

Abstract
N-glycosylation of eukaryotic proteins is widespread and vital to survival. The pentasaccharide unit −Man3GlcNAc2– lies at the protein-junction core of all oligosaccharides attached to asparagine side chains during this process. Although its absolute conservation implies an indispensable role, associated perhaps with its structure, its unbiased conformation and the potential modulating role of solvation are unknown; both have now been explored through a combination of synthesis, laser spectroscopy, and computation. The proximal −GlcNAc-GlcNAc– unit acts as a rigid rod, while the central, and unusual, −Man-β-1,4-GlcNAc– linkage is more flexible and is modulated by the distal Man-α-1,3– and Man-α-1,6– branching units. Solvation stiffens the ‘rod’ but leaves the distal residues flexible, through a β-Man pivot, ensuring anchored projection from the protein shell while allowing flexible interaction of the distal portion of N-glycosylation with bulk water and biomolecular assemblies.
Introduction
N-glycosylation of proteins is a highly conserved process in all eukaryotes whereby complex oligosaccharides (glycans) are cotranslationally appended to the side-chain asparagine γ-nitrogen of the consensus motif NXS/T (Figure 1).1,2N-Glycans display a rich structural diversity3 and are vital for both the correct folding of nascent glycoproteins, where interactions with the chaperone lectins calnexin and calreticulin are key,4 and also for the correct functioning of the mature glycoproteins, where glycans play crucial roles in such processes as protein targeting, intercellular signaling, and host–pathogen recognition.5,6 Common to these diverse roles and the many structural possibilities for N-glycans is the conserved presence of an invariant core pentasaccharide (Man3GlcNAc2−) (Figure 1a). This presence in all mammalian N-glycoproteins (regardless of protein location or function) and the highly conserved nature of N-glycosylation among eukaryotes suggests an indispensable (and perhaps general) role for the core pentasaccharide in facilitating the proper functioning of N-glycans.4,7,8 Despite its ubiquity, however, the underlying physical reasons for its conservation are not yet fully understood.9
Figure 1.

The pivotal role of the core pentasaccharide in N-glycoproteins. (a) The conserved core pentasaccharide motif, (Man3GlcNAc2 shown in gray box), is attached to asparagine residues of glycoproteins (N, S, and T denote asparagine, serine, and threonine, X denotes any amino acid but not proline). Proteins are cotranslationally modified with a tetradecasaccharide which is tailored by glycosyl-hydrolase and transferase enzymes to create diverse glycans with varying antennae but all based upon the conserved core pentasaccharide (gray box). (b) The structures and symbol representations of the core pentasaccharide 4 and the building blocks 1–3 from key regions of 4 used in this study. The naming convention A–E is used in this manuscript to identify the individual glycosyl residues. The site of the benign chromophore used in this study mimics the location of the protein scaffold or truncated glycan (shown by a red star).
NMR measurements of model glycopeptides in aqueous solution, coupled with molecular dynamics investigations,10,11 have exposed the influence of the proximal chitobiose (−GlcNAc-GlcNAc−) stem on the local peptide conformation and suggested an explanation of nature’s choice of N-acetyl-d-glucosamine (GlcNAc) over the more plentiful d-glucose. Does the chosen chitobiose stem play other structural roles? Does the notably rare β-mannoside (Man) central linkage between this stem and the trimannoside (Man3) unit impart any unique structural or functional benefits and do these units (and other building blocks) function independently or in concert? Similar NMR and molecular dynamic investigations of the naturally occurring Man9GlcNAc2N-glycan in aqueous solution have suggested enduring structural features in the core pentasaccharide, incorporating locally bound water molecules.12,13 Does the core pentasaccharide present any favored water pockets and are its shape and stiffness influenced by solvent-mediated interactions?
One approach to these various questions is to explore the structural mechanics of the individual building blocks, in isolation and then linked together, using a combination of synthetic oligosaccharide assembly, mass- and conformer-selected infrared laser spectroscopy under molecular beam conditions, molecular mechanics, and quantum chemical calculations. This allows determination of their preferred (inherent) conformational structures when isolated in the gas phase to provide basic structural benchmarks; then interrogation of the locations and structural consequences of their microhydration; and finally, their response to a bulk, aqueous environment.
Early stages of this approach have previously been applied to the NXS sequon14 and the trimannoside unit.15 We now report the culmination of this strategy, applied here to dissect the unbiased structure of the entire core pentasaccharide and then to understand the effects of hydration upon it. Target oligosaccharide building blocks (1–4, Figure 1b) were designed in a form that provided a chromophore required for detection through mass-selected ultraviolet (UV) photoionization.16 Syntheses of the chitobiose (GlcNAc-β-1,4-GlcNAc) stem 1, the connecting disaccharide (Man-β-1,4-GlcNAc) 2, the extended trisaccharide (Man-β-1,4-GlcNAc-β-1,4-GlcNAc) 3, and finally the complete core pentasaccharide (Man3GlcNAc2) 4, all as their phenyl (Ph) glycosides, were achieved on the gram-scales required for their gas-phase interrogation. Their conformational preferences have been determined in the gas phase using infrared ion depletion (IRID) laser spectroscopy16 coupled with molecular mechanics, density functional and ab initio theoretical calculations. Those of their discretely hydrated (and ‘blocked’) complexes were determined through theoretical calculation. Comparisons with their preferred conformations in aqueous solution, reported for chitobiose17 and the trimannoside (Man3)18−20 and computed here for the core pentasaccharide through molecular mechanics calculations, reveal a unique insight into the inner workings of the core pentasacccharide and the molecular scaffolding provided by it and its components.
