

Abstract
A novel β-glucosidase (BglPm) was identified from Paenibacillus mucilaginosus KCTC 3870T which has ginsenoside converting activity. The gene, termed bglPm, consists of 1,260 bp and belongs to glycoside hydrolase family 1 (GH1). After being overexpressed and purified from Escherichia coli, the enzymatic properties of BglPm were investigated. The enzyme exhibited an optimal activity at 45°C and pH 7.5 and showed high bioconversion ability for major ginsenoside Rb1 and Rd into ginsenoside F2. Thus, it was used for mass production of relatively high pure F2 from relatively abundant protopanaxadiol type ginsenosides mixture (PPDGM) with combined usage of ginsenoside Rc-hydrolyzing enzyme. Scale-up of production using 250 g of the PPDGM resulted in 152 g of F2 with 80.1% chromatography purity and 95.7% recovery. These results suggest that this enzyme would be useful in the preparation of pharmacologically active ginsenoside F2 in the functional food and pharmaceutical industries.
Introduction
The root of Panax ginseng C.A. Meyer is commonly used in Northeast Asian countries as a traditional medicine for more than 2000 years and has become better known in the West during the past few decades [1], [2], [3]. Therapeutic effects of ginseng such as anti-neoplastic, anti-stress, and anti-oxidant activities have been attributed mainly to the primary active ingredients-ginsenosides [4], [5], [6], [7]. More than 180 different ginsenosides have been identified, and they can be categorized as protopanaxadiol (PPD), protopanaxatriol (PPT), and oleanane saponins based on the structure of the aglycon [5], [6], [8]. Ginsenosides Rb1, Rb2, Rc, Rd, Re, and Rg1 are major ginsenosides that make up more than 80% of total ginseng ginsenosides [4], [5], [9], and minor ginsenosides (Rg3, Rh2, F2, C-K, Rg2, Rh1, and F1) that are deglycosylated from the major ginsenosides exist in smaller amounts or are absent in ginseng. The deglycosylated minor ginsenosides have some chemical reactivity that the major ginsenosides do not. Furthermore, emerging evidence has demonstrated that the minor ginsenosides have more important pharmaceutical effects, such as anti-cancer, anti-diabetic, anti-oxidative, and anti-aging effects, than the glycosylated major ginsenosides [10], [11], [12], [13].
As a minor ginsenoside, F2 accounts for less than 0.01% in raw ginseng and red ginseng (a heat-treated ginseng with more minor ginsenosides) [14], and thus isolation of F2 from natural products is difficult. F2 has been produced via bioconversion of PPD type ginsenosides [i.e. Rb1, gypenoside XVII (Gyp XVII), Rd etc.] using fungal β-glucosidase or recombinant β-glucosidase derived from bacteria [15], [16]. Owing to the difficulty of usage of research material, a few pharmaceutical activities have thus far been surveyed using F2, which was also gained using biotransformation. F2 exerted effects against malignant brain tumor and breast cancer stem cells [17], [18]. Thus, it is imperative to develop mass production of F2 for its application as a functional material for cosmetics, functional health supplements, and drugs. Although some researchers have identified ginsenoside bioconversion enzymes which can produce F2 from major ginsenosides [19], they only conducted simple enzyme characterizations without further scale-up or process engineering.
Attempts to produce gram-scale ginsenosides have been made using microbial method. The major ginsenoside Rd has been produced on a gram-scale from the pure ginsenoside Rb1 using Paecilomyces bainier 229-7 [20]. Thus, it is timely to design and develop a means of mass production of minor ginsenosides to meet industrial demand and fulfill their original purpose of application as a recombinant enzyme. Recently, minor ginsenoside Rg3(S) was produced successfully as 100 g unit with relatively high purity using two recombinant glycoside hydrolases in series by our group [21].
