

Abstract
Background
Birch bark has a long lasting history as a traditional medicinal remedy to accelerate wound healing. Recently, the efficacy of birch bark preparations has also been proven clinically. As active principle pentacyclic triterpenes are generally accepted. Here, we report a comprehensive study on the underlying molecular mechanisms of the wound healing properties of a well-defined birch bark preparation named as TE (triterpene extract) as well as the isolated single triterpenes in human primary keratinocytes and porcine ex-vivo wound healing models.
Methodology/Principal Findings
We show positive wound healing effects of TE and betulin in scratch assay experiments with primary human keratinocytes and in a porcine ex-vivo wound healing model (WHM). Mechanistical studies elucidate that TE and betulin transiently upregulate pro-inflammatory cytokines, chemokines and cyclooxygenase-2 on gene and protein level. For COX-2 and IL-6 this increase of mRNA is due to an mRNA stabilizing effect of TE and betulin, a process in which p38 MAPK and HuR are involved. TE promotes keratinocyte migration, putatively by increasing the formation of actin filopodia, lamellipodia and stress fibers. Detailed analyses show that the TE components betulin, lupeol and erythrodiol exert this effect even in nanomolar concentrations. Targeting the actin cytoskeleton is dependent on the activation of Rho GTPases.
Conclusion/Significance
Our results provide insights to understand the molecular mechanism of the clinically proven wound healing effect of birch bark. TE and betulin address the inflammatory phase of wound healing by transient up-regulation of several pro-inflammatory mediators. Further, they enhance migration of keratinocytes, which is essential in the second phase of wound healing. Our results, together with the clinically proven efficacy, identify birch bark as the first medical plant with a high potential to improve wound healing, a field which urgently needs effective remedies.
Introduction
The skin primarily functions as a protective barrier against the environment. Therefore, loss of its integrity immediately results in starting complex processes to restore the epidermal barrier function. The repair of wounds is an extremely complex biological process basically divided into three overlapping phases: inflammation, new tissue formation, and remodeling [1]–[4]. The first stage is characterized by hemostasis and by initiating a controlled inflammatory response. Release of various pro-inflammatory cytokines including chemokines and growth factors orchestrates the attraction of macrophages and granulocytes to the wound area to remove dead tissue and to prevent bacterial infection [5]–[8]. The second stage includes migration and proliferation of keratinocytes ( = reepithelialization) and fibroblasts ( = formation of granulation tissue and extracellular matrix) as well as angiogenesis. During the third stage old collagen is replaced, apoptosis takes place and a scar is formed. Hence wound healing requires the integration of many complex cellular and molecular events which can be targeted at many points, leading either to accelerated or delayed healing, the latter can lead to chronic wounds [4], [9], [10].
Besides of the conventional remedies, phytomedicines turned out to be an interesting alternative or addendum to beneficially influence the different stages of wound healing. In this context, extracts from birch bark (Betula alba, syn. B. pendula, Betulaceae family) have gained more and more interest. Birch bark has a long lasting history as a traditional medicinal remedy known already by the North American Indians who wrapped their wounds with birch bark to accelerate wound healing [11]. Recently, efficacy of birch bark preparations has also been proven clinically. A case report described the successful treatment of necrotizing herpes zoster when using a birch bark emulsion [12]. Moreover, birch bark extract was found to be effective in the treatment of two patients suffering from a second degree burning [13]. Notably, an open, blind-evaluated, controlled, prospective, randomized phase II clinical trial including 24 patients revealed that a birch bark preparation significantly accelerated reepithelialization in split thickness skin graft donor sites [14]. All these studies were conducted with a preparation containing an n-heptane dry extract from the outer bark of birch (TE: triterpene extract) [15]. About 97% of the extract is composed of pentacyclic triterpenes with about 87% of betulin as main compound accompanied by minor amounts of lupeol, betulinic acid, oleanolic acid and erythrodiol (for structures see Fig. S1B). Hence these pentacyclic triterpenes are considered as the mainly effective constituents of birch bark. The individual triterpenes themselves have not yet been evaluated for their wound healing efficacy.
Given the positive effect of TE in in-vivo wound healing we wanted to elucidate the underlying molecular mechanisms of its wound healing properties as well as of the isolated single triterpenes (betulin, lupeol, betulinic acid, oleanolic acid and erythrodiol). We demonstrate that TE and betulin influence the inflammatory phase of wound healing by upregulating pro-inflammatory cytokines, chemokines and cyclooxygenase-2 (COX-2) in human primary keratinocytes. Exemplarily, we confirm upregulation in the ex-vivo pig wound healing model for IL-6 and COX-2. We provide evidence for COX-2 and IL-6 that their mRNA increase is due to an mRNA stabilizing effect, a process in which p38 MAPK and HuR (human antigen R) are essentially involved. We demonstrate that TE, betulin, lupeol and erythrodiol increase the formation of actin filopodia, lamellipodia and stress fibers, processes that are dependent on the activation of Rho GTPases. Finally, we show in the porcine ex-vivo model that TE improves epidermal regeneration and accelerates the repair of the epidermal barrier function.
Results
TE and betulin exhibits wound healing effects in a porcine ex-vivo wound healing model (WHM)
The efficacy of a birch bark preparation (TE-oleogel) has already been proven in humans [14], but the influence of the vehicle control or the main constituent, the triterpene betulin, was not studied separately. Therefore, we systematically investigated the wound healing progress in a porcine ex-vivo wound healing model (WHM) using an oleogel with the same composition as used in [14] (10% TE, 90% sunflower oil) compared to sunflower oil alone and sunflower oil in ethylcellulose to mimic the higher viscosity of oleogel compared to oil. We found a significant acceleration of reepithelialization with TE-oleogel compared to the controls after 48 h (Fig. 1A). There was also a significant improvement compared to vaseline (data not shown). Additionally, we analyzed whether the use of TE dissolved in PBS also resulted in a beneficial effect on wound healing in the WHM. This approach allowed us to have a rational basis for our further experiments on the elucidation of the underlying molecular mechanisms of the wound healing properties of TE, because these studies were performed with primary human keratinocytes in culture where the oleogels or the oils can not be used. Indeed, we could show significantly accelerated wound healing with 10 μg/ml TE in PBS compared to PBS alone 48 h after wounding (Fig. 1B). We also observed a beneficial effect with betulin (8.69 μg/mL), which was studied in a concentration as it occurs in 10 μg/ml TE, but the effect was less than that one of TE and statistically not significant. Therefore, it can be assumed that betulin, the main constituent of TE, is not exclusively responsible for the effect observed (Fig. 1B).
Figure 1. Wound healing progress in WHM.

Effect of TE and betulin on reepithelialization in the porcine ex-vivo WHM. (A) Reepithelialization in WHM treated with 10% TE in sunflower oil (oleogel) compared to sunflower oil alone, sunflower oil with ethylcellulose and untreated control 48 h after wounding. (B) Reepithelialization in WHM treated with 10 µg/ml TE and betulin in the concentration as it occurs in 10 µg/ml TE in PBS compared to PBS 48 h after wounding. Reepithelialization of the various models was normalized to untreated control (A) or PBS (B), respectively. Mean ± SEM. (C) Effect of TE and betulin in PBS on barrier regeneration in WHM normalized to barrier regeneration in WHM treated with PBS alone. WHM were treated 72 h after wounding for 24 h. Mean ± SEM. A: n = 7; B: n = 5–7; C: n = 4–5. *p<0.05 and ***p<0.001.
TE promotes formation of the skin barrier
A major goal of wound healing is the restoration of the skin barrier to protect the body from the invasion of pathogens. Therefore, we investigated whether 10 μg/ml TE and 8.69 μg/mL betulin, respectively, improve skin barrier function by using a dye penetration assay in our ex-vivo WHM (Fig. 1C). Application of TE and betulin 72 h after wounding for 24 h resulted in an improved skin barrier function. This was the same when TE was used directly after wounding for 4 days (data not shown).
TE and betulin increase mRNA of proinflammatory mediators in primary human keratinocytes
Disruption of the epidermal barrier induces the release of various pro-inflammatory mediators, such as cytokines, enzymes or growth factors from keratinocytes and platelets [2], [5], [6]. Both events promote the recruitment of granulocytes and macrophages to the site of injury. These cells augment the inflammatory phase by induction of further proinflammatory mediators [2], [16]. In this context, COX-2, IL-6 and IL-8 have been shown to be upregulated and to play crucial roles in reepithelialization and angiogenesis [17]–[20]. To evaluate whether this initiating phase can be accelerated by increasing the amount of these pro-inflammatory mediators, primary human keratinocytes were treated with two different concentrations of TE (1 and 5 μg/mL) and its main triterpenes, betulin, lupeol, and betulinic acid, respectively, and their impact on mRNA expression at different time points was evaluated. The triterpenes were studied in concentrations in which they occur in 5 μg/mL of the studied extract. mRNA of COX-2 significantly increased after an 8 h treatment with 5 μg/mL TE (6.3-fold ±1.2) and 4.34 μg/mL ( = 9.81 μM) betulin (Bet, 3.5-fold ±0.4) (Fig. 2A). Treatment with 1 μg/mL of TE resulted in lower levels of COX-2 and were significant after 24 h (3.4-fold ±0.3). The effect on IL-6 mRNA expression was much more pronounced with a fold increase of 172.4±83.3 for TE (5 μg/mL) and of 84.3±29.9 for betulin (4.34 μg/mL) after 8 h (Fig. 2B). Again, the effect was dose-dependent, i.e. TE in the concentration of 1 μg/mL caused lower effects (12.7-fold ±4.9 after 8 h), which were time dependently upregulated (82.1-fold ±26.8 for 12 h, 181.9-fold ±22.9 for 24 h) and significant after 24 h. Studies on IL-8 mRNA expression resulted in a 57.1±8.3 fold increase at a concentration of 5 μg/mL TE and of 37.6±12.6 at 4.34 μg/mL of betulin after 8 h. The lower concentration of 1 μg/mL TE gave less high levels of IL-8 mRNA with significance at 24 h (fold increase of 33.7±8.6) (Fig. 2). The triterpenes lupeol and betulinic acid exhibited no effects on the mRNA of these three mediators.
Figure 2. Birch bark (TE) and betulin differently influence mRNA of COX-2, IL-6 and IL-8 in human primary keratinocytes.

COX-2 (A), IL-6 (B) and IL-8 (C) mRNA are upregulated in response to birch bark (TE) and betulin (Bet), but not to lupeol (Lup) and betulinic acid (BA). Time course of mRNA expression in response to TE (1 and 5 µg/mL) measured by qRT-PCR. The isolated triterpenes (Bet, Lup, BA) were measured in that concentration in which they occur in 5 µg/mL TE extract. Values represent means of at least three independent experiments ± SEM. *p<0.05, **p<0.01, ***p<0.001 versus control (DMSO).