Methods
Experimental Section
Full details of all synthetic procedures and characterization are provided in the Supporting Information (SI). The carbohydrates were vaporized by laser desorption into an expanding supersonic jet of argon before passing through a 2 mm skimmer to create a collimated molecular beam which then intersected tunable UV and IR laser beams in the extraction region of a linear time-of-flight mass spectrometer (Jordan). Conformer-specific, mass-selected spectra were recorded in the UV and IR using UV–UV and IR–UV ion dip (IRID) double resonance spectroscopy.14
Computational
The calculations began with completely unrestricted and exhaustive surveys of the conformational landscapes of each of the carbohydrate ‘building blocks’ and their singly hydrated complexes, using a molecular mechanics method (MMFFs force field).21 Structures with relative energies <15 kJ mol–1, together with additional representative structures that might have a significant population in the cooled adiabatic expansion (see SI), were reoptimized through density functional theory calculations (B3LYP/6-311+G*) using the Gaussian 09 program package22 (and two supercomputers, i2BASQUE and SGIker, employing up to 96 processors per calculation) to provide a more accurate energy ranking of the lowest-energy structures and their associated harmonic vibrational spectra. Zero-point corrected relative energies were computed through subsequent single point ab initio calculations (MP2/6-311++G**), and final optimizations were based upon comparisons with the experimental spectra themselves, to provide feedback and guide the ‘fine-tuning’ of the predicted structures.
The core pentasaccharide conformational structure was also explored exhaustively, using the molecular mechanics OPLS200523 and GLYCAM06/AMBER24 force fields. (OPLS2005 possessed the fewest low-quality stretch, bend, and torsional parameters when benchmarked for the carbohydrate segments against all other native force fields found in the Macromodel software; GLYCAM06/AMBER (not included in Macromodel) is referenced specifically, for carbohydrate structures). All stereogenic centers were preserved in the computational search, and the ring-opening method of Still25 was used to explore additional ring conformations (full details of all calculations are given as SI).
Results and Discussion
Design of the Glycan Targets
The target core pentasaccharide 4 and its building blocks 1–3 (Figure 1b) were synthesized as phenyl β-glycosides, to provide a UV chromophore,16 installed at the structurally benign reducing terminus. The target sugars were all required in gram-scale quantities to provide sufficient material for spectroscopic investigation in the gas phase. While such scales have been accomplished for certain smaller mono- and disaccharides, large-scale synthesis is rare on larger glycans, such as the core pentasaccharide, and required the development of efficient synthetic routes. This dictated a global approach to all targets that would allow flexible reuse of key building blocks and intermediates in more than one target (Figure 2).
Figure 2.

Schematic of synthetic strategy toward the core pentasaccharide and its building blocks. Synthesis of the core pentasaccharide 4 and its building blocks 1–3 shown using symbol representations. The chromophore used in this study, phenyl, is shown by a red star. For full synthetic details and structures see Figure S1 for the steps describing the conversion of 5 to 1–3 and intermediate 9; Figure S2 for conversion of 18, 19, and 9 onward to 4. All glycosylations were accomplished with >98% stereoselectivity for α- (αG) or β- (βG) glycosidic linkages through the use of participatory C-2 ester substituents (Ac or Lev).
An additional key synthetic challenge was the formation of the central β-d-mannoside linkage in 2–4, notoriously difficult in carbohydrate chemistry.26−29 Several elegant methods have been reported for the direct stereoselective synthesis of β-d-mannosides,30−38 however selectivities can sometimes be low, giving unwanted α-mannoside side products32 that are difficult to remove through purification. Notably, <90% β-selectivity is the best reported to date for direct β-mannosylation of the OH-4 position of a GlcNAc precursor that would correspond to the synthesis of the core pentasaccharide.34,39,40 Since this would place severe limitations on the effective construction of grams of the chromophore-equipped targets, an alternative strategy, with potential for greater selectivity for the β-mannoside linkage, was chosen. This took advantage of neighboring group participation of a levulinate ester stereodirecting group (Lev) at the C-2 position of an unnatural glucoside residue to allow the formation of a corresponding β-glucoside linkage. The levulinate ester may be selectively deprotected. Subsequent stereospecific inversion of the configuration at C-2 (I in Figure 2) would then allow conversion of this unnatural residue from the β-d-gluco to the natural β-d-manno configuration. We chose to accomplish this critical inversion through SN2 displacement at a late stage of our syntheses, envisaging that, despite the strategic risk, this might afford more rigid intermediates that, by virtue of their reduced conformational flexibility, would be less prone to unwanted decomposition (e.g., elimination) pathways.
Synthesis of the Proximal (GlcNAc21), Central (Man-GlcNAc 2), and Extended Stem (Man-GlcNAc-GlcNAc 3) Building Blocks
A suitably chromophoric ‘stem’ unit of the core pentasaccharide was prepared as the phenyl β-chitobioside 1 (Figures 2 and S1) from parent monosaccharide sugar d-glucosamine 5. A common thioglycoside divergent precursor 6(41) was prepared with participatory phthalimide N-2 protection in six steps from d-glucosamine. 6 was divergently elaborated to both donor 7(42) and acceptor 8(43) (through OH-4-regioselective reductive benzylidene ring opening); use of a trichloroacetimidate donor group in 7 allowed its activation by TMSOTf without disruption (or aglycon exchange) of the S-ethyl (−SEt) group in acceptor 8 (Figures 2 and S1) to assemble 9. The phenyl chromophore was then introduced using phenol through activation of the −SEt group in 9 with N-iodosuccinimide (NIS) and TMSOTf; global deprotection of resulting phenyl β-glycoside 10, afforded the free, chromophore-tagged stem (GlcNAc)2 disaccharide unit 1.
The central disaccharide Man-β-1,4-GlcNAc unit 2 contains the unusual and challenging β-d-mannoside linkage. An acceptor 12 already containing the phenyl chromophore was prepared from 6 (using phenol and NIS/TMSOTf-mediated activation followed by regioselective reductive benzylidene ring opening to reveal OH-4). 12 was glycosylated using the thio-d-glucoside donor 11 containing a C-2 Lev stereodirecting group (Figures 2 and S1); activation (NIS/TMSOTf) gave disaccharide 13 with excellent (>98%) β-d-stereoselectivity. The crucial C-2 epimerization (vide supra) from β-d-gluco- to β-d-manno-configuration (13 → 14, Figure S1) was accomplished in 80% yield over three steps (selective Lev cleavage, triflate formation, and ultrasound-assisted SN2 displacement with acetate).44−47 The necessity for ultrasound treatment to promote triflate displacement was consistent with the expected stability afforded by the conformational restriction of the glucoside 13 by its 4,6-O-benzylidene acetal; this stability also ensured minimal side reaction through competing elimination. Global deprotection of 14 yielded chromophore-tagged 2 in 70% yield over four steps.