In the present study, a ginsenoside-transforming β-glucosidase was cloned from Paenibacillus mucilaginosus KCTC 3870T. The recombinant protein, BglPm, was purified and the enzymatic properties were investigated. This enzyme showed strong ginsenoside-transformation ability, especially major ginsenoside Rb1 and Rd into minor ginsenoside F2. Furthermore, enhanced production of F2 from relatively abundant protopanaxadiol type ginsenosides mixture (PPDGM) from ginseng extraction was performed using recombinant BglPm and another α-L-arabinofuranosidase (Abf22-3) with ginsenoside-Rc transformation activity from Leuconostoc sp. 22-3 which has been cloned by our group [22]. BglPm displayed excellent F2-production activities and can be used for mass production of relatively pure compound from abundant PPDGM and may prompt the pharmacological studies and applications of rare ginsenoside F2.
Methods
2.1. Materials
The PPD type ginsenosides mixture (PPDGM) from the root of Panax quinquefolius [comprised of Rb1: 53.8%, Rc: 15.8%, Rb2: 2.8%, Rb3: 4.8%, Rd: 16.7%, Rg3(S): 1.7% and other ginsenosides: 4.4% based on mole ratio] from Hongjiou Biotech Co. Ltd. (China) was used as the substrate in the current investigation. Ginsenosides standards which are over 98% purity such as Rb1, Rc, Rb2, Rd, Rg3(S), Rh2(S), F2, compound K (C-K), PPD, Rg1, Re, Rg2(S), Rh1(S) and PPT were purchased from Nanjing Zelang Medical Technology Co., Ltd. (China). Methanol and acetonitrile with HPLC grade were obtained from Merck (Darmstadt, Germany). Gypenoside XVII (Gyp XVII), compound O (C-O), compound Mc1 (C-Mc1), and compound Mx1 (C-Mx1) were prepared as described by An et al. (2010) and Wang et al. (2011). The other chemicals used in this study were a minimum of analytical reagent grade, and the sources are noted individually in the Methods section. Recombinant α-L-arabinofuranoside (Abf22-3) was prepared as described [22]. The genomic DNA from Paenibacillus mucilaginosus KCTC 3870T, Escherichia coli BL21 (DE3), and pGEX 4T-1 plasmid (GE Healthcare, USA) were used as β-glucosidase gene, host, and expression vector sources, respectively. P. mucilaginosus KCTC 3870T was grown in aerobic conditions at 37°C on nutrient agar (NA, BD, USA). The recombinant E. coli for protein expression was cultivated in a Luria-Bertani (LB) medium supplemented with ampicillin (100 mg/l).
2.2. Analysis of BglPm sequence
Database homology search was performed with BLAST program provided by NCBI. Furthermore, the multiple amino acid sequence alignment and the conserved patterns of discrete amino acid sequences of BglPm and known the most homologous β-glucosidases were performed by using ClustalW program (http://embnet.vital-it.ch/software/ClustalW.html).
2.3. Molecular cloning, expression, and purification of recombinant BglPm
The genomic DNA from P. mucilaginosus KCTC 3870T was extracted using a genomic DNA extraction kit (Solgent, Korea). The gene encoding β-glucosidase was amplified from the genomic DNA as a template via a polymerase chain reaction (PCR) using Pfu DNA polymerase (Solgent, Korea). The sequence of the oligonucleotide primers used for the gene cloning was based on the DNA sequence of β-glucosidase (GenBank accession number: AEI42200). Forward (5′- G GTT CCG CGT GGA TCC GAA TAT ATT TTT CCA CAG CAA TTT -3′) and reverse (5′- G ATG CGG CCG CTC GAG TTA CAG CAC TTT CGT GGA TGC GAT -3′) primers were designed as primers to introduce the BamHI and XhoI restriction sites (underlined), respectively, and were synthesized by Macrogen Co. Ltd. (Korea). The amplified DNA fragment obtained from the PCR was purified and inserted into the pGEX 4T-1 GST fusion vector digested with BamHI and XhoI using an EzCloning Kit (Enzynomics Co. Ltd., Korea). The resulting recombinant pGEX-bglPm was transformed into E. coli BL21(DE3). The E. coli BL21(DE3) harboring the recombinant plasmid was grown in an LB-ampicillin medium at 37°C until the culture reached an OD600 of 0.6, at which point the protein expression was induced through the addition of 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The bacterial cells were incubated for a further 18 h at 30°C and were then harvested via centrifugation at 13,000 rpm for 15 min at 4°C. The cells were washed twice with a solution consisting of 50 mM sodium phosphate, 5 mM EDTA, and 1% Triton X-100 (pH 7.0); then, they were resuspended in 50 mM sodium phosphate (pH 7.0). The cells were disrupted via ultrasonication (Vibra-cell, Sonics & Materials, CT, USA). The intact cells and debris were removed via centrifugation at 13,000 rpm for 15 min at 4°C in order to obtain the crude cell extract. The GST tag was purified using the GST·bind agarose resin (Elpisbiotech Co. Ltd, Korea). The homogeneity of the protein was assessed using 10% SDS-PAGE and an EZ-Gel staining solution (Daeillab Co. Ltd., Korea).