TE and betulin differently influence mRNA of further mediators in human primary keratinocytes
Subsequently, the influence of TE on further molecules involved in the wound healing process was studied. Whereas mRNA levels of IL-1β remained unchanged when primary keratinocytes were treated with 1 μg/mL TE for 4, 6, 8, 12 and 24 h (Fig. S2A), levels of TNF-α mRNA increased significantly at 12 h and decreased again at 24 h (Fig. S2B). As mentioned above, these cytokines are important for initiating the wound healing process. Transforming growth factor-β (TGF-β) was significantly affected by TE resulting in an increase of about 2.6-fold ±0.4 at 8 h (Fig. S2C). This mediator regulates the recruitment of inflammatory cells and macrophages and the formation of granulation tissue by increasing the expression of genes associated with extracellular matrix formation [2], [3]. The effect of TE and betulin on mRNA of the transcription factor NF-E2-related factor 2 (Nrf2), the antimicrobial peptide human beta-defensin 3 (hBD3) as well as the matrix metalloproteinases (MMPs) MMP-2 and MMP-9 were not significant (Fig. S2).
TE increases mRNA levels of IL-6 and COX-2 in the WHM
To control whether upregulation of pro-inflammatory mediators found in primary keratinocytes can also be demonstrated in a 3D wound, we exemplarily determined the effect of TE on mRNA level of IL-6 and COX-2 in the porcine ex-vivo wound healing model. After treatment with 10 µg/mL TE for 6 and 48 h, mRNA was isolated and quantified by qRT-PCR. A 10 time higher concentration was used to consider the different condition concerning bioavailability in the ex-vivo model and also to be in line with the wound healing experiments (Fig 1B). An increase of IL-6 and COX-2 mRNA 6 h after wounding, which significantly decreased after 48 h was observed (Fig. 3).
Figure 3. TE upregulates mRNA of IL-6 and COX-2 in an ex-vivo wound healing model 6 h after wounding and treatment with 10 µg/ml TE, after 48 h the levels decreased to normal levels.
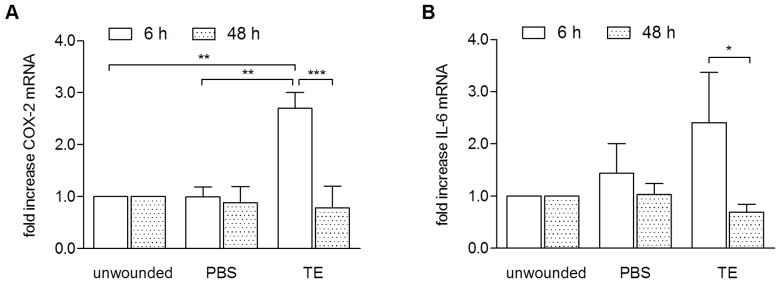
Values represent means of at least three independent experiments ± SEM. *p<0.05, **p<0.01, ***p<0.001 as indicated.
TE and betulin increase protein levels of various pro-inflammatory mediators
To confirm enhanced levels of pro-inflammatory mediators induced by TE and betulin also on the protein level, firstly the effect on IL-6 and IL-8 release was studied in human primary keratinocytes by ELISA. Supernatants were collected after 24 h and 48 h of incubation with either TE (1 and 5 μg/mL) or betulin (0.87 μg/mL). In both cases enhanced levels of IL-6 and IL-8 were measured compared to control. For IL-6 release highest levels were obtained with 5 μg/mL TE after 48 h (67±34 pg/mL) and with 0.87 μg/mL betulin (51±27 pg/mL) after 24 h (Fig. 4A). IL-8 release was concentration dependent with TE (maximum level of 350±128 pg/mL with 5 μg/mL and 48 h). Lupeol did not influence IL-6 or IL-8 level (data not shown). Protein levels of COX-2 after TE and betulin treatment were determined by Western blot analysis (Fig. 4C). TE and betulin led to an increase in COX-2 protein after 24 h for both tested concentrations (1 and 5 μg/mL TE and 0.87 and 4.34 μg/mL betulin, respectively).
Figure 4. TE and betulin (Bet) enhance release of IL-6 (A) and IL-8 (B), as well as formation of COX-2 protein (C) in primary human keratinocytes.

Measurement of IL-6 (A) and IL-8 (B) release in response to TE (1 and 5 µg/mL) and betulin (0.87 µg/mL, which is in 1 µg/mL TE) measured by ELISA. Values represent means of at least three independent experiments ± s.d. (*p<0.05, **p<0.01 and ***p<0.001 versus control). (C) COX-2 protein was measured by Western blot analysis after 24 h treatment with TE (1 and 5 µg/mL) and the respective betulin concentration (0.87 and 4.34 µg/mL). DMSO (0.1%, v/v) served as a solvent control. A representative Western blot is shown, n = 3.
To confirm the data mentioned above with a different method and to gain a more comprehensive overview on further mediators being important in the inflammatory wound healing phase, a Bio-Plex® Cytokine assay was performed. This assay allows the simultaneous measurement of released pro-inflammatory cytokines and growth factors. Only one concentration of TE (1 μg/mL), betulin (0.87 μg/mL = 1.96 μM) and lupeol (0.04 μg/mL = 0.09 μM) was tested at 24 h, conditions proven to be suitable from the previous experiments. Altogether, the effect on 27 different mediators including pro-inflammatory [IL-1β, IL-6, interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α)] and anti-inflammatory [interleukin-1 receptor antagonist (IL-1ra), IL-4, IL-10, IL-13] cytokines, cytokines with various functions (IL-2, IL-5, IL-7, IL-9, IL-12, IL-15, IL-17), chemokines [IL-8, eotaxin, IFN-γ-induced protein-10 (IP-10), monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1α (MIP-1α), MIP-1β, regulated upon activation, normal T-cell expressed, and secreted (RANTES)] and growth factors [basic fibroblast growth factor (basic FGF), granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), platelet derived growth factor-bb (PDGF-bb), vascular endothelial growth factor (VEGF)] was evaluated (Fig. 5 and S3). We could confirm the increase of IL-6 and IL-8 already described above. Significant upregulating effects induced by TE and betulin were also observed for INF-γ, TNF-α, IP-10, MIP-1α and β, RANTES, and basic FGF with IP-10 being released to the highest amount (Fig. 5). The effect on the production of other cytokines and growth factors was not significant (Fig. 5) or negligible (Fig. S3). Lupeol, which was studied at a concentration of 0.04 μg/mL (as it occurs in 1 μg/mL TE), had no effect on the production of these mediators (data not shown).
Figure 5. TE and betulin enhance the production of various cytokines, chemokines and growth factors in human keratinocytes after 24 h of incubation.

Cells were treated either with TE (1 µg/mL) or betulin (0.87 µg/mL, which is in 1 µg/mL TE) for 24 h. Protein levels of the indicated mediators were determined in the supernatant with the Bio-Plex® Cytokine Assay. Values represent means of two independent experiments ± s.d. *p<0.05, **p<0.01 and ***p<0.001 versus control.
TE and betulin have no influence on NF-κB DNA binding, but on mRNA stability
The transcription factor nuclear factor κB (NF-κB) was shown to be involved in the transcriptional regulation of many inflammatory cytokines and chemokines [21]. To elucidate the putative mechanism for elevated amounts of mediators on the gene and protein level by TE and its main ingredient betulin, we firstly studied the impact on NF-κB-DNA binding in an electrophoretic mobility shift assay (EMSA). However, no increased NF-κB DNA binding activity was observed when human primary keratinocytes were incubated either with 1 µg/mL TE or 0.87 µg/mL betulin ( = 1.96 µM) for different time points in contrast to TNF-α which served as positive control (Fig. S4). Betulinic acid (0.04 µg/mL = 0.08 µM) and lupeol (0.04 µg/mL = 0.09 µM) were also inactive under these conditions (data not shown).
Increased mRNA levels of cytokines, chemokines and other pro-inflammatory mediators may also be due to increased stability of mRNA as it has been shown for IL-6, COX-2 and IL-8 [22]–[24]. We therefore exemplarily studied the effect of TE and betulin on IL-6 and COX-2 mRNA stabilization. At first the half-life time of IL-6 and COX-2 mRNA were measured in the presence of actinomycin D (ActD). Figure 6 shows the decay of COX-2 (A) and IL-6 mRNA (C) over time and a half-life time of 74.4 min for COX-2 and 66.0 min for IL-6 calculated by one phase decay non linear regression (Fig. 6E). Treatment of human keratinocytes with TE (1 µg/mL) and betulin (0.87 µg/mL) for 24 h prolongs the half-life time of COX-2 mRNA to 226.9 min by TE and 196.5 min by betulin (Fig. 6A and B). mRNA of IL-6 was also stabilized and resulted in a T1/2 of 157.7 min for TE and 260.3 min for betulin (Fig. 6C, D and E).
Figure 6. TE and betulin increase the mRNA half-life time of COX-2 (A, B) and IL-6 (C, D) by modulating RNA stability involving p38 MAPK.

Primary human keratinocytes were treated either with TE (1 µg/mL) (A, C) or betulin (0.87 µg/mL) (B, D) for 24 h or left untreated followed by ActD (5 µg/mL) for the indicated times with or without the p38 MAPK inhibitor LN950 (100 nM). COX-2 and IL-6 mRNA levels were quantified by qRT-PCR. Results are expressed as % of initial (0 h) mRNA, decay curves are applied and RNA half-life times were calculated (E). Values represent means of at least three independent experiments ± s.d. (F) TE and betulin lead to increased phospho-p38 MAPK levels in Western blot. Primary human keratinocytes were treated either with TE (1 and 5 µg/mL) or betulin (0.87 and 4.34 µg/mL, which is in 1 and 5 µg/mL TE, respectively) for 0.5 and 1 h. IL-1β (20 ng/mL) was used as a positive control. Phosphorylated p38 MAPK (p-p38 MAPK) and total p38 MAPK levels were analyzed. The result of the Western blot was reproduced and one representative Western blot is shown.
p38 MAPK and HuR are involved in the mRNA stabilizing effect of TE and betulin
Since p38 MAPK is known to be primarily involved in COX-2 and IL-6 mRNA stability [24]–[26] we next studied the potential involvement of p38 MAPK in TE and betulin enhanced mRNA stability. Therefore, human primary keratinocytes were treated with TE or betulin and subsequently with the p38 MAPK inhibitor LN950 and ActD and the half-life times for COX-2 and IL-6 mRNA were determined, respectively. Combined treatment of p38 MAPK inhibitor and TE substantially decreased T1/2 of COX-2 mRNA to 100.7 min, even though not to the level of the control. Combination of p38 MAPK inhibitor with betulin even led to a lower T1/2 as compared to the control (51.6 min). Concerning IL-6, the mRNA half-life time was strongly reduced in the presence of TE and betulin after addition of the p38 MAPK inhibitor. For TE, this resulted in a half-life time lower than the control (31.4 min compared to 66.0 min), whereas the combination with betulin led to a slightly higher T1/2 of 70.3 (Fig. 6A–E).