Utilizing the intended divergency of common synthetic intermediates (Figure 2), a strategically similar synthetic route gave 3; this reused intermediates used for 1 and 2. Thus, Lev-bearing donor 11 (used for 2) was again used for the ultimate installation of the challenging β-d-mannoside. 11 was combined with (GlcNAc)2-disaccharide 10 (used for 1) in which the phenyl chromophore was already present; 10 was converted to a suitable acceptor 15 through regioselective 4,6-O-benzylidene opening to reveal OH-4B (Figure S1). 11 and 15 gave trisaccharide 16 containing the desired β(1,4)-linkage (>98% stereoselectivity). Creation of the desired β-d-mannoside linkage (16 → 17) successfully employed the same inversion-epimerization strategy used for 2; global deprotection gave 3 in a very good yield of 55% over 7 steps.
Synthesis of the Chromophore-Tagged Core Pentasaccharide 4
Consistent with our strategy, the most challenging target, the core pentasaccharide 4, was created (Figures 2 and S2) using previously generated intermediate (GlcNAc)2S-ethyl disaccharide 9. This (GlcNAc)2 disaccharide was elaborated to a suitable acceptor 20, again through regioselective protecting group manipulation to reveal OH-4′ and coupled in a convergent manner with disaccharide donor 22. 22 itself was assembled from d-manno trichloroacetimidate 18(48) and the thioglucoside diol 19.49 Although 19 contains two potential sites for reaction (OH-2 and OH-3), TMSOTf-activated glycosylation proceeded with both excellent regio- (OH-3 over OH-2) and stereo- (>98% α-manno) selectivities to give 21(50) in 76% yield. Advantageously, this left free the OH-2; direct installation of the stereodirecting Lev group onto OH-2 gave 22.
Unusually, our intended disaccharide 20 + disaccharide 22 [2 + 2] glycosylation utilized a donor and an acceptor both containing the same −SEt anomeric substituent; ordinarily this would give rise to unwanted coincident activation during glycosylation. However, by using an iterative glycosylation strategy,51 thioglycoside 22 was successfully preactivated first (with triflic anhydride and diphenylsulfoxide in the presence of stabilizing base DTBMP, allowing its successful use as a donor). Subsequent addition of 20 then led to its reaction only as an acceptor nucleophile (without −SEt activation) and gave tetrasaccharide 23, in good yield and with excellent stereoselectivity for the critical central β-linkage (>98%). This therefore directly provided 23 bearing a subsequently useful −SEt group, thereby avoiding functional group manipulations at this reducing terminus.
Before installation of the final α(1,6)-d-mannosyl unit of the core pentasaccharide, we performed the critical inversion of the central unit from β-d-gluco- to β-d-manno-configuration (23 → 24) (Figures 2 and S2) as for blocks 2 and 3 (chemoselective Lev deprotection, triflate formation, and ultrasound-mediated SN2 displacement with acetate), again in excellent yields. Next, benzylidene removal revealed OH-4 and OH-6 in 25. Regioselective OH-6 glycosylation with mannosyl donor 18 (used for 22) gave pentasaccharide 26; here the use of a compatible trichloroacetimidate donor again led to retention of the useful −SEt group at the reducing terminus. Acetylation and subsequent NIS/TMSOTf-mediated activation of the −SEt with phenol installed the chromophore into protected, phenyl β-pentasaccharide 27. Three high-yielding deprotection reactions gave the final chromophore-tagged, core pentasaccharide 4 in gram-scale quantities.
The Gas-Phase IRID Spectra and Structures of Building Blocks 1–3
The R2PI spectrum of 1, the chitobiose (GlcNAcB-β1,4-GlcNAcA) stem that links the core pentasaccharide to protein, presented two overlapping components (shown later in Figure 5 and in Figure S3). Their associated IRID spectra were in best accord with its two lowest-energy conformers, termed 1-trans and 1-cis (Figure 3a), in which the two coplanar pyranose rings are anti- or syn-periplanar, respectively (see also Figure S5).
Figure 5.
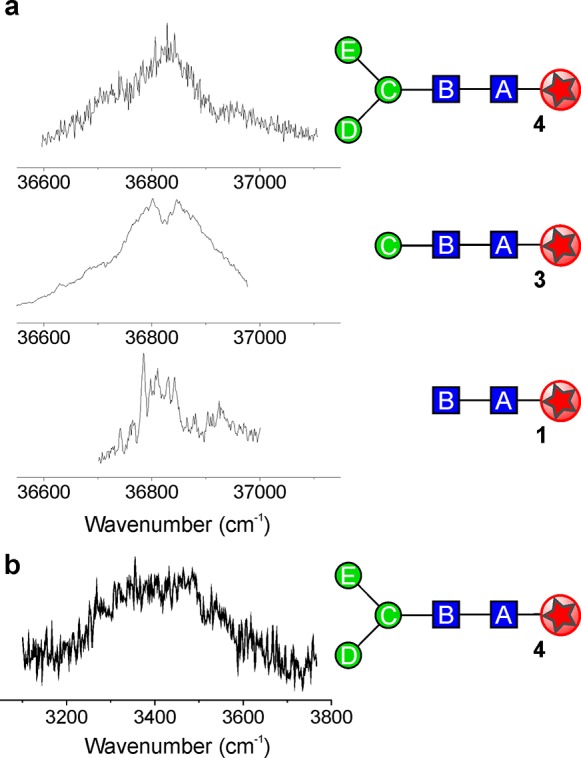
Spectra of the core pentasaccharide. (a) Resonant two-photon ionization (R2PI) spectra of the chitobiose stem, 1, the trisaccharide linker, 3, and the complete core pentasaccharide, 4. (b) The IRID spectrum of 4.
Figure 3.