2.4. Effect of pH, temperature, metal ions and chemical reagent on enzyme activity
The specific activity of purified BglPm was determined using p-nitrophenyl-β-D-glucopyranoside (pNPGlc) as a surrogate substrate in 50 mM sodium phosphate buffer, pH 7.5 at 37°C. Reactions were stopped after 10 minutes (min) by the addition of Na2CO3 at a final concentration of 0.5 M, and the release of p-nitrophenol was measured immediately using a microplate reader at 405 nm (Bio-Rad model 680; Bio-Rad, Hercules, CA). One unit of activity was defined as the amount of protein required to generate 1 µmol of p-nitrophenol per min. Specific activity was expressed as units per milligram of protein. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL), with bovine serum albumin (Sigma) as the standard. All assays were performed in triplicate.
The effect of pH on enzymatic activity was determined using 2.0 mM pNPGlc as a substrate in the following buffers (each at 50 mM): KCl-HCl (pH 2.0), glycine-HCl (pH 3.0), sodium acetate (pH 4.0 and 5.0), sodium phosphate (pH 6.0, 7.0 and 7.5), Tris-HCl (pH 8.0, and 9.0) and glycine-sodium hydroxide (pH 10). The pH stability of recombinant BglPm was determined by measuring enzymatic activity after incubation in each buffer (containing 2.0 mM pNPGlc in 50 mM potassium buffer as a substrate) for 12 h at 4°C. The results are expressed as a percentage of the activity obtained at the optimum pH. The effect of temperature on enzymatic activity was tested by incubating the enzyme at various temperatures ranging from 4 to 65°C at optimum pH for 10 min in 50 mM potassium phosphate buffer containing 2.0 mM pNPGlc. The thermostability of the enzyme was examined by incubating the enzyme in 50 mM potassium phosphate buffer for 30 min at different temperatures. After cooling the sample on ice for 10 min, activity was determined using pNPGlc as the substrate.
The effects of metals and other chemicals on BglPm activity were also determined. BglPm activity was tested in the presence of 1 or 10 mM (final concentration) of HgCl2, MnCl2, CaCl2, CoCl2, MgCl2, EDTA, NaCl, KCl, CuCl2, SDS, dithiothreitol (DTT), or β-mercaptoethanol for 30 min at 37°C. The remaining activity was determined using pNPGlc as a substrate, and activities are expressed as a percentage of the activity obtained in the absence of the compound.
Substrate preference was examined using 2.0 mM chromogenic o-nitrophenyl (ONP) and p-nitrophenyl (PNP) as substrates at 37°C for 10 min, with one activity unit being defined as the release of 1 µmol o-nitrophenol or p-nitrophenol per min. The following substrates were tested: PNP-β-D-glucopyranoside, PNP-β-D-galactopyranoside, PNP-β-D-fucopyranoside, PNP-N-acetyl-β-D-glucosaminide, PNP-β-L-arabinopyranoside, PNP-β-D-mannopyranoside, PNP-β-D-xylopyranoside, PNP-α-D-glucopyranoside, PNP-α-L-arabinofuranoside, PNP-α-L-arabinopyranoside, PNP-α-L-rhamnopyranoside, PNP-α-D-mannopyranoside, PNP-α-D-xylopyranoside, ONP-β-D-glucopyranoside, ONP-β-D-galactopyranoside, ONP-β-D-fucopyranoside and ONP-α-D-galactopyranoside (all from Sigma).