Altogether, these results clearly show that p38 MAPK is involved in TE and betulin-induced COX-2 and IL-6 mRNA stability. These data are supported by increased cellular levels of activated p38 MAPK ( = p-p38) (Fig. 6F) after treatment with TE (1 and 5 µg/mL) and betulin (0.87 and 4.34 µg/mL).
Subsequently, the effect on HuR, a mRNA stabilizing factor which is known to take part in the posttranscriptional regulation of genes with AU-rich elements (ARE) present in their 3′-untranslated regions [27], was studied. The genes for COX-2 and IL-6 also have this feature and a participation of HuR in COX-2 mRNA stabilization was previously demonstrated [28]. Therefore, the influence on HuR translocation from the nucleus into the cytosol, where it exerts its stabilizing function, was exemplarily assessed with TE by Western blotting. Figure 7A shows that 1 µg/mL TE increased cytosolic localization of HuR. IL-1β (20 ng/mL) was used as control due to its influence on cytoplasmic HuR [29]. Whereas levels of nuclear HuR seem to remain unchanged, total HuR was slightly upregulated after IL-1β (3 h) and TE (1 h). To explain the missing decrease of nuclear HuR levels, qRT-PCR was performed to study whether TE upregulated HuR gene expression. Indeed, TE rapidly enhanced mRNA of HuR after 10 min, which rapidly decreased after 20 min (Fig. 7B). However, the fold increase was low, so that it is questionable that the missing change in the nuclear HuR levels can be solely explained by the new synthesis of HuR. It has to be considered that the change in cytosolic HuR levels may be too small to have an impact on nuclear levels.
Figure 7. TE increases the amount of cytosolic HuR determined by Western blot analysis.
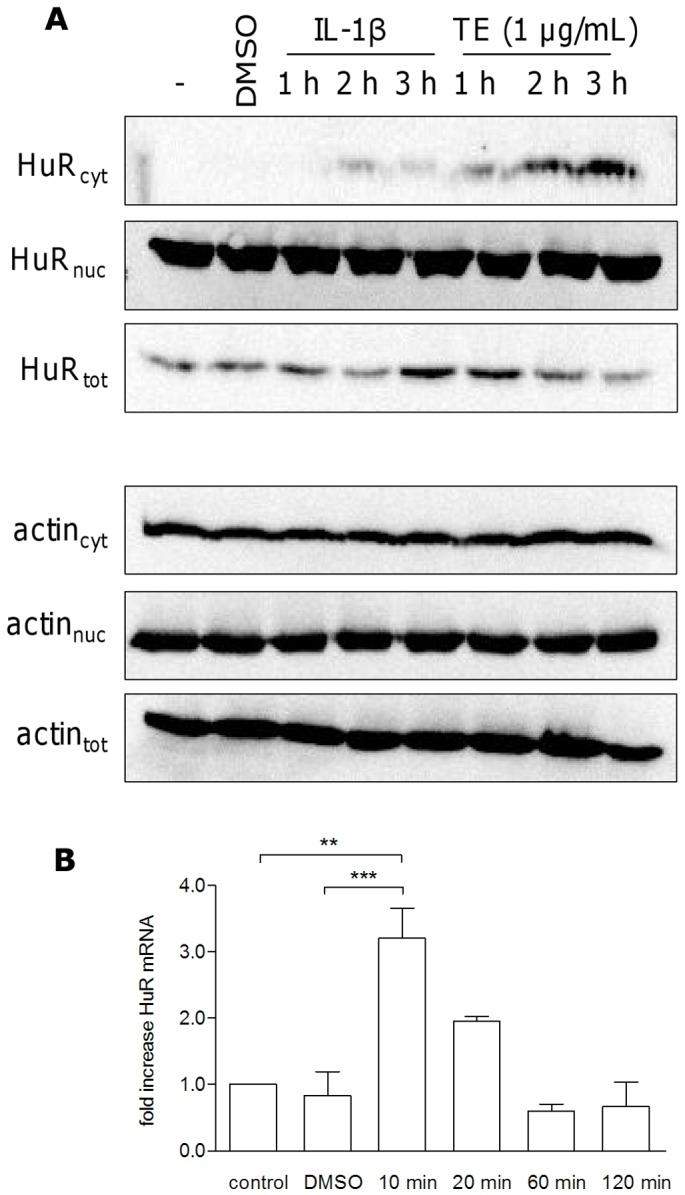
(A). Treatment of human primary keratinocytes with 1 µg/mL TE for 1 to 3 h showed a time dependent increase in cytosolic HuR. IL-1β (20 ng/mL) was used as a positive control, the hyphen indicates untreated cells. Actin was used as a loading control. The result of the Western blot was reproduced and one representative Western blot is shown. (B) TE increases HuR mRNA analysed by qRT-PCR. Primary human keratinocytes were treated with TE (1 µg/mL) for various times. Values represent means of at least two independent experiments ± SEM. **p<0.01 and ***p<0.001 versus control or DMSO.
TE activates the transcription factor STAT3
As mentioned above, treatment of human primary keratinocytes with TE increases IL-6 production (Fig. 4 and 5). This cytokine is an important activator of the transcription factor signal transducer and activator of transcription (STAT3) which plays an essential role in the proliferation and migration of keratinocytes [30]. To study the effect of TE- induced IL-6 on STAT3 activation a Western Blot was performed after different time points. Increase of STAT3 phosphorylation was observed after 12 h and 16 h (Fig. 8).
Figure 8. TE (1 µg/mL) induces phosphorylation of the transcription factor STAT3 ( = pSTAT3).
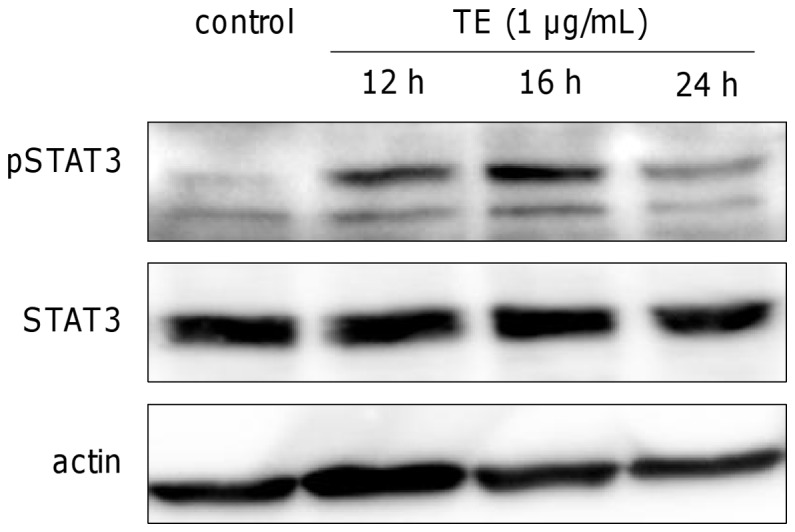
The amount of total STAT3 and actin served as loading controls. The result of the Western blot was reproduced and one representative Western blot is shown.
TE and betulin do not increase proliferation in primary human keratinocytes and in the WHM
Because of the putative effect of STAT3 on proliferation, we investigated the effect of 1 µg/mL and 5 µg/mL TE, as well as the respective concentrations of betulin, on proliferation of primary human keratinocytes. We did not observe any proliferative effect with TE, betulin or with lupeol and betulinic acid, which were also included in the study (Fig. S5A). This was also confirmed in the WHM (Fig. S5B). There was no significant influence on proliferation in all parts of the WHM even though there was a trend of decreased number of proliferative cells after treatment with 10 µg/mL TE and betulin (8.69 µg/mL).
TE enhances migration of primary human keratinocytes in the scratch assay
Reepithelialization comprises proliferation and migration. We observed increased reepithelialization in the TE-treated WHM (Fig. 1) but no proliferative effects (Fig. S5). Hence we studied the impact on migration in scratch assay experiments with primary human keratinocytes using ibidi® culture inserts. Cells were treated with 1 µg/mL TE and 10 ng/mL hepatocyte growth factor (HGF), as a positive control [31] and microscopic images were taken immediately after wounding (t = 0 h) and during an incubation time of up to 24 h. Both stimuli showed a significant increase of closure of the scratch (64% ±15 for HGF and 52% ±24 for TE) compared to untreated cells (31%±17) 8 h after wounding (Fig 9A and B).
Figure 9. TE enhances migration of primary human keratinocytes in the scratch assay.

Cells were incubated with 1 µg/mL TE, 10 ng/mL HGF or treated with DMSO, as control. Images were taken immediately after wounding (0 h) and after 8 h incubation (A). The scale bar is of 100 µm width. Representative images of repeated experiments are shown. (B) Closed area in % after 8 h (1 µg/mL TE, 10 ng/mL HGF or untreated control) compared to the scratch area at time point zero (0 h). Values represent mean of closed areas of 4 independent experiments ± s.d. ***p<0.001 versus control.
TE and its triterpenes affect the actin cytoskeleton
The reorganization of the actin cytoskeleton in terms of controlled polymerization and depolymerization is involved in cell motility. It provides a driving force for migration of cells, such as keratinocytes, at the wound edge, which is necessary for wound closure [32]. Filopodia, lamellipodia and stress fibers are different kinds of actin structures, which contribute to cell migration.
To study whether TE and its main triterpenes influence the actin cytoskeleton, primary human keratinocytes were treated with different concentrations of TE and the respective isolated triterpenes for two hours and stained with fluorophore labelled phalloidin. Subconfluent cells were used, as they phenotypically and biochemically resemble the active keratinocytes from the basal layer at the wound margin migrating on the newly formed granulation tissue until recovering the injured skin [33], [34]. Treatment with TE at concentrations of 0.5 and 1 µg/mL induced formation of stress fibers (S), filopodia and lamellipodia (F) (Fig. S6). Interestingly, even lower concentrations of TE (0.51, 5.1 and 51 ng/mL) induced the formation of filopodia and lamellipodia (F) as well as stress fibers (S) to a greater extent compared to the higher concentrations (Fig. 10). Long, subtle and spiky filopodia and a very dense, stripe-like actin meshwork (lamellipodia) were observed at the leading edge of the cell compared to the solvent control DMSO, where only few short filopodia were visible. As positive control for the formation of filopodia and lamellipodia, cytotoxic necrotizing factor 1 from Escherichia coli (CNF1) was used which directly activates Rac and Cdc42. For the induction of stress fibers, CNFY (cytotoxic necrotizing factor from Yersinia pseudertuberculosis) was applied which selectively activates RhoA [35].
Figure 10. TE, betulin and lupeol influence the actin cytoskeleton of primary human keratinocytes.

Cells were incubated on glass coverslips for 2/mL TE and 1, 10 and 100 nM betulin and lupeol, respectively. The actin cytoskeleton was stained with phalloidin-rhodamine. 0.1% (v/v) DMSO was used as solvent control and CNF1 and CNFY as positive controls. Rows labeled with F show the impact on filopodia and lamellipodia and S the impact on stress fiber formation. A white arrow indicates the leading edge of the cell. Representative pictures of repeated experiments (n = 5) are shown.