The IR ion depletion (IRID) spectra, computed vibrational spectra and structures of 1, 2, and 3. σ1,2, ... and σNH indicate the OH and NH vibrational mode assignments; relative energies (at 0 K) and free energies (at 298 K), kJ mol–1, are shown in brackets. Structural assignments are based first on the correspondence between the experimental (IRID) and computed vibrational spectra and second on their relative energies, favoring the most stable conformer(s). (a) The computed vibrational spectrum of the trans conformation of 1 predicted as the global minimum, corresponded best with the experimental IRID spectrum associated with the major conformer (1-trans) while the lowest-lying cis conformation corresponded to the IRID signature of the minor conformer (1-cis). (b) The IRID and computed vibrational spectra associated with the three lowest-energy conformations of ManC-β1,4-GlcNAcB2; the two lowest (which differ only in the orientation of the hydroxymethyl group on the mannopyranoside ring) both display 2-cis conformations. The commonly observed B-ring acetamido → OH-3 interaction in the two units 1 and 2 lends further support to the ‘building block’ approach adopted here. (c) The calculated spectrum associated with the lowest energy, cis–cis conformer of 3 is in good qualitative agreement with the IRID spectrum of the trisaccharide 3, ManC-β1,4-GlcNAcB-β1,4-GlcNAcA.
The glucopyranoside rings in the 1-trans conformer are supported by two inter-ring hydrogen bonds, OH-3A → OH-6B(O-5B), while a third, stronger OH-3B → O=C(NH)B bond pulls the B-ring acetamido group into the plane of the pyranose ring. The A-ring acetamido group remains ‘free’ with its amide plane presenting a near perpendicular orientation. The 1-cis conformer (Figure 3a), which has a similar relative energy at 0 K but a higher free energy at 298 K, is supported by a strong OH-6B → OH-6A hydrogen bond; a further inter-ring interaction, NHB → OH-3A contributes to a cooperative chain, OH-4B → OH-3B → O=C(NH)B → OH-3A → O=C(NH)A, to provide additional stabilization. Since both acetamido groups are now able to interact with their neighboring OH-3A/OH-3B groups, both are rotated toward the plane of their respective pyranose rings.
The disaccharide unit −ManC-β1,4-GlcNAcB– 2 straddles the unusual β-mannoside linkage. Its IRID spectrum is in good accord with a superposition of the vibrational spectra associated with its two lowest-energy conformers (2-cis), but is quite unlike that of the lowest-lying 2-trans conformer (9.9 kJ mol–1 higher, Figure 3b). The 2-cis conformation is supported by two inter-ring hydrogen bonds, OH-2C → O-3B and OH-6C → O-6B, and the B-ring acetamido group is again hydrogen bonded to its nearest neighbor, OH-3B → O=C(NH)B.
The IRID spectrum of the −ManC-β1,4-GlcNAcB-β1,4-GlcNAcA– trisaccharide 3, (Figure 3c), which extends from ∼3200 to ∼3600 cm–1, displays three broad maxima below 3500 cm–1 and congested weaker features at higher wavenumbers, indicating contributions from strong and weakly hydrogen-bonded OH groups. Although poorly resolved, its contour is in qualitative correspondence with the IR spectrum associated with its minimum-energy structure. This presents cis-oriented ManC-GlcNAcB and GlcNAcB-GlcNAcA segments, linked together by two very strong hydrogen bonds that connect the terminal ManC and the central GlcNAcB units, and a third that links GlcNAcB with GlcNAcA. The next conformation lies 12.2 kJ mol–1 higher in energy; it only presents a single inter-residue hydrogen bond, and its calculated vibrational spectrum does not reproduce the experimental band at low wavenumber (Figure S11).
Hydrated and ‘Blocked’ Conformations of 1–3
The structural information obtained from IRID spectroscopy was developed further through computation, to explore the effect of explicit hydration and ‘blocking’ (to cap hydroxyls that would be absent in extended oligosaccharides). The lowest-energy computed structures of the monohydrate 1·H2O and also 2·H2O (Figures 4a,b, S6, and S8) now both present trans conformations, supported by the bound water molecule which bridges across the two pyranose rings. In 1·H2O this creates an extended cooperative chain, OH-4B→OH-3B → O=C(NH)B → H2O → OH-6A → O-1A, which greatly strengthens the inter-ring binding by linking NHB to O-6A and, as a consequence, also strengthens the supporting inter-ring bond, OH-3A → OH-6B(O-5B) (r[OH3A···O6B] 2.16 → 2.07 Å). Its relative energy now lies 4.5 kJ mol–1 below that of the lowest-lying cis hydrate, cf. 0.6 kJ mol–1 in the ‘bare’ unsolvated disaccharide 1 (Figure S6). This contrasts markedly with hydrated cellobiose (Glc-β1,4-Glc) where a cis conformation is retained.52 Explicit hydration therefore locks the trans conformation of 1, enhancing the rigidity of the ‘stem’. NMR studies of chitobiose in an aqueous environment at 298 K also identify a very similar average conformation in solution.17 The two acetamido groups continue to adopt a trans relative disposition, consistent with the maintenance of a rigid conformation about the inter-ring glycosidic bond,17 reinforced perhaps by transiently bound, bridging water molecules.53,54
Figure 4.