2.5. Determination of kinetic parameters
Kinetic studies were performed with freshly purified enzymes using pNPGlc at 0.1–10.0 mM, Rb1, Gyp XVII and Rd at concentrations from 0.2 mM to 5.0 mM. One unit of activity was defined as the amount of protein required to generate 1 µmol of p-nitrophenol or to convert 1 µmol of Rb1 or Gyp XVII or Rd per minute. All assays were performed in triplicate. The parameters, Km and Vmax, were determined using the enzyme kinetics program described by Cleland [23].
2.6. Biotransformation activity of PPD ginsenosides using BglPm
The initial biotransformation experiments using the major ginsenosides Rb1 and Rd as substrates revealed that the GST-fused enzyme does not affect the activities of BglPm. Therefore, the fusion protein (GST-BglPm) was used to determine the specificity and selectivity of the enzymes for the hydrolysis of the glucose moieties attached at the C3 and C20 sites in the seven PPD ginsenosides. The enzyme solutions at a concentration of 0.1 mg/ml in 50 mM of sodium phosphate buffer (pH 7.5) were reacted with an equal volume of Rb1, Rb2, Rb3, Rc, Rd, Gyp XVII and Rg3(S) solution at a concentration of 0.1% (wt/vol) in 50 mM of sodium phosphate buffer (pH 7.5) at 37°C. The samples were taken at regular intervals and analyzed via TLC or HPLC after pretreatment (see analytic methods).
2.7. Optimization of concentration of the substrate
In order to determine the optimal condition for the biotransformation of PPDGM to F2, the substrate concentration of PPDGM in the reaction was optimized. The final crude BglPm concentration was fixed to 10 mg/ml (32 U/ml of pNPGlc activity) and reacted with an equal volume of 20, 50, 100 and 150 mg/ml of PPDGM in order to have 10, 25, 50 and 75 mg/ml as the final substrate concentrations. These 4 types of optimization reactions were performed in a 50 ml conical tube with a 10 ml working volume at 150 rpm for 12 h at 37°C. The samples were taken at regular intervals and analyzed via HPLC.
2.8. Scaled-up biotransformation of PPDGM
2.8.1. Preparation of two recombinant enzymes using high cell density culture
For the production of recombinant Abf22-3, a Luria-Bertani (LB) medium supplemented by ampicillin (100 µg/ml) was used for the cultivation of E. coli harboring pMBP-abf22-3 in a 10 L stirred-tank reactor (Biotron GX, Hanil science Co., Korea) with a 5 L working volume at 500 rpm. The pH value of the medium was adjusted to be 7.0 using 100 mM sodium phosphate buffer. The culture was incubated at 37°C until the culture reached an OD of 3.0 at 600 nm. The protein expression was induced through the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) with a final concentration of 0.1 mM with feeding 2% (w/v) glucose. The bacterial cells were incubated for a further 24 h at 18°C and were then harvested via centrifugation at 5,000 rpm for 20 min (Component R, Hanil science Co Ltd., Korea) at 4°C.
For the production of the recombinant BglPm, the LB medium supplemented with ampicillin (100 µg/ml) was used to cultivate the E. coli harboring pGEX-bglPm in a 10 L stirred-tank reactor (Biotron GX, Hanil science Co., Korea) with a 5 L working volume at 500 rpm. The pH value of the medium was adjusted to 7.0 using 100 mM of sodium phosphate. The culture was incubated at 37°C until the culture reached an OD of 3.0 at 600 nm. The protein expression was induced through the addition IPTG with a final concentration of 0.1 mM. The bacterial cells were incubated for a further 18 h at 30°C and were then harvested via centrifugation at 5,000 rpm for 20 min (Component R, Hanil science Co Ltd., Korea) at 4°C.
The cells suspended in 100 mM of phosphate buffer (pH 6.0 for Araf22-3 and pH 7.5 for BglPm) were disrupted via sonication (Vibra-cell, Sonics & Materials, CT, USA), and then the intact cells and debris were removed via centrifugation at 5,000 rpm for 20 min (Component R, Hanil science Co Ltd., Korea) at 4°C in order to obtain the supernatants of the crude enzymes. The crude recombinant Abf22-3 was diluted to the desired concentration with 100 mM of sodium phosphate buffer (pH 6.0) and was used to biotransform the PPDGM. The crude recombinant BglPm was lyophilized for application in the biotransformation reactor following the reaction with the recombinant Abf22-3.
