article] [PubMed] [Google Scholar]