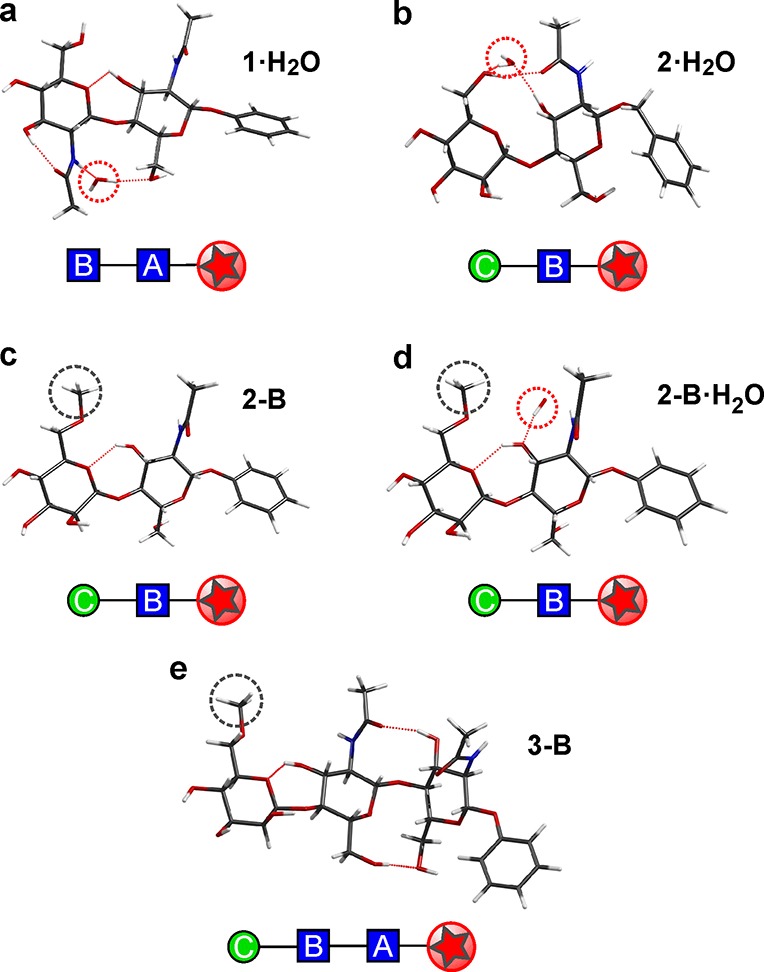
Hydrated and ‘blocked’ structures of the core pentasaccharide building blocks. The computed lowest-energy structures of (a,b), the monohydrates 1·H2O and 2·H2O and (c,d) the “blocked” subunits 2-B and 2-B·H2O, in which the 6-OHC group is modified to the methyl ether, and (e) the blocked subunit 3-B. Note the effect of blocking; in 2-B·H2O it removes the water bridge present in 2·H2O (and 1·H2O), and in 2-B and 3-B it switches the conformation about the ManC-β1,4C-GlcNAcB linkage from cis to trans. Gray dotted circles indicate ‘blocked’ sites, explored here through the use of a methyl ether capping group. Red dotted circles indicate a water binding site.
The central glycosyl residue in the core pentasaccharide, ManC, is itself glycosylated on hydroxyl groups OH-6 and OH-3, thereby precluding donor hydrogen bonds from these positions.15,55 The dramatic structural consequences of this are revealed in the finding that the lowest-energy conformer of the ‘blocked’ Man (6-OMe)C-β1,4-GlcNAcB– disaccharide 2-B (and also its hydrate, 2-B·H2O) adopts a trans orientation, in contrast to the strongly preferred cis conformation of 2 (compare Figures 3b and 4c); the lowest-energy 2-B-cis conformer now lies ∼5 kJ mol–1 above the global minimum. Unlike 2-cis (Figure 3b), the trans conformer of 2-B is only supported by a single, weak inter-ring hydrogen bond, OH-3B → O-5C. The trans conformation is also retained in the hydrate (Figure 4d) where the water molecule localizes on the GlcNAcB ring, inserted between the acetamido group and O-3 rather than forming an inter-ring bridge, again in contrast to hydrated chitobiose.52 The reduced inter-ring bonding increases the flexibility about the ManC-GlcNAcB glycosidic linkage, to provide a fluxional ‘pivot’.
Similarly, in 3-B the blocked ManC OH-6 group lacks the hydrogen bond found in 3 that linked the ManC and GlcNAcB units (Figure 4e). The ManC-GlcNAcB segment again switches from cis to trans to create a more flexible local structure about the β-ManC ‘pivot’, very similar to that of 2-B, and the trisaccharide adopts a more extended structure, with r[O-4C···O-1A] increasing from 11.2 to 14.5 Å.
Gas-Phase Spectra and Structures of the Core Pentasaccharide 4
The R2PI spectrum of the core pentasaccharide 4 (Figure 5a) is centered at the same wavenumber as the trans conformer of 1 and 3 (also trans about GlcNAcB-GlcNAcA), providing circumstantial evidence for a possible preference for a trans GlcNAcB-GlcNAcA conformation in 4 also. Since the pentasaccharide contains 14 hydroxy and 2 acetamido groups, structures that optimize global OH···O and NH···O hydrogen bonding are to be expected in the gas phase: not surprisingly, its IRID spectrum (Figure 5b) presents a broad red-shifted quasi-continuum, ranging from ∼3100 to ∼3700 cm–1. This suggests a highly congested set of overlapping bands associated with a large number of both strong, and weak, hydrogen bonded OH and acetamido groups.
Molecular mechanics simulations, undertaken partly in response to the limitations of the experiment (see also Supporting Information Methods and Results and Figures S13 and S14), also predicted a preference for a large number of hydrogen bonds (defined by r[OH···O] < 2.5 Å, θ[OH···O] > 120°) in the isolated pentasaccharide, see Figure 6a(i), and also its triply hydrated complex, Figure 6a(ii), with 6–8 being the optimal number for the isolated molecule (using the OPLS2005 force field, Figure S13). The simulations in bulk water, Figure 6a(iii), predicted disruption of the intramolecular networks, leaving only 2–4 hydrogen bonds in the low-energy ensemble (Figure S13).
Figure 6.

The spectra and structures of the core pentasaccharide 4. (a) The lowest-energy structures of the core pentasaccharide calculated on the OPLS2005 and GLYCAM06/AMBER potential energy surfaces: (i) the isolated molecule; (ii) the triply hydrated complex (the water molecules were initially located at binding sites based upon the preferences of singly hydrated 1, 2, and the trimannosyl ManE(ManD)ManC– head unit15); and (iii) in bulk water, (hydrogen bonds shown in red). (iv) An overlay of the “open” conformer of the trimannosyl ManE(ManD)ManC– head unit and the core pentasaccharide in (v); (v) the preferred aqueous structure of the high mannose glycan, Man9GlcNAc2, determined through NMR measurements and molecular dynamics simulations.12,13 Red dots represent transiently bound water molecules. (b) Distributions of the longest intramolecular distances (for conformers with energies <30 kJ mol–1) in the core pentasaccharide, predicted by molecular mechanics (OPLS2005) simulations: isolated, unsolvated (red), explicitly hydrated (green), and in bulk water (black).