2.8.2. PPDGM transformation via crude Abf22-3 and BglPm in series
The scaled-up biotransformation was performed in a 10 L stirred-tank reactor (Biotron GX, Hanil science Co., Korea) with a 5.0 L working volume at 50 rpm for 24 h. The reaction was performed under optimal conditions in pH 6.0 at 37°C. The reaction started with a composition of 50 mg/ml of substrate ginsenosides (PPDGM; total 250 g) as final concentration and 1.0 L of crude recombinant Abf22-3 (10 mg/ml) in 100 mM of phosphate buffer (pH 6.0). After 6 hours when the ginsenoside Rc was nearly completed converted to Rd, pH was adjusted to 7.5 using 0.5 M NaOH and lyophilized recombinant BglPm was added. Samples were collected at regular intervals and were analyzed by HPLC in order to determine the biotransformation of the ginsenoside F2 from Rb1, Rc, and Rd.
2.9. Purification of biotransformed F2
Following the 5 L reaction of PPDGM with Abf22-3 and BglPm, the mixture was cooled at 4°C and centrifuged at 5,000 rpm for 15 min (Component R, Hanil science Co Ltd., Korea). The biotransformed ginsenoside F2 in the supernatants and precipitates was processed separately in order to purify the samples. The precipitate was also dissolved in 5.0 L of 70% ethanol solution twice and filtered through a filter paper (Advantec, Japan). The ethanol extracts were combined and adjusted to be a 45% ethanol solution. The column chromatography [3,170(L)×128(D) mm; Doointech, Korea] packed with HP20 resin (Mitsubishi, Japan) was adopted in order to remove the impurities, except the ginsenosides. The supernatants and 45% ethanol solution were loaded onto the column together. The free sugar molecules and unwanted hydrophilic compounds from the HP-20 that were adsorbed in beads were washed with 6 bed volumes (BV) of water, and finally the adsorbed ginsenosides were eluted using 6 BV of 95% ethanol (extra pure grade; SK Chemicals, Korea). The ethanol eluent was evaporated in vacuo. The resulting powder was dissolved in 100% methanol and analyzed via HPLC.
2.10. Analytic methods
2.10.1. Thin layer chromatography (TLC) analysis
The thin layer chromatography (TLC) was performed using 60F254 silica gel plates (Merck, Germany) with CHCl3-CH3OH-H2O (65∶35∶10, lower phase) as the solvent. The spots on the TLC plates were identified through comparisons with standard ginsenoside after visualization was made by spraying 10% (vol/vol) H2SO4, followed by heating at 110°C for 5 min.
2.10.2. High performance liquid chromatography (HPLC) analysis
The HPLC analysis of the ginsenosides was performed using an HPLC system (Younglin Co. Ltd, Korea) with a quaternary pump, automatic injector, single wavelength UV detector (model 730D), and Younglin's AutoChro 3000 software for peak identification and integration. The separation was performed on a Prodigy ODS(2) C18 column (5 µm, 150×4.6 mm i.d.; Phenomenex, USA) with a guard column (Eclipse XDB C18, 5 µm, 12.5×4.6 mm i.d.). The mobile phases were A (acetonitrile) and B (water). The gradient elution started with 32% solvent A and 68% solvent B, and was changed to the following: from 0–8 min, A was increased from 32 to 65%; from 8–12 min, A was increased from 65 to 100%; from 12–15 min, A was constant at 100%; from 15–15.1 min, A was decreased from 100 to 32%; from 15.1–25 min, A was constant at 32% The flow rate was 1.0 ml/min, and the detection was performed by monitoring the absorbance at 203 nm and with an injected volume of 25 µl.