The single triterpenes of birch bark extract behaved differently. Betulin was tested at concentrations of 1, 10 and 100 nM, corresponding to the amount of this triterpene in 0.51, 5.1 and 51 ng/mL of TE. Lupeol, betulinic acid, oleanolic acid and erythrodiol were tested at concentrations of 1, 10 and 100 nM, due to comparability reasons to betulin. Betulin and lupeol strongly polarized the cells by formation of broad lamellipodia and spiky filopodia at the leading edge (indicated by white arrows) even at the lowest nanomolar concentration (Fig. 10). Stress fiber assembly within the cell body, particularly at the lowest concentration (1 nM), was also increased by both triterpenes. No effect could be observed for betulinic acid and oleanolic acid at any tested concentrations, whereas erythrodiol (1–100 nM) increased the formation of stress fibers, filopodia and lamellipodia compared to the solvent control DMSO (see Fig. S6).
TE, betulin and lupeol activate Rho GTPases involved in regulation of the actin cytoskeleton
Members of the Rho family of GTPases, especially RhoA, Rac1 and Cdc42 are essentially involved in the regulation of the actin cytoskeleton [36]–[39]. Activation of Cdc42 polarizes the cell [40], leads to the formation of filopodia [41] and also affects lamellipodia [42], whereas activation of Rac results in lamellipodia extension [37], [42], [43], which is the driving force of cell migration. The formation of actin-myosin containing stress fibers, which contract the cell body, as well as the stress-fiber associated focal adhesions, the anchorage point to the substrate, is finally mediated by RhoA [43].
Due to the strong effects of TE, betulin and lupeol on different cytoskeletal actin structures (Fig. 10 and S6) we examined their influence on the above mentioned Rho GTPases using exemplarily 5.1 ng/mL of TE, 10 nM betulin (corresponding concentration in 5.1 ng/mL TE), and 10 nM lupeol. Figure 11A shows the effect of TE, betulin and lupeol on RhoA in primary human keratinocytes assessed by pulldown experiments and subsequent Western blot analysis. The active form of RhoA, GTP-RhoA, was precipitated with rhotekin-loaded beads after 3 h of incubation with the respective compounds. TE, betulin and lupeol, were able to slightly activate RhoA. Cytotoxic necrotizing factor from Escherichia coli (CNFY) (300 nM), which constitutively activates RhoA by deamination of glutamine 63, that arrests RhoA in the active, GTP-bound state, was used as positive control [44]. Slight effects on Cdc42 were only observed at a ten-fold higher concentration of TE (51 ng/mL) and betulin (100 nM), but not for lupeol (100 nM) (Fig. 11B). CNF1 (300 nm) which also deaminates Cdc42 and Rac1 at glutamine 61 was used as positive control [45]. There was no clear activation of Rac1 with TE and the triterpenes (Fig. 11C).
Figure 11. Influence of TE (5.1 or 51 ng/mL), betulin (10 or 100 nM) and lupeol (10 or 100 nM) on the activity of the Rho GTP-binding proteins RhoA (A), Cdc42 (B) and Rac1 (C) after 3 h incubation measured by pulldown experiments in primary human keratinocytes.

CNFY and CNF1 were used as positive controls. (D) Influence of the calcium channel blocker verapamil (100 µM) on the activation of RhoA. Verapamil was added 10 min prior to treatment with TE (5.1 ng/mL) and lupeol (10 nM). Each experiment was reproduced and a representative Western blot is shown.
Calcium is a multifaceted second messenger connecting receptor activation to downstream signaling pathways and is also involved in the regulation of Rho GTPases [46]–[48]. Since TE has been shown to increase intracellular calcium levels in primary human keratinocytes [49], we exemplarily examined the role of calcium in TE and lupeol mediated RhoA activation by using verapamil, a blocker of voltage dependent calcium channels [50]. Primary human keratinocytes were pretreated with 100 µM verapamil 10 minutes prior to incubation with either TE (5.1 ng/mL) or lupeol (10 nM) (see Fig. 11). Reduction of the activation of RhoA by TE and lupeol, when using the inhibitor, shows that calcium is likely involved in RhoA activation.
Discussion
The aim of this study was to elucidate the underlying molecular mechanism of the clinically proven wound healing effect of a birch bark extract (TE), which consists mainly of different pentacyclic triterpenes. We used primary human keratinocytes as a model, because they are the main cell population of the epidermis and are essential for the rapid coverage of the wound with a neo-epidermis. We demonstrate that TE influenced the inflammatory phase as well as the new tissue formation phase of the wound healing process. Regarding pro-inflammatory effects only betulin, the main constituent of TE, was shown to be responsible, whereas an influence on cell migration, measured by rearrangement of the actin cytoskeleton, could additionally be observed for lupeol and erythrodiol. These processes may contribute to the improvement of the epidermal regeneration and acceleration of the repair of the epidermal barrier function by TE proven in the porcine ex-vivo model. Kinetics of wound healing have not been evaluated in the ex-vivo model yet, but would be an interesting task in the future.
Related to the inflammatory phase TE and betulin led to a time and concentration dependent increase of COX-2 and IL-6 mRNA in primary human keratinocytes. This result was confirmed in the porcine ex-vivo wound healing model, and on the protein level of primary human keratinocytes. The importance of COX-2 in the wound healing process was shown by Futagami et al., who observed delayed reepithelialization of rat skin wounds after application of a COX-2 specific inhibitor [17]. COX-2 knock-out mice exhibited diminished tensile strength after dermal wounding compared to wild type [51]. However, the role of COX-2 in wound healing has not yet been doubtlessly clarified. Several studies demonstrated that COX-2 inhibition had no or only a minor negative effect on wound healing [52]–[54]. Given that the birch bark extract had already shown promising wound healing properties in patients [14] the observed slight increase of COX-2 expression after TE treatment may be beneficial in wound healing.
The importance of IL-6 in wound healing was demonstrated in experiments with IL-6 knock-out mice, which displayed impaired wound healing resulting in decelerated reepithelialization, diminished angiogenesis, collagen deposition and leucocyte infiltration [18], [55]. IL-6 was also evidenced to be involved in epidermal barrier repair, as topical application of IL-6 enhanced epidermal barrier repair in wild-type mice, whereas inhibition of the agonist IL-6/sIL-6 receptor complex, delayed barrier repair in wild mice [56]. We also observed an improved skin barrier function after treatment with TE and betulin, which may be linked to increased levels of IL-6. Pantothenate, a common self-medication for wound healing, also showed an upregulation of IL-6 in cultured human fibroblasts [57]. This was discussed by the authors as a contribution to wound healing properties of pantothenate. IL-6 is also a well-known activator of the JAK/STAT signaling cascade [58]. STAT3 activation commonly results in expression of genes both related to migration and proliferation [59]. However, in case of the birch bark extract, only cell migration seems to be affected, since we detected no increase in the number of proliferative cells. Beside IL-6, the Rho GTPase RhoA, which was slightly activated by TE, could additionally contribute to the observed STAT3 activation as previously shown by Reipschläger et al. [60].
Further studies were performed to elucidate the mechanism of increased mRNA levels of COX-2 and IL-6 under TE and betulin treatment. The transcription factor NF-κB, which is a potent inducer of many pro-inflammatory genes [21], was not involved. Instead, we found a prolonged mRNA half-life. p38 MAPK has been shown to participate in mRNA expression and stability of many proinflammatory mediators including COX-2 and IL-6 [24]–[27]. We demonstrated its involvement using a specific p38 MAPK inhibitor, which reduced the prolonged half-life time. Accordingly, TE and betulin slightly increased the amount of phosphorylated p38 MAPK as shown by Western blot analysis in primary human keratinocytes.
p38 MAPK functions by activating or inhibiting mRNA stability or destability factors like HuR and TTP [61], [62]. HuR exerts its stabilizing function only when localized in the cytosol [63]. Our data suggest that TE led to an increase of the cytosolic HuR fraction. Since p38 MAPK is known to induce the nuclear export of this factor [64], [65], the observed HuR accumulation may be the result of p38 MAPK activation. Nucleo-cytoplasmatic shuttling of HuR normally implicates a decrease in nuclear HuR levels, which we could not observe. The missing reduction in nuclear HuR can possibly be explained by rapid de novo synthesis, as we measured an increase in HuR mRNA level. However, it cannot be excluded that the differences in protein level are too small to be detectable by Western blot analysis.
In addition to COX-2 and IL-6, IL-8 and TNF-α were shown to be upregulated on gene and protein level by TE and betulin in primary human keratinocytes. By simultaneous measurement of different pro-inflammatory cytokines, chemokines and growth factors with the Bio-Plex® cytokine assay we achieved a more comprehensive overview about other affected mediators. Notably, among the mediators especially the tested chemokines (IL-8, IP-10, MIP-1β, and RANTES) were remarkably upregulated by TE and betulin treatment in keratinocytes. The chemokine IL-8 is known to be significantly upregulated in healing skin wounds and can be found in the wound fluid [20], [66], where it acts as a potent chemoattractant recruiting and activating inflammatory cells [67], [68]. Topical application of IL-8 has been shown to enhance wound healing through accelerated reepithelialization [20].
In summary, our results show that TE and betulin upregulates various mediators being involved in the inflammatory phase of the wound healing process. Pro-inflammatory effects of some of the investigated triterpenes have already been previously reported. Similar to our results, Zdzisinska et al. demonstrated a moderate induction of TNF-α by betulin in primary human whole blood cell cultures [69]. Betulinic acid was shown to induce TNF-α production in mouse macrophages [70]. However, it has also been demonstrated that betulin, lupeol and betulinic acid exert potent anti-inflammatory effects in-vivo and in-vitro [71]–[73]. Thus, it seems to strongly depend on the concentration, time of incubation, examined cell or tissue type (primary or cancer cells), and the experimental setup (stimulated vs. unstimulated cell systems) [74], whether these triterpenes exhibit inflammatory or anti-inflammatory effects.
Whereas a temporary inflammation is necessary for the wound healing process, an excessive and prolonged inflammatory phase is deleterious for proper wound healing and leads to chronic wounds. Hence, a termination of the inflammatory response is indispensable [1], [3], [6], [75]. In the WHM we clearly demonstrated that the increase in COX-2 and IL-6 mRNA was only temporary. Moreover, COX-2 is not only involved in pro-inflammatory effects, in later stages of wound healing the enzyme promotes the formation of anti-inflammatory prostaglandins, e.g. PGD2 and PGJ2 derivatives, which contribute to resolution of inflammation [76], [77]. Additionally, TGF-β, which was slightly upregulated by TE, can contribute to a termination of the inflammatory phase by induction of IL-1ra and reduction of TNF-α, IL-8, MCP-1 und MIP-1α [78]. Taken together, we assume that TE and betulin enhance the beginning of the inflammatory phase of wound healing by a transient upregulation of pro-inflammatory mediators, which does not result in a prolonged inflammation.