As a consequence, the unsolvated pentasaccharide 4, preferentially adopted compact structures with the distal Man3 head unit folded back, supported by hydrogen-bonded interactions with the proximal GlcNAc2 unit, which presented a cis conformation about the −GlcNAcB-GlcNAcA– linkage (although trans conformations, generally associated with more extended structures, were also present at higher relative energies). The addition of water molecules in the gas phase, Figure 6a(ii), disrupted the intramolecular hydrogen-bonding pattern. The lowest-energy conformers of the triply hydrated complex, Figure 6a(ii), presented more extended structures, now with a trans configuration about the −GlcNAcB-GlcNAcA– linkage (Figure 4), supported by water bridging across the inter-ring bond and extended cooperative hydrogen-bonded networks (which were conserved in both low and high energy structures).
In bulk water, the intramolecular hydrogen bonds were largely ‘washed out’; the (near identical) lowest-energy structures predicted by OPLS2005 and GLYCAM06/AMBER, Figure 6a(iii), again presented a trans GlcNAcB-GlcNAcA– stem, but now part of a fully extended structure in which the distal Man3 unit was completely unfurled. Strikingly, apart from the difference in the dihedral angle ω(H1-C1-O6-C6) between ManC and ManE, the preferred structure of the solvated core pentasaccharide was very similar to that of the core unit in the high-mannose glycan, Man9GlcNAc2, determined through coupled NMR measurements and molecular dynamics simulation using the GLYCAM93/AMBER force field, Figure 6a(iv,v).12,13,18−20 There were also remarkable similarities between the conformations of the distal Man3– head unit in bulk water, found here, and the isolated (hydrated) Man3 unit, determined in solution18−20 and predicted through investigations in the gas phase,15 Figure 6a(iv), and also between the conformation (and flexibility) of the ManC-GlcNAcB segments in the ‘blocked’ units 2-B and 3-B and in the core pentasaccharide 4.
The analysis of intramolecular distances (using the OPLS2005 simulations), shown in Figures 6b and S14, provided a way of estimating molecular size56 and hence the favorability of extended versus compact structures. The longest end-to-end distribution for the unsolvated, isolated core pentasaccharide reflected flexibility, but the most favored distance (∼16.7 Å) indicated a compact structure corresponding to a geometric cross-section (σ ∼ 230 Å2) similar to the gas kinetic cross-section (σ ∼ 260 Å2) reported for the doubly sodiated core pentasaccharide ion,3 which is also likely to be compact. Addition of three water molecules resulted in a broad distribution with peaks at ∼21.5 and 23.5 Å, indicating a more extended but still flexible structure. In bulk water, however, the structures were exclusively extended and relatively inflexible, with a narrow spread of end-to-end distances peaked at ∼23.4 Å. The experiments and simulations have revealed a folded ‘naked’ core pentasaccharide that uncurled as it became hydrated, with conserved structural motifs taking shape, in particular the trans conformation of the proximal chitobiose unit, to provide an extended, well-defined structure in aqueous solution.
Discussion and Conclusions
A recent bioinformatics analysis57 of thousands of glycoproteins listed in the PDB58 found that many N-glycans showed significantly similar substructures close to the protein, suggesting their use as fragments in glycan modeling (and supporting the approach employed in the present, experimentally based study). The N-glycans also displayed a rigid protein-proximal GlcNAcB-GlcNAcA stem.
The acetamido groups of the stem appear to play a key role; in the gas phase the preferred trans conformation of 1 is strengthened by microhydration through an inter-ring bridging water molecule linking the GlcNAcB acetamide to O-6A. In bulk water, molecular dynamic simulations predict the absence of a direct inter-ring bond between these groups, which suggests the trans chitobiose stem is held in a rigid extended conformation through ‘filling’ of a conserved water pocket at the local bridging site. The other GlcNAcA acetamide is predicted to be unbonded, leaving it free to act as a hydrogen-bond donor/acceptor and ‘anchor’ the glycan to the peptide backbone of the glycoprotein.10,11,59 Hexoses (such as the more abundant d-glucose, Glc) that lack this acetamide could not form such strong hydrogen bonds (if they were to play the roles of GlcNAcA and GlcNAcB) and would not match the rigidity of the GlcNAc2 stem nor the potential for anchoring with the protein backbone.10,11,59
The next structural feature, provided by the central β-mannoside ManC-GlcNAcB, also contains a glycosidic linkage formed through OH6C that precludes formation of the OH6C → O6B inter-ring hydrogen bond found in the isolated unit 2. Instead of the compact cis conformation adopted by 2, supported by two inter-ring hydrogen bonds, the ‘deletion’ of one of them has the key effect of greatly increasing the flexibility of the β-mannoside linkage, which adopts a trans conformation that is unaffected by discrete hydration. Its inherent flexibility allows the β-mannosyl residue to act effectively as a pivot between the rigid chitobiose stem and the outer trimannosyl head.
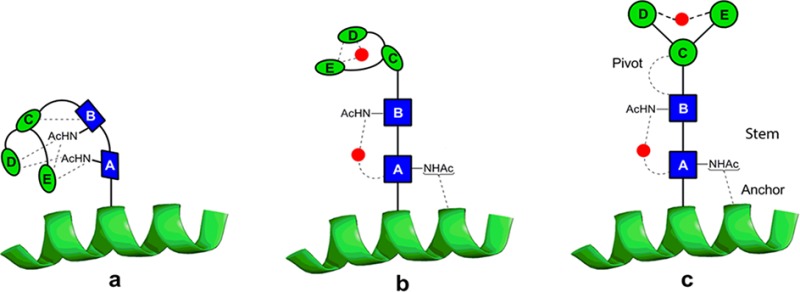
Unsolvated structures of 4 are compacted by many intramolecular hydrogen bonds (Figure 6a(i)). However, discrete hydration sees the structure begin to unfurl with a water molecule stabilizing the rigid chitobiose stem (vide supra), while the trimannosyl head unit folds over to encapsulate water in another pocket. In a fully aqueous environment, a picture emerges (Figure 7) of an N-linked glycan accommodating an extended core pentasaccharide structural unit that incorporates a rigid proximal chitobiose stem, anchored at one end to the adjoining protein10,11,59 and at the other connected through a more flexible β-mannoside pivot to the branched mannosyl D and E arms.