Results and Discussion
3.1. Analysis of BglPm sequence
The β-glucosidase gene (bglPm) consisting of 1,260 bp encoding 419 amino acids with a molecular mass of 47.72 KDa and a theoretical pI value of 5.27 (http://web.expasy.org/compute_pi/). BglPm has homology to the protein domain of glycoside hydrolase family 1 (GH1). The Carbohydrate-Active enZymes database (http://www.cazy.org) describes more than 3,000 uncharacterized and 271 characterized GH1 members that are widespread across numerous organisms. In characterized GH1 members, a protein blast (Blastp) search in the databases of NCBI indicated that the protein has the highest similarity (74%) with a β-glucosidase from metagenomic library of mangrove soil. Multiple sequence alignments of BglPm with ginsenoside-transforming and characterized glycoside hydrolases from GH1 allowed the identification of the active site (Fig. 1). Amino acid sequence comparisons revealed that BglPm shared many conserved amino acid residues with other known glycoside hydrolases from GH1, which confirmed that BglPm belonged to GH1, and they all shared the same catalytic central conserved regions (NEP and ENG domains, Fig. 1). Furthermore, Glu 158 and Glu 335, which located in both conserved regions was thought to be the active site of BglPm (Fig. 1, the arrowhead denoted the active site). Glu 158 was identified as the acid base catalyst, while Glu 335 corresponded to the catalytically active nucleophile compared with other known family 1 glycoside hydrolases.
Figure 1. Sequence alignment of BglPm and related ginsenoside-transforming or characterized glycoside hydrolase family 1 enzymes.

The conserved regions for the catalytic central in glycoside hydrolase family 1 are boxed, and the conserved catalytic amino acids are marked with an asterisk. The two putative conserved motifs were shown in box, and the predicted GH1 active site residues (general acid/base and nucleophile residue) were marked by an arrowhead. Full species names and Genbank IDs of the glycoside hydrolases family 1 are as follows, Paenibacillus mucilaginosus KCTC 3870T β-glucosidase [P. muc (This study)], AEI42200; Uncultured bacteria β-glucosidase (U. bac), ABI18350; Thermus caldophilus β-glycosidase (T. cal), AAO15361; Sulfolobus solfataricus P2 β-glycosidase (S. sol), AAK43121; Sulfolobus acidocaldarius β-glycosidase (S. aci), WP_011278657; Sphingomonas sp. 2F2 β-glucosidase (S. 2F2), ADY18331; Pyrococcus furiosus β-glucosidase (P. fur), AAC25555; Micrococcus antarcticus β-glucosidase (M. ant), ACM66669; Uncultured bacterium β-glucosidase (U. bact), AFN70956; Cellulomonas biazotea cellobiase (C. bia), AEM45802.
3.2. Overexpression, and purification of recombinant BglPm
The bglPm was amplified via PCR and then inserted into the pGEX 4T-1 vector. In order to maximize the yield of the fusion protein in a soluble form, different induction conditions were tested and it was found that an induction with 0.1 mM IPTG at 30°C for 18 h cultivation after induction produced the maximum level of soluble active fusion enzyme (data not shown). The recombinant enzyme was purified by GST•bind agarose resin, and then supernatant from cell lysates as well as purified protein were applied to SDS–PAGE (Fig. 2). The molecular mass of the GST-BglPm calculated via an amino acid sequence was 74.6 Kda which is similar masses with the migration in SDS-PAGE. In addition, the recombinant GST-BglPm contains 42.5±0.3% of total soluble protein in E. coli lysate. This high expression level in soluble form makes it more possible to industrial application.
Figure 2. SDS-PAGE analysis of the recombinant BglPm after purification using the GST-bind agarose resin.