Furthermore, our studies provide evidence that birch bark extract influences the second stage of wound healing, the new tissue formation phase, by increasing cell migration of primary human keratinocytes shown in a scratch assay experiment. Migration strongly depends on morphological polarization of the cells and includes membrane protrusions at the leading edge of the cell with formation of new attachments to the substrate, contraction of the cell body and finally release of the rear [79]. These processes require a coordinated interaction of cytoskeletal elements, especially the assembly and disassembly of actin filaments, which lead to the formation of distinct cellular structures such as filopodia, lamellipodia and stress fibers [80]–[82]. Compounds, which directly or indirectly affect these actin structures, are interesting in context of supporting and enhancing wound healing processes. We clearly showed that TE in micromolar and even in nanomolar concentrations strongly influenced the actin cytoskeleton resulting in polarization of the cells, formation of lamellipodia and filopodia at the leading edge and stress fibers in the cell body. Formation of stabilized filopodia and stress fibers was additionally confirmed by visualizing vinculin with fluorescence labeled antibodies (data not shown). This protein accumulates in newly formed focal complexes in filopodia and in focal adhesions in the whole cell body [83], [84]. The influence on the actin cytoskeleton in nanomolar concentrations could be confirmed with the isolated triterpenes betulin, lupeol and erythrodiol. Notably, in mouse melanoma cells, Hata et al. observed just the opposite effect, that means a disassembly of stress fibers under lupeol treatment, which suppressed cell migration at concentrations of 1 to 20 µM [85]. Martin et al. also showed a disassembly of stress fibers for erythrodiol (25 to 50 µM) in a human astrocytoma cell line [86]. These contrasting results may be explained by the fact that cancer cell lines were used by these groups. In accordance with this conclusion. Martin et al. observed disrupted stress fibers in a human astrocytoma cell line after oleanolic acid treatment [87], whereas the same compound showed no alteration of cytoskeletal structures in primary human dermal fibroblasts [88], supporting our findings in primary keratinocytes.
Stress fiber formation is regulated by activation of the Rho GTPase RhoA [41], [43]. Consequently, we could demonstrate an increase in GTP-RhoA by TE, betulin and lupeol, indicating that activation of RhoA might contribute to the observed stress fiber formation. It is known that calcium is involved in the regulation of Rho GTPases and other downstream players, like actin-binding proteins, which directly control actin polymerization and depolymerization [46]–[48], [89], [90]. Accordingly, we could also demonstrate the involvement of calcium in TE- and lupeol-mediated RhoA activation using verapamil, a blocker of voltage dependent L-type calcium channels [50], as a significant reduction of GTP-bound RhoA by verapamil treatment was observed. An increase in intracellular calcium using a comparable birch bark extract was also previously reported by Woelfle [49]. This group additionally demonstrated an augmented expression of TRPC6 (transient receptor potential superfamily of cation channels 6) after stimulation with their birch bark extract, and discussed that this effect may be due to the observed calcium influx. Considering both results on Ca2+ influx, it can be concluded that TRPC6 may not be exclusively and entirely responsible for calcium influx, but that activation of L-type calcium channels can be generated by TRPC6 mediated membrane depolarization, as reported by Soboloff et al. [91].
Changes in the actin cytoskeleton and Rho GTPase activation are closely related to mitogen activated protein kinase pathways [92]. Thus, Rho GTPases can lead to phosphorylation of p38 MAPK via influencing upstream kinases [38]. Therefore, the activation of p38 by TE and betulin may not only affect mRNA stabilization, but also be involved in the observed assembly and disassembly of actin filaments. TE and betulin also activated the Rho GTPase Cdc42, which is known to stimulate filopodia formation [41], [43], but here higher concentrations were required compared to RhoA activation. Interestingly, lupeol, which also increased the amount of filopodia, did not activate Cdc42. Additionally, no activation of Rac1 by TE, betulin or lupeol was observed, despite increased formation of lamellipodia. These observations may be explained by spatial or temporary activation or the involvement of other Rho GTPases like Rif [93].
In summary our results contribute to the understanding of the molecular mechanism of the clinically proven wound healing effect of birch bark. Our results, together with the proven efficacy, identify birch bark as the first medical plant with a high potential to improve wound healing, a field which urgently needs effective remedies. Moreover, birch bark is a successful example that traditional medicinal plants can become rational drugs.
Materials and Methods
Foreskin samples used for keratinocyte culture were obtained following medical circumcisions. Its use was approved by the ethics committee of the Aerztekammer Hamburg, Germany (060900) and Freiburg, Germany (45/03). The ethics committee waived the need for consent, respectively. Porcine skin was obtained from a local slaughterhouse. The animals were slaughtered for human consumption and the ears were harvested following slaughtering with the permission of the slaughterhouse (Hoose, Hammor).
Tissues, antibodies, reagents and cell culture consumables
The birch bark extract (TE), betulin, lupeol, betulinic acid, erythrodiol and oleanolic acid were received from Birken AG, Niefern-Öschelbronn, Germany. Porcine skin was obtained from a local slaughterhouse. All the pigs were of the same age (6 months) and race (crossbred Yorkshire/Deutsches Edelschwein). The number of pigs or keratinocyte samples used in the various experiments is denoted in the respective figure legends.
NF-κB oligonucleotide: Promega, Mannheim, Germany; [γ33P]ATP: Hartmann Analytic, Braunschweig, Germany; T4 polynucleotide kinase: New England Biolabs, Frankfurt, Germany; Antibiotic-Antimycotic® 100×, phalloidin-rhodamine, Prolong Gold® Antifade reagent, Keratinocyte SFM® and supplements (human recombinant EGF, bovine pituitary extract), FCS, Trypsin/EDTA 0,05%/0,02%: Life Technologies, Darmstadt, Germany; Ciprobay®: Bayer, Leverkusen, Germany; LightCycler® 480 Probes Master, penicillin-streptomycin, poly(dI-dC), Complete® EDTA free protease inhibitor cocktail, PhosStop®, 0.45 μm PVDF membrane: Roche, Mannheim, Germany; Quantikine® Human IL-6 and IL-8 ELISA, ParameterTM PGE2 ELISA: R&D Systems, Minneapolis, USA; RNeasy® Plus Mini Kit and QuantiTect® Reverse Transcription Kit: Qiagen, Hilden, Germany; primer and probes for quantitative RT-PCR, HGF, phosphatase inhibitor cocktail 2: Sigma, Steinheim, German; ActD: Enzo Life Sciences, Lörrach, Germany; LN950: Prof. S. Laufer, University of Tübingen, Germany; Bio-Plex® Cytokine Assay and Bradford Quick Start Dye: Bio-Rad Laboratories, Munich, Germany; ibidi culture inserts with μ-dishes: ibidi, Munich, Germany; DS-QiMc: Nikon Instruments Inc., Tokyo, Japan; GST-Rhotekin and GST-PAK (immobilized to glutathione-sepharose beads, Pharmacia Biotech, Cambridge, USA): Prof. G. Schmidt, Albert-Ludiwgs-University Freiburg, Germany; HRP juice: pjk, Kleinblittersdorf, Germany; pSTAT3, STAT3, p38 and pp38 MAPK antibody: Cell Signaling, Danvers, USA; HuR, RhoA and Rac1 antibody: Santa Cruz, USA; Cdc42 antibody: Millipore, Bedford, USA; COX-2 antibody: Cayman, Chemical Company, USA; anti rabbit and anti mouse antibody: Jackson Immuno Research, Newmarket, UK; Ki67-antibody: MIB; Dako, Hamburg, Germany.
Isolation and cultivation of primary normal human keratinocytes (NHK)
Normal human skin keratinocytes (NHK) from foreskin explants were isolated and cultivated as described in Protocol S1.
Ex-vivo porcine wound healing model and reepithelialization (granted patent DE 10317400)
Pig ears were removed directly after slaughtering, delivered immediately to the laboratory, washed, and disinfected. Subsequently, punch biopsies (ø 6 mm diameter) were taken from the plicae of the ears and fat, subcutis and parts of the dermis were removed. Wounds were formed by removing epidermis and upper dermis from the central area of the biopsies (ø 3 mm). Finally, the models were placed dermis down on gauze in culture dishes and incubated air-liquid interface with Dulbecco's modified Eagle's medium supplemented with hydrocortisone, 2% fetal calf serum, penicillin, and streptomycin at 37°C with 5% CO2. The substances were applied immediately after wounding, for substances diluted in PBS application was repeated after 24 h. After 48 h, models were shock frozen in liquid nitrogen and stored at −80°C. Subsequently, cryostat sections were gained from the central parts of the wound healing models and stained with hematoxylin and eosin [94]. Reepithelialization was evaluated by measuring the distance between the wound margin to the tip of the regenerated epidermis with an axiophot II Zeiss microscope and the measurement tool of Openlab 5.0.2 software (Improvision, Coventry, UK). Means of left and right wound margins were calculated. Pigs with completely closed wounds in controls were excluded because an improvement of reepithelialization compared to PBS could not be unambiguously evaluated in these wounds.
Ex-vivo barrier assay
The WHM was established as described above. For the investigation of the early effect of the substances on barrier regeneration they were applied directly after wound healing. After 4 days the upper surface of the models was incubated for 5 min in methanol, for 5 min in PBS and for 15 min in toluidine blue (0.1% in PBS). Excessive dye was removed and models were shock frozen. For the investigation of late effects on barrier regeneration the substances were applied 72 h after wounding and toluidine blue staining was performed after another 24 h of incubation. Subsequently sections of the middle of the models were generated and the distance of regenerated epidermis without dye penetration starting from the wound margin was measured with an axiophot II Zeiss microscope and Openlab 5.0.2 software (Improvision; Coventry, UK).
RNA isolation, cDNA synthesis and quantitative Real-Time PCR
Total RNA from normal human keratinocytes was isolated using the RNeasy® Plus Mini Kit from Qiagen and converted to single strand cDNA using the QuantiTect® Reverse Transcription Kit according to the manufactures instructions. The analysis of mRNA expression profiles was performed with quantitative real time PCR as described in detail in Protocol S1.
Determination of the mRNA half-life time
For determination of the mRNA half-life time of IL-6 and COX-2 primary human keratinocytes were incubated with the transcription inhibitor ActD (5 µg/mL) for the indicated times. Subsequently, total RNA was harvested and IL-6 and COX-2 expression was quantified by qRT-PCR at different time points, respectively. The amount of mRNA without ActD treatment (time point zero) was referred to as 100% and the amount at different time points as percentage of remaining mRNA. From the calculated curves (one phase decay curve using GraphPad Prism 5) the time of 50% mRNA decay was deduced. To measure mRNA stability in response to TE and betulin treatment keratinocytes were stimulated either with TE or betulin in the presence of ActD (5 μg/mL). IL-6 and COX-2 mRNA expression were again quantified by qRT-PCR after the indicated times and calculation was performed as described in Protocol S1. To analyze the influence of p38 MAPK on the half-life time the p38 MAPK inhibitor LN950 (100 nM) was added simultaneously with ActD. Means of three independent experiments are presented.