Figure 7.
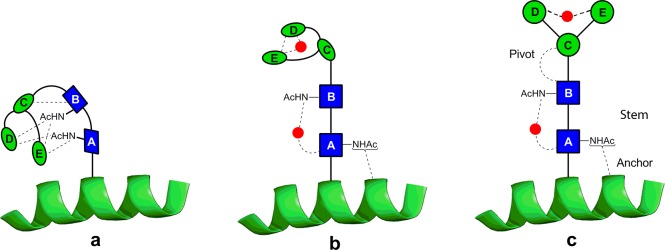
An extrapolated structural model for the N-glycan core in an aqueous environment. In the absence of water (a), the pentasaccharide would adopt a compact conformation, but the interactions between the building blocks of the core pentasaccharide and water (b) lead, in solution (c), to a rigid chitobiose stem that is anchored at one end to the peptide10,11,59 and at the other, through a flexible ‘pivot’, to a β-mannosyl structure that projects the information-rich distal head of the glycan for interaction with other partners. The red dots indicate favored water-bridging sites identified here or in previous investigations.15
This combines both structural integrity and freedom of movement to allow interaction with binding partners. The extended display of the N-glycan would leave the distal branch-tip sugars free to function as potential ligands for receptors and also provide the potential for ‘levered’ interactions promoting a conformational ‘response’ in the underlying protein fold. The latter could explain the apparent ability of the calnexin/calreticulin chaperone system to bind the tips of the sugars found on N-glycoproteins60 while still being able to stimulate proper folding of the underlying N-glycoprotein. The selection of chitobiose, with its two acetamido groups rather than cellobiose for the stem, appears to be critical, facilitating hydrogen-bonded interactions with neighboring amino acid residues10,11 and suggesting a mechanism by which the core pentasaccharide influences the structural stability of the adjacent peptide chain and its folding kinetics.7 The central ManE(ManD)ManC– unit, which unfurls upon hydration, presents the information-rich outer extremities of N-glycans to the environment (for solubility, recognition, or transport).
Acknowledgments
We are grateful to the following for their support: the Leverhulme Trust for the award of an Emeritus Fellowship (J.P.S.); the Royal Society and the Wolfson Foundation for a Research Merit Award (B.G.D.); the EPSRC (B.G.D., J.P.S.); the Spanish Ministry (MICINN) for a project CTQ2011-22923 and “Ramón y Cajal” Contract (E.J.C.); the STFC for the provision of equipment from the Laser Loan Pool; the Oxford Supercomputing Centre and SGIker at UPV-EHU. We are especially grateful for the advice and assistance provided Professor Jesus Jiménez-Barbero and Professor David Clary and thank Dr. Mark Wormald for helpful discussions. Experiments performed in Orsay were supported by Triangle de la Physique, contract 2010-079T.
Supporting Information Available
Synthetic methods; NMR data; spectroscopic and computational methods; computed relative energies, vibrational frequencies, and structural data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Departamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco, (UPV–EHU), Apartado 644, E-48940, Bilbao, Spain.
Author Present Address
∇ Institut für Experimentalphysik, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany.
Author Contributions
# These authors contributed equally
The authors declare no competing financial interest.
Supplementary Material
References
- Weerapana E.; Imperiali B. Glycobiology 2006, 16, 91r. [DOI] [PubMed] [Google Scholar]
- Yan A. X.; Lennarz W. J. J. Biol. Chem. 2005, 280, 3121. [DOI] [PubMed] [Google Scholar]
- Plasencia M. D.; Isailovic D.; Merenbloom S. I.; Mechref Y.; Clemmer D. E. J. Am. Soc. Mass Spectrom. 2008, 19, 1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse B. R.; Hebert D. N. Biochim. Biophys. Acta, Mol. Cell Res. 2010, 1803, 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J. N.; Wormald M. R.; Sim R. B.; Rudd P. M.; Dwek R. A. Annu. Rev. Immunol. 2007, 25, 21. [DOI] [PubMed] [Google Scholar]
- Walsh S. T. R. Biochemistry 2010, 49, 8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson S. R.; Culyba E. K.; Hsu T. L.; Wong C.-H.; Kelly J. W.; Powers E. T. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormald M. R.; Dwek R. A. Structure 1999, 7, R155. [DOI] [PubMed] [Google Scholar]
- Schwarz F.; Aebi M. Curr. Opin. Struct. Biol. 2011, 21, 576. [DOI] [PubMed] [Google Scholar]
- O’Connor S. E.; Imperiali B. Chem. Biol. 1996, 3, 803. [DOI] [PubMed] [Google Scholar]
- O’Connor S. E.; Imperiali B. Chem. Biol. 1998, 5, 427. [DOI] [PubMed] [Google Scholar]
- Woods R. J.; Pathiaseril A.; Wormald M. R.; Edge C. J.; Dwek R. A. Eur. J. Biochem. 1998, 258, 372. [DOI] [PubMed] [Google Scholar]
- Wormald M. R.; Petrescu A. J.; Pao Y. L.; Glithero A.; Elliott T.; Dwek R. A. Chem. Rev. 2002, 102, 371. [DOI] [PubMed] [Google Scholar]
- Cocinero E. J.; Stanca-Kaposta E. C.; Gamblin D. P.; Davis B. G.; Simons J. P. J. Am. Chem. Soc. 2009, 131, 1282. [DOI] [PubMed] [Google Scholar]
- Stanca-Kaposta E. C.; Gamblin D. P.; Cocinero E. J.; Frey J.; Kroemer R. T.; Fairbanks A. J.; Davis B. G.; Simons J. P. J. Am. Chem. Soc. 2008, 130, 10691. [DOI] [PubMed] [Google Scholar]
- Simons J. P. Mol. Phys. 2009, 107, 2435. [Google Scholar]
- Yu F.; Prestegard J. H. Biophys. J. 2006, 91, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almond A.; Duus J. O. J. Biomol. NMR 2001, 20, 351. [DOI] [PubMed] [Google Scholar]
- Sayers E. W.; Prestegard J. H. Biophys. J. 2000, 79, 3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F.; Al-Hashimi H. M.; Craighead J. L.; Prestegard J. H. J. Am. Chem. Soc. 2001, 123, 485. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. J. Comput. Chem. 1999, 20, 730. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; M. A. R.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T. Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2010.