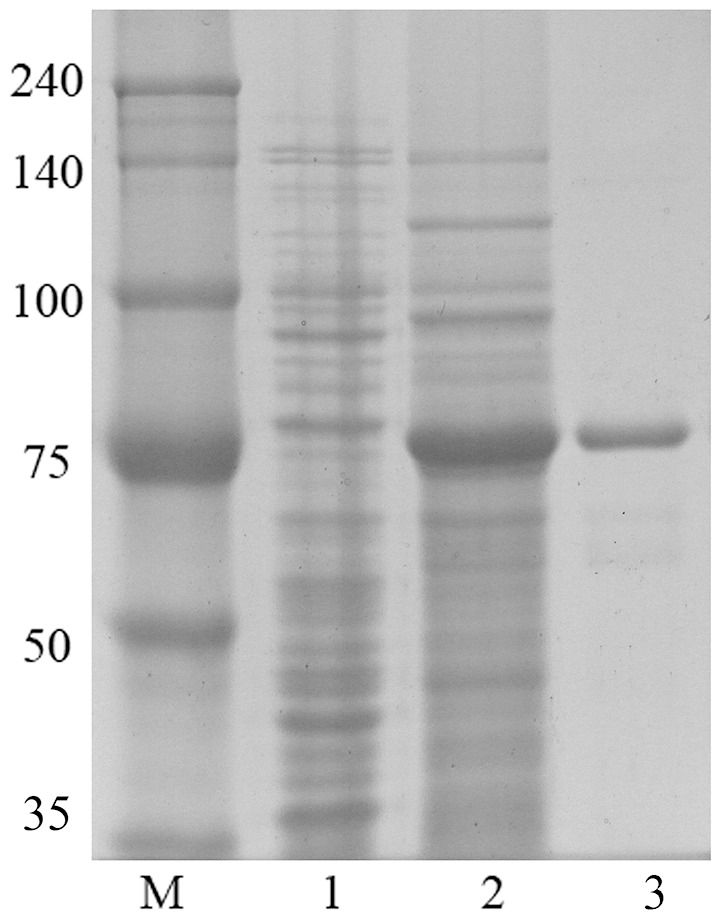
Lanes: M, molecular weight standard; 1, insoluble fraction of the crude extract of the induced recombinant BL 21 (DE3) cells; 2, soluble fraction of the crude extract of the induced recombinant BL 21 (DE3); 3, GST-BglPm after purification with the GST-bind agarose resin.
3.3. Characterization of recombinant BglPm
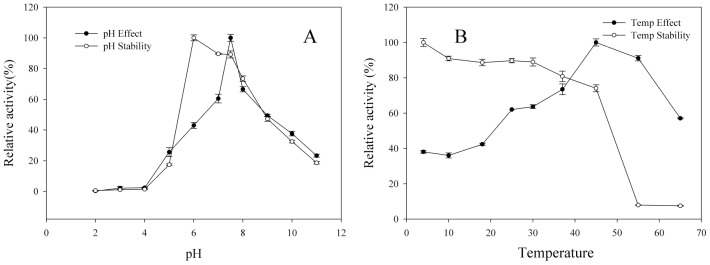
BglPm had optimal pH activity at 7.5 and stability at pH 6.0 to 7.5 in a sodium phosphate buffer at 37°C; from pH 8.0, the enzyme activity decreased swiftly, while at pH 5.0 the enzyme activity decreased to 17.4% (Fig. 3A). The optimal temperature activity was 45°C; at 30°C and 37°C, the enzyme has 63.7% and 73.5% of relative activity, respectively, while thermostability was decreased from 37°C and at 45°C the enzyme has 53.1% of relative activity, and not detected at 55°C (Fig. 3B). Though BglPm has optimum temperature at 45°C for pNPGlc, ginsenoside-conversion reaction was occurred at 37°C for extension of stable transformation activity.
Figure 3. Effects of pH (A) and temperature (B) on the activity and stability of recombinant BglPm.
The effects of metal ions, EDTA, β-mercaptoethanol, and SDS on BglQM activity were also investigated (Table 1). BglPm activity was not affected by β-mercaptoethanol, which is well known thiol group inhibitors. These results suggested that sulfhydryl groups may not be involved in the catalytic center of the enzyme. The enzyme was not affected by 1 mM and 10 mM Na+, K+, Mn2+, Ca2+, Zn2+, Co2+, and Cu2+. The chelating agent EDTA did not inhibit BglPm activity, which indicated that divalent cations are not required for enzymatic activity. The enzyme activity appeared to be 100% inhibited in the presence of sodium dodecyl-sulfate (SDS) and strongly inhibited in the presence of 10 mM Hg2+ and also inhibited in the presence of 10 mM Mg2+, which caused a 43% activity drop. However, no dramatic positive effects on the activity of the BglPm were found for the tested ion (Table 1).
Table 1. Effects of metal ions and chemical agents on the activity of purified recombinant BglPm.
| Relative activity | ||
| 1 