IL-6 and IL-8 ELISA
Cells were seeded into 6-well plates at a density of 200 000 cells/well. After 24 h, medium was changed to medium without supplements and incubated for additional 24 h before test compounds were added. Upon incubation for various times, cell culture supernatants were transferred to reaction tubes, centrifuged for 5 min at 10.000 rpm and aliquoted in new reaction tubes. A Quantikine® Human IL-6 and IL-8 ELISA were used according to the manufacturer's instruction, respectively.
Bio-Plex® Cytokine Assay
To simultaneously determine the influence of the tested compounds on a variety of cytokines, chemokines and growth factors, the Bio-Plex® Human Cytokine Assay was used. After a period of 24 h of incubation supernatants were obtained according to the method described for the IL-6 and IL-8 ELISA. Measurement of the supernatants was performed according to the manufactureŕs instruction.
Scratch Assay
Monolayer wounding assays were used to evaluate migration of keratinocytes as described before [95]. Briefly, NHK cells were grown to confluence in ibidi μ-dishes containing culture inserts. Cells were incubated in Keratinocyte SFM® medium (with rhEGF, BPE and penicillin-streptomycin) containing 1 μg/mL TE or 10 ng/mL HGF. Live cell imaging migration was recorded in the PFS system on a Nikon Eclipse Ti microscope with a digital sight DS-QiMc (Nikon Instruments Inc., Tokyo, Japan), coupled to an ibidi-heating chamber. The migratory activity was measured by calculating the percentage of closed areas.
Determination of proliferation in WHM and cultured keratinocytes
Proliferative (Ki67-positive) cells were detected by MIB-1 in WHM by immunofluorescence staining as described previously [96] and in Protocol S1. Proliferation of cultured human keratinocytes was measured with the BdrU-ELISA kit from Roche (Mannheim, Germany) as described in Protocol S1.
Staining of the actin cytoskeleton
To analyze the effect of TE and the single triterpenes on the actin cytoskeleton, cells were stained with phalloidin-rhodamine and analyzed by fluorescence microscopy as described in Protocol S1.
Preparation of the protein extracts for Western Blotting and EMSA
Whole cell, cytosolic and nuclear extracts from primary human keratinocytes were prepared as described in detail in Protocol S1.
Electrophoretic mobility shift assay (EMSA)
Nuclear protein was resolved through non-denaturing 6% polyacrylamide gel electrophoresis and NF-κB-DNA binding was detected according to the Protocol S1.
Rho-GTPase pulldown experiments
Cell extracts were obtained as described by [60]. Briefly, cells were scraped in lysis buffer containing 10% glycerol, 50 mM Tris pH 7.4, 100 mM NaCl, 1% NP-40, 2 mM MgCl2 and 1 mM PMSF. After centrifugation at 4°C, the supernatant was used for determination of total Rho GTPase content and for pulldown experiments. For pulldown analysis of RhoA and Cdc42/Rac1, GST-Rhotekin or GST-PAK immobilized to gluthatione-sepharose beads were incubated with the supernatant for 1 h under gently shaking at 4°C. Subsequently, the beads were washed and separated by centrifugation. The Rho GTPases were subsequently analyzed by immunoblot.
Immunoblot analysis
The protein concentration was determined by using the Bradford Quick Start Dye according to the manufacturer's instruction. For Western blot analysis, 50 µg of protein was separated by a 10–12% SDS-PAGE and transferred to a 0.2 or 0.45 μm PVDF membrane. After blocking the membrane with 5% milk powder in TBST (TBS buffer containing 0.1% Tween 20) or 3% BSA in TBST for HuR, it was incubated with the respective antibody overnight at 4°C under gently shaking. The proteins were detected by using horseradish peroxidase labeled secondary antibodies and an enhanced chemiluminescence detection system.
Antibody concentrations: pSTAT3 (Tyr705) and STAT3: 1∶1000 in 5% BSA in TBST; pp38 and p38 MAPK: 1∶1000 in 5% BSA in TBST; HuR: 1∶500 in 5% milk in TBST; COX-2: 1∶1000 in 5% milk in TBST; RhoA: 1∶400 in TBST; Rac1: 1∶2000 in TBST; Cdc42: 1∶500 in 3% milk in PBS; anti rabbit: 1∶5000 in 5% BSA in TBST; anti mouse: 1∶5000 in 3% milk in TBST.
Statistical analysis
Values are expressed as means ± standard deviation (s.d.) or ± standard error of the mean (SEM). Statistical analyses of data sets were performed by using ANOVA followed by Dunnett's post-test. P-values were calculated and p<0.05 was considered significant. Wherever significance has been proven, it is indicated by *p<0.05, **p<0.01 and ***p<0.001.
Supporting Information
Quantitative analysis by gas chromatography and structures of triterpenes in birch bark. A: Quantitative composition of TE analyzed by gas chromatography [15]: betulin (86.85%), lupeol (3.94%), erythrodiol (0.77%), oleanolic acid (0.62%), betulinic acid (3.52%). In brief, 0.2 mg of TE was diluted in 500 µL tetrahydrofuran. This solution was silylated with 100 µL of silylating mixture (Silylating Mixture Fluka III; Buchs, Switzerland) at 80°C for 30 min and subsequently 1 µL was injected. GC analysis was carried out using a ZB-35 column (Phenomenex, 30 m×250 µm ×0.25 µm; Aschaffenburg, Germany) with hydrogen as carrier gas at a constant pressure of 12 psi. Detection was done with a flame ionization detector (FID) and a hydrogen flow of 50 mL/min and an air flow of 450 mL/min. Extern standards of the relevant components were used for quantification. Coefficients of variation of independent experiments were below 5% (w/w). Each measurement was performed in triplicate. B: Structural formula of the investigated triterpenes occurring in birch bark extract (Betula pendula).
(TIF)
TE (1 µg/mL) time dependently upregulates mRNA of the pro-inflammatory cytokines IL-1β (A) and TNF-α (B), TGF-β (C), the transcription factor Nrf2 (D), the antimicrobial peptide hBD3 (E) and the matrix-metalloproteinase MMP-2 (mRNA of MMP-9 was not upregulated) (F) in human primary keratinocytes. Time course of mRNA expression measured by qRT-PCR. Values represent means of at least 2 independent experiments ± SEM. *p<0.05 and ***p<0.001 as indicated.
(TIF)
TE and betulin have either none or a negligible effect on the production of the cytokines IL-1β, −4, −5, −7, −9, −10, −12, −13, −15, −17 and growth factors GM-CSF and VEGF in human keratinocytes after 24 h incubation. Cells were treated with TE (1 µg/mL) or betulin (0.87 µg/mL, which is in 1 µg/mL TE) for 24 h. Protein levels of the indicated mediators were determined in the supernatant by the Bio-Plex® Cytokine Assay. Values represent means of two independent experiments ± s.d.
(TIF)
TE and betulin do not influence NFκB DNA binding in primary human keratinocytes. Lane 1: Cells with TNF-α (4 ng/mL, for 30 min) as positive control, lane 2: unstimulated control, lanes 3–5: cells treated with TE (1 µg/mL) and lanes 6–8 treated with betulin (0.87 µg/mL, which is in 1 µg/mL TE) for the indicated times (2, 4, 6 h). Equal amounts of protein from total cell extracts were analyzed for NFκB-DNA binding activity in an electrophoretic mobility shift assay (EMSA). ○: NFκB-DNA complexes, •: non-specific binding to the probe, ◂: unbound oligonucleotide. The result of the EMSA was reproduced and one representative EMSA is shown.
(TIF)
TE and betulin do not increase proliferation in primary human keratinocytes and in the WHM. (A) BrdU-ELISA with primary human keratinocytes treated for 48 h with TE and the compounds at the respective concentrations. Values represent the means of at least 6 independent experiments ± SEM. (B) Proliferation in WHM 48 h after wounding and treatment with TE and the compounds. Values represent means of at least 7 independent experiments + SEM.
(TIF)
Influence of TE (0.5 and 1 µg/mL), betulinic acid, oleanolic acid and erythrodiol (1, 10, and 100 nM) on the actin cytoskeleton of primary human keratinocytes. Cells were incubated on glass coverslips for 2 h with TE and the respective triterpenes for the indicated concentrations and the actin cytoskeleton was stained with phalloidin-rhodamine. 0.1% (V/V) DMSO was used as solvent control and CNF1 and CNFY as positive controls. Rows labelled with F show the impact on filopodia and lamellipodia and S the impact on stress fiber formation. A white arrow indicates the leading edge of the cell. Representative pictures of repeated experiments (n = 4) are shown.
(TIF)
The file contains all experimental data which is not shown in the manuscript.
(DOC)
Acknowledgments
The authors thank Dr. Barbara Silakowski and Dr. Karsten Strauß from BioRad, Munich, Germany, for carrying out the Bio-Plex ® Cytokine assay, Prof. Dr. Stefan Laufer, University Tübingen, Germany, for the supply with the p38 MAPK inhibitor, Birken AG for providing the birch bark extract and the triterpenes and Dr. Melanie Laszczyk-Lauer, Birken AG, for fruitful discussions.
Funding Statement
This study was funded by Aif and Birken AG. The article processing charge was funded by the open access publication fund of the Albert-Ludwigs-University Freiburg. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gurtner GC, Werner S, Barrandon Y, Longaker MT (2008) Wound repair and regeneration. Nature 453: 314–321. [DOI] [PubMed] [Google Scholar]
- 2. Kondo T, Ishida Y (2010) Molecular pathology of wound healing. Forensic Sci Int 203: 93–98. [DOI] [PubMed] [Google Scholar]
- 3. Schreml S, Szeimies RM, Prantl L, Landthaler M, Babilas P (2010) Wound healing in the 21st century. J Am Acad Dermatol 63: 866–881. [DOI] [PubMed] [Google Scholar]
- 4. Velnar T, Bailey T, Smrkolj V (2009) The wound healing process: an overview of the cellular and molecular mechanisms. J Int Med Res 37: 1528–1542. [DOI] [PubMed] [Google Scholar]
- 5. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M (2008) Growth factors and cytokines in wound healing. Wound Repair Regen 16: 585–601. [DOI] [PubMed] [Google Scholar]
- 6. Behm B, Babilas P, Landthaler M, Schreml S (2011) Cytokines, chemokines and growth factors in wound healing. J Eur Acad Dermatol Venereol 26: 812–820. [DOI] [PubMed] [Google Scholar]
- 7. Martin P, Leibovich SJ (2005) Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 15: 599–607. [DOI] [PubMed] [Google Scholar]
- 8. Widgerow AD (2012) Cellular resolution of inflammation-catabasis. Wound Repair Regen 20: 2–7. [DOI] [PubMed] [Google Scholar]
- 9. Goldman R (2004) Growth factors and chronic wound healing: past, present, and future. Adv Skin Wound Care 17: 24–35. [DOI] [PubMed] [Google Scholar]
- 10. Blakytny R, Jude E (2006) The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med 23: 594–608. [DOI] [PubMed] [Google Scholar]
- 11.Hiller K, Melzig MF (2010) Lexikon der Arzneipflanzen und Drogen. Spektrum Akademischer Verlag.
- 12. Weckesser S, Laszczyk MN, Müller ML, Schempp CM, Schumann H (2010) Topical treatment of necrotising herpes zoster with betulin from birch bark. Forsch Komplementmed 17: 271–273. [DOI] [PubMed] [Google Scholar]
- 13. Schempp C, Huyke C (2005) Behandlung von Verbrennungen 2. Grades mit Birkencreme. Der Merkurstab 5: 402. [Google Scholar]
- 14. Metelmann HR, Brandner J, Schumann H, Bross F, Hoffmann M, et al. (2011) Accelerating the aesthetic benefit of wound healing by triterpene. J Craniomaxillofac Surg 40: 150–154. [DOI] [PubMed] [Google Scholar]
- 15. Laszczyk M, Jäger S, Simon-Haarhaus B, Scheffler A, Schempp CM (2006) Physical, chemical and pharmacological characterization of a new oleogel-forming triterpene extract from the outer bark of birch (Betulae cortex). Planta Med 72: 1389–1395. [DOI] [PubMed] [Google Scholar]
- 16. Eming SA, Krieg T, Davidson JM (2007) Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 127: 514–525. [DOI] [PubMed] [Google Scholar]
- 17. Futagami A, Ishizaki M, Fukuda Y, Kawana S, Yamanaka N (2002) Wound healing involves induction of cyclooxygenase-2 expression in rat skin. Lab Invest 82: 1503–1513. [DOI] [PubMed] [Google Scholar]
- 18. Lin ZQ, Kondo T, Ishida Y, Takayasu T, Mukaida N (2003) Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol 73: 713–721. [DOI] [PubMed] [Google Scholar]
- 19. Lee JL, Mukhtar H, Bickers DR, Kopelovich L, Athar M (2003) Cyclooxygenases in the skin: pharmacological and toxicological implications. Toxicol Appl Pharmacol 192: 294–306. [DOI] [PubMed] [Google Scholar]
- 20. Rennekampff HO, Hansbrough JF, Kiessig V, Dore C, Sticherling M, et al. (2000) Bioactive interleukin-8 is expressed in wounds and enhances wound healing. J Surg Res 93: 41–54. [DOI] [PubMed] [Google Scholar]
- 21. Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853–6866. [DOI] [PubMed] [Google Scholar]
- 22. Al Ghouleh I, Magder S (2012) NADPH oxidase-derived superoxide destabilizes lipopolysaccharide-induced interleukin 8 mRNA via p38, extracellular signal-regulated kinase mitogen-activated protein kinase, and the destabilizing factor tristetraprolin. Shock 37: 433–440. [DOI] [PubMed] [Google Scholar]
- 23. Harper KA, Tyson-Capper AJ (2008) Complexity of COX-2 gene regulation. Biochem Soc Trans 36: 543–545. [DOI] [PubMed] [Google Scholar]
- 24. Zhao W, Liu M, Kirkwood KL (2008) p38alpha stabilizes interleukin-6 mRNA via multiple AU-rich elements. J Biol Chem 283: 1778–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dean JL, Brook M, Clark AR, Saklatvala J (1999) p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem 274: 264–269. [DOI] [PubMed] [Google Scholar]
- 26. Mifflin RC, Saada JI, Di Mari JF, Valentich JD, Adegboyega PA, et al. (2004) Aspirin-mediated COX-2 transcript stabilization via sustained p38 activation in human intestinal myofibroblasts. Mol Pharmacol 65: 470–478. [DOI] [PubMed] [Google Scholar]
- 27. Dean JL, Sully G, Clark AR, Saklatvala J (2004) The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal 16: 1113–1121. [DOI] [PubMed] [Google Scholar]
- 28. Fernau NS, Fugmann D, Leyendecker M, Reimann K, Grether-Beck S, et al. (2010) Role of HuR and p38MAPK in ultraviolet B-induced post-transcriptional regulation of COX-2 expression in the human keratinocyte cell line HaCaT. J Biol Chem 285: 3896–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di Mari JF, Saada JI, Mifflin RC, Valentich JD, Powell DW (2007) HETEs enhance IL-1-mediated COX-2 expression via augmentation of message stability in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol 293: G719–G728. [DOI] [PubMed] [Google Scholar]
- 30. Schaefer M, Werner S (2007) Transcriptional control of wound repair. Annu Rev Cell Dev Biol 23: 69–92. [DOI] [PubMed] [Google Scholar]
- 31. Singh A, Nascimento JM, Kowar S, Busch H, Boerries M (2012) Boolean approach to signalling pathway modelling in HGF-induced keratinocyte migration. Bioinformatics 28: i495–i501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Etienne-Manneville S (2004) Actin and microtubules in cell motility: which one is in control? Traffic 5: 470–477. [DOI] [PubMed] [Google Scholar]
- 33. Mansbridge JN, Knapp AM (1987) Changes in keratinocyte maturation during wound healing. J Invest Dermatol 89: 253–263. [DOI] [PubMed] [Google Scholar]
- 34. Rys-Sikora KE, Konger RL, Schoggins JW, Malaviya R, Pentland AP (2000) Coordinate expression of secretory phospholipase A(2) and cyclooxygenase-2 in activated human keratinocytes. Am J Physiol Cell Physiol 278: C822–C833. [DOI] [PubMed] [Google Scholar]
- 35. Knust Z, Schmidt G (2010) Cytotoxic necrotizing factors (CNFs) – A growing toxin family. Toxins (Basel) 2: 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hall A (1994) Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol 10: 31–54. [DOI] [PubMed] [Google Scholar]
- 37. Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509–514. [DOI] [PubMed] [Google Scholar]
- 38. Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269. [DOI] [PubMed] [Google Scholar]
- 39. Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701. [DOI] [PubMed] [Google Scholar]
- 40. Etienne-Manneville S (2004) Cdc42 – the centre of polarity. J Cell Sci 117: 1291–1300. [DOI] [PubMed] [Google Scholar]
- 41. Nobes CD, Hall A (1995) Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53–62. [DOI] [PubMed] [Google Scholar]
- 42. Aspenstrom P, Fransson A, Saras J (2004) Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J 377: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ridley AJ (2001) Rho GTPases and cell migration. J Cell Sci 114: 2713–2722. [DOI] [PubMed] [Google Scholar]
- 44. Hoffmann C, Pop M, Leemhuis J, Schirmer J, Aktories K, et al. (2004) The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J Biol Chem 279: 16026–16032. [DOI] [PubMed] [Google Scholar]
- 45. Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, et al. (1999) Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect Immun 67: 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Masiero L, Lapidos KA, Ambudkar I, Kohn EC (1999) Regulation of the RhoA pathway in human endothelial cell spreading on type IV collagen: role of calcium influx. J Cell Sci 112: 3205–3213. [DOI] [PubMed] [Google Scholar]
- 47. Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, et al. (2003) Calcium signaling regulates translocation and activation of Rac. J Biol Chem 278: 39413–39421. [DOI] [PubMed] [Google Scholar]
- 48. Jin M, Guan CB, Jiang YA, Chen G, Zhao CT, et al. (2005) Ca2+-dependent regulation of rho GTPases triggers turning of nerve growth cones. J Neurosci 25: 2338–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Woelfle U, Laszczyk MN, Kraus M, Leuner K, Kersten A, et al. (2010) Triterpenes promote keratinocyte differentiation in vitro, ex vivo and in vivo: a role for the transient receptor potential canonical (subtype) 6. J Invest Dermatol 130: 113–123. [DOI] [PubMed] [Google Scholar]
- 50. Fleckenstein A (1977) Specific pharmacology of calcium in myocardium, cardiac pacemakers, and vascular smooth muscle. Annu Rev Pharmacol Toxicol 17: 149–166. [DOI] [PubMed] [Google Scholar]
- 51. Laulederkind SJ, Thompson-Jaeger S, Goorha S, Chen Q, Fu A, et al. (2002) Both constitutive and inducible prostaglandin H synthase affect dermal wound healing in mice. Lab Invest 82: 919–927. [DOI] [PubMed] [Google Scholar]
- 52. Hardy MM, Blomme EA, Lisowski A, Chinn KS, Jones A, et al. (2003) Selective cyclooxygenase-2 inhibition does not alter keratinocyte wound responses in the mouse epidermis after abrasion. J Pharmacol Exp Ther 304: 959–967. [DOI] [PubMed] [Google Scholar]
- 53. Müller-Decker K, Hirschner W, Marks F, Fürstenberger G (2002) The effects of cyclooxygenase isozyme inhibition on incisional wound healing in mouse skin. J Invest Dermatol 119: 1189–1195. [DOI] [PubMed] [Google Scholar]
- 54. Blomme EA, Chinn KS, Hardy MM, Casler JJ, Kim SH, et al. (2003) Selective cyclooxygenase-2 inhibition does not affect the healing of cutaneous full-thickness incisional wounds in SKH-1 mice. Br J Dermatol 148: 211–223. [DOI] [PubMed] [Google Scholar]
- 55. Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, et al. (2000) Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J 14: 2525–2531. [DOI] [PubMed] [Google Scholar]
- 56. Wang XP, Schunck M, Kallen KJ, Neumann C, Trautwein C, et al. (2004) The interleukin-6 cytokine system regulates epidermal permeability barrier homeostasis. J Invest Dermatol 123: 124–131. [DOI] [PubMed] [Google Scholar]
- 57. Wiederholt T, Heise R, Skazik C, Marquardt Y, Joussen S, et al. (2009) Calcium pantothenate modulates gene expression in proliferating human dermal fibroblasts. Exp Dermatol 18: 969–978. [DOI] [PubMed] [Google Scholar]
- 58. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S (2011) The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813: 878–888. [DOI] [PubMed] [Google Scholar]
- 59. Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, et al. (2005) Stat3 regulates genes common to both wound healing and cancer. Oncogene 24: 3397–3408. [DOI] [PubMed] [Google Scholar]
- 60. Reipschlaeger S, Kubatzky K, Taromi S, Burger M, Orth J, et al. (2012) Toxin-induced RhoA activity mediates CCL1-triggered STAT signaling 9. J Biol Chem 287: 11183–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liao WL, Wang WC, Chang WC, Tseng JT (2011) The RNA-binding protein HuR stabilizes cytosolic phospholipase A2α mRNA under interleukin-1beta treatment in non-small cell lung cancer A549 Cells. J Biol Chem 286: 35499–35508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tudor C, Marchese FP, Hitti E, Aubareda A, Rawlinson L, et al. (2009) The p38 MAPK pathway inhibits tristetraprolin-directed decay of interleukin-10 and pro-inflammatory mediator mRNAs in murine macrophages. FEBS Lett 583: 1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Doller A, Pfeilschifter J, Eberhardt W (2008) Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 20: 2165–2173. [DOI] [PubMed] [Google Scholar]
- 64. Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A (2009) p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum Mol Genet 18: 4035–4045. [DOI] [PubMed] [Google Scholar]
- 65. Lafarga V, Cuadrado A, Lopez dS, I, Bengoechea R, Fernandez-Capetillo O, et al. (2009) p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol 29: 4341–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nuutila K, Siltanen A, Peura M, Bizik J, Kaartinen I, et al. (2012) Human skin transcriptome during superficial cutaneous wound healing. Wound Repair Regen 20: 830–839. [DOI] [PubMed] [Google Scholar]
- 67. Remick DG (2005) Interleukin-8. Crit Care Med 33: S466–S467. [DOI] [PubMed] [Google Scholar]
- 68. Roupe KM, Nybo M, Sjobring U, Alberius P, Schmidtchen A, et al. (2010) Injury is a major inducer of epidermal innate immune responses during wound healing. J Invest Dermatol 130: 1167–1177. [DOI] [PubMed] [Google Scholar]
- 69. Zdzisinska B, Rzeski W, Paduch R, Szuster-Ciesielska A, Kaczor J, et al. (2003) Differential effect of betulin and betulinic acid on cytokine production in human whole blood cell cultures. Pol J Pharmacol 55: 235–238. [PubMed] [Google Scholar]
- 70. Yun Y, Han S, Park E, Yim D, Lee S, et al. (2003) Immunomodulatory activity of betulinic acid by producing pro-inflammatory cytokines and activation of macrophages. Arch Pharm Res 26: 1087–1095. [DOI] [PubMed] [Google Scholar]
- 71. Lin YC, Cheng HY, Huang TH, Huang HW, Lee YH, et al. (2009) Analgesic and anti-inflammatory activities of Torenia concolor Lindley var. formosana Yamazaki and betulin in mice. Am J Chin Med 37: 97–111. [DOI] [PubMed] [Google Scholar]
- 72. Recio MD, Giner RM, Manez S, Gueho J, Julien HR, et al. (1995) Investigations on the steroidal antiinflammatory activity of triterpenoids from Diospyros leucomelas . Planta Medica 61: 9–12. [DOI] [PubMed] [Google Scholar]
- 73. Fernandez MA, de las Heras B, Garcia MD, Saenz MT, Villar A (2001) New insights into the mechanism of action of the anti-inflammatory triterpene lupeol. J Pharm Pharmacol 53: 1533–1539. [DOI] [PubMed] [Google Scholar]
- 74. Laszczyk MN (2009) Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med 75: 1549–1560. [DOI] [PubMed] [Google Scholar]
- 75. Martin P, Leibovich SJ (2005) Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 15: 599–607. [DOI] [PubMed] [Google Scholar]
- 76. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, et al. (1999) Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 5: 698–701. [DOI] [PubMed] [Google Scholar]
- 77. Lanaro A, Lalenti A, Maffia P, Pisano B, Di RM (2001) Role of cyclopentenone prostaglandins in rat carrageenin pleurisy. FEBS Lett 508: 61–66. [DOI] [PubMed] [Google Scholar]
- 78. Ashcroft GS (1999) Bidirectional regulation of macrophage function by TGF-beta. Microbes Infect 1: 1275–1282. [DOI] [PubMed] [Google Scholar]
- 79. Lauffenburger DA, Horwitz AF (1996) Cell migration: a physically integrated molecular process. Cell 84: 359–369. [DOI] [PubMed] [Google Scholar]
- 80. Ridley AJ (2011) Life at the leading edge. Cell 145: 1012–1022. [DOI] [PubMed] [Google Scholar]
- 81. Biname F, Pawlak G, Roux P, Hibner U (2010) What makes cells move: requirements and obstacles for spontaneous cell motility. Mol Biosyst 6: 648–661. [DOI] [PubMed] [Google Scholar]
- 82. Lee SH, Dominguez R (2010) Regulation of actin cytoskeleton dynamics in cells. Mol Cells 29: 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Geuijen CA, Sonnenberg A (2002) Dynamics of the α6β4 integrin in keratinocytes. Mol Biol Cell 13: 3845–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schäfer C, Borm B, Born S, Mohl C, Eibl EM, et al. (2009) One step ahead: role of filopodia in adhesion formation during cell migration of keratinocytes. Exp Cell Res 315: 1212–1224. [DOI] [PubMed] [Google Scholar]
- 85. Hata K, Hori K, Murata J, Takahashi S (2005) Remodeling of actin cytoskeleton in lupeol-induced B16 2F2 cell differentiation. J Biochem 138: 467–472. [DOI] [PubMed] [Google Scholar]
- 86. Martin R, Ibeas E, Carvalho-Tavares J, Hernandez M, Ruiz-Gutierrez V, et al. (2009) Natural triterpenic diols promote apoptosis in astrocytoma cells through ROS-mediated mitochondrial depolarization and JNK activation. PLoS One 4: e5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Martin R, Carvalho-Tavares J, Ibeas E, Hernandez M, Ruiz-Gutierrez V, et al. (2007) Acidic triterpenes compromise growth and survival of astrocytoma cell lines by regulating reactive oxygen species accumulation. Cancer Res 67: 3741–3751. [DOI] [PubMed] [Google Scholar]
- 88. Wojciak-Kosior M, Paduch R, Matysik-Wozniak A, Niedziela P, Donica H (2011) The effect of ursolic and oleanolic acids on human skin fibroblast cells. Folia Histochem Cytobiol 49: 664–669. [DOI] [PubMed] [Google Scholar]
- 89. Vandekerckhove J (1990) Actin-binding proteins. Curr Opin Cell Biol 2: 41–50. [DOI] [PubMed] [Google Scholar]
- 90. Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, et al. (2007) Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem 282: 7833–7843. [DOI] [PubMed] [Google Scholar]
- 91. Soboloff J, Spassova M, Xu W, He LP, Cuesta N, et al. (2005) Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J Biol Chem 280: 39786–39794. [DOI] [PubMed] [Google Scholar]
- 92. Kustermans G, Piette J, Legrand-Poels S (2008) Actin-targeting natural compounds as tools to study the role of actin cytoskeleton in signal transduction. Biochem Pharmacol 76: 1310–1322. [DOI] [PubMed] [Google Scholar]
- 93. Goh WI, Sudhaharan T, Lim KB, Sem KP, Lau CL, et al. (2011) Rif-mDia1 interaction is involved in filopodium formation independent of Cdc42 and Rac effectors. J Biol Chem 286: 13681–13694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vockel M, Pollok S, Breitenbach U, Ridderbusch I, Kreienkamp HJ, et al. (2011) Somatostatin inhibits cell migration and reduces cell counts of human keratinocytes and delays epidermal wound healing in an ex vivo wound model. PLoS One 6: e19740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Busch H, Camacho-Trullio D, Rogon Z, Breuhahn K, Angel P, et al. (2008) Gene network dynamics controlling keratinocyte migration. Mol Syst Biol 4: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pollok S, Pfeiffer AC, Lobmann R, Wright CS, Moll I, et al. (2011) Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J Cell Mol Med 15: 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantitative analysis by gas chromatography and structures of triterpenes in birch bark. A: Quantitative composition of TE analyzed by gas chromatography [15]: betulin (86.85%), lupeol (3.94%), erythrodiol (0.77%), oleanolic acid (0.62%), betulinic acid (3.52%). In brief, 0.2 mg of TE was diluted in 500 µL tetrahydrofuran. This solution was silylated with 100 µL of silylating mixture (Silylating Mixture Fluka III; Buchs, Switzerland) at 80°C for 30 min and subsequently 1 µL was injected. GC analysis was carried out using a ZB-35 column (Phenomenex, 30 m×250 µm ×0.25 µm; Aschaffenburg, Germany) with hydrogen as carrier gas at a constant pressure of 12 psi. Detection was done with a flame ionization detector (FID) and a hydrogen flow of 50 mL/min and an air flow of 450 mL/min. Extern standards of the relevant components were used for quantification. Coefficients of variation of independent experiments were below 5% (w/w). Each measurement was performed in triplicate. B: Structural formula of the investigated triterpenes occurring in birch bark extract (Betula pendula).
(TIF)
TE (1 µg/mL) time dependently upregulates mRNA of the pro-inflammatory cytokines IL-1β (A) and TNF-α (B), TGF-β (C), the transcription factor Nrf2 (D), the antimicrobial peptide hBD3 (E) and the matrix-metalloproteinase MMP-2 (mRNA of MMP-9 was not upregulated) (F) in human primary keratinocytes. Time course of mRNA expression measured by qRT-PCR. Values represent means of at least 2 independent experiments ± SEM. *p<0.05 and ***p<0.001 as indicated.
(TIF)
TE and betulin have either none or a negligible effect on the production of the cytokines IL-1β, −4, −5, −7, −9, −10, −12, −13, −15, −17 and growth factors GM-CSF and VEGF in human keratinocytes after 24 h incubation. Cells were treated with TE (1 µg/mL) or betulin (0.87 µg/mL, which is in 1 µg/mL TE) for 24 h. Protein levels of the indicated mediators were determined in the supernatant by the Bio-Plex® Cytokine Assay. Values represent means of two independent experiments ± s.d.
(TIF)
TE and betulin do not influence NFκB DNA binding in primary human keratinocytes. Lane 1: Cells with TNF-α (4 ng/mL, for 30 min) as positive control, lane 2: unstimulated control, lanes 3–5: cells treated with TE (1 µg/mL) and lanes 6–8 treated with betulin (0.87 µg/mL, which is in 1 µg/mL TE) for the indicated times (2, 4, 6 h). Equal amounts of protein from total cell extracts were analyzed for NFκB-DNA binding activity in an electrophoretic mobility shift assay (EMSA). ○: NFκB-DNA complexes, •: non-specific binding to the probe, ◂: unbound oligonucleotide. The result of the EMSA was reproduced and one representative EMSA is shown.
(TIF)
TE and betulin do not increase proliferation in primary human keratinocytes and in the WHM. (A) BrdU-ELISA with primary human keratinocytes treated for 48 h with TE and the compounds at the respective concentrations. Values represent the means of at least 6 independent experiments ± SEM. (B) Proliferation in WHM 48 h after wounding and treatment with TE and the compounds. Values represent means of at least 7 independent experiments + SEM.
(TIF)
Influence of TE (0.5 and 1 µg/mL), betulinic acid, oleanolic acid and erythrodiol (1, 10, and 100 nM) on the actin cytoskeleton of primary human keratinocytes. Cells were incubated on glass coverslips for 2 h with TE and the respective triterpenes for the indicated concentrations and the actin cytoskeleton was stained with phalloidin-rhodamine. 0.1% (V/V) DMSO was used as solvent control and CNF1 and CNFY as positive controls. Rows labelled with F show the impact on filopodia and lamellipodia and S the impact on stress fiber formation. A white arrow indicates the leading edge of the cell. Representative pictures of repeated experiments (n = 4) are shown.
(TIF)
The file contains all experimental data which is not shown in the manuscript.
(DOC)