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. J. Phys. Chem. B 2001, 105, 6474. [Google Scholar]
- Kirschner K. N.; Yongye A. B.; Tschampel S. M.; González-Outeiriño J.; Daniels C. R.; Foley B. L.; Woods R. J. J. Comput. Chem. 2008, 29, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still W. C.; Galynker I. Tetrahedron 1981, 37, 3981. [Google Scholar]
- Demchenko A. V. Curr. Org. Chem. 2003, 7, 35. [Google Scholar]
- Fairbanks A. J. Synlett 2003, 1945. [Google Scholar]
- Gridley J. J.; Osborn H. M. I. J. Chem. Soc., Perkin Trans. 1 2000, 1471. [Google Scholar]
- Paulsen H. Angew. Chem., Int. Ed. Engl. 1990, 29, 823. [Google Scholar]
- Barresi F.; Hindsgaul O. J. Am. Chem. Soc. 1991, 113, 9376. [Google Scholar]
- Barresi F.; Hindsgaul O. Can. J. Chem. 1994, 72, 1447. [Google Scholar]
- Crich D.; Chandrasekera N. S. Angew. Chem., Int. Ed. Engl. 2004, 43, 5386. [DOI] [PubMed] [Google Scholar]
- Crich D.; Sun S. X. J. Org. Chem. 1997, 62, 1198. [Google Scholar]
- Crich D.; Sun S. X. Tetrahedron 1998, 54, 8321. [Google Scholar]
- Cumpstey I.; Fairbanks A. J.; Redgrave A. J. Org. Lett. 2001, 3, 2371. [DOI] [PubMed] [Google Scholar]
- Ito Y.; Ogawa T. Angew. Chem., Int. Ed. Engl. 1994, 33, 1765. [Google Scholar]
- Ito Y.; Ohnishi Y.; Ogawa T.; Nakahara Y. Synlett 1998, 1102. [Google Scholar]
- Seward C. M. P.; Cumpstey I.; Aloui M.; Ennis S. C.; Redgrave A. J.; Fairbanks A. J. Chem. Commun. 2000, 1409. [Google Scholar]
- Crich D.; Sun S. X. J. Am. Chem. Soc. 1998, 120, 435. [Google Scholar]
- Dudkin V. Y.; Miller J. S.; Danishefsky S. J. Tetrahedron Lett. 2003, 44, 1791. [Google Scholar]
- Nagorny P.; Fasching B.; Li X. C.; Chen G.; Aussedat B.; Danishefsky S. J. J. Am. Chem. Soc. 2009, 131, 5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte M. D.; Horst D.; Wiertz E. J. H. J.; van der Marel G. A.; Overkleeft H. S. J. Org. Chem. 2009, 74, 605. [DOI] [PubMed] [Google Scholar]
- Macindoe W. M.; Nakahara Y.; Ogawa T. Carbohydr. Res. 1995, 271, 207. [DOI] [PubMed] [Google Scholar]
- Gunther W.; Kunz H. Carbohydr. Res. 1992, 228, 217. [DOI] [PubMed] [Google Scholar]
- Kunz H.; Gunther W. Angew. Chem., Int. Ed. Engl. 1988, 27, 1086. [Google Scholar]
- Matsuo I.; Isomura M.; Ajisaka K. J. Carbohydr. Chem. 1999, 18, 841. [Google Scholar]
- Watt G. M.; Boons G. J. Carbohydr. Res. 2004, 339, 181. [DOI] [PubMed] [Google Scholar]
- Yamazaki F.; Sato S.; Nukada T.; Ito Y.; Ogawa T. Carbohydr. Res. 1990, 201, 31. [DOI] [PubMed] [Google Scholar]
- Soderberg E.; Westman J.; Oscarson S. J. Carbohydr. Chem. 2001, 20, 397. [Google Scholar]
- Rising T. W. D. F.; Claridge T. D. W.; Davies N.; Gamblin D. P.; Moir J. W. B.; Fairbanks A. J. Carbohydr. Res. 2006, 341, 1574. [DOI] [PubMed] [Google Scholar]
- Codee J. D. C.; Litjens R. E. J. N.; den Heeten R.; Overkleeft H. S.; van Boom J. H.; van der Marel G. A. Org. Lett. 2003, 5, 1519. [DOI] [PubMed] [Google Scholar]
- Cocinero E. J.; Gamblin D. P.; Davis B. G.; Simons J. P. J. Am. Chem. Soc. 2009, 131, 11117. [DOI] [PubMed] [Google Scholar]
- Almond A. Carbohydr. Res. 2005, 340, 907. [DOI] [PubMed] [Google Scholar]
- Almond A.; Sheehan J. K. Glycobiology 2003, 13, 255. [DOI] [PubMed] [Google Scholar]
- Carcabal P.; Hünig I.; Gamblin D. P.; Liu B.; Jockusch R. A.; Kroemer R. T.; Snoek L. C.; Fairbanks A. J.; Davis B. G.; Simons J. P. J. Am. Chem. Soc. 2006, 128, 1976. [DOI] [PubMed] [Google Scholar]
- Remmert S.; Hollis H.; Parish C. A. Bioorg. Med. Chem. 2009, 17, 1251. [DOI] [PubMed] [Google Scholar]
- Jo S.; Lee H. S.; Skolnick J.; Im W. PLoS Comput. Biol. 2013, 9, e1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. Nucleic Acids Res. 2000, 28, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosques C. J.; Tschampel S. M.; Woods R. J.; Imperiali B. J. Am. Chem. Soc. 2004, 126, 8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi M.; Bernasconi R.; Clerc S.; Molinari M. Trends Biochem. Sci. 2010, 35, 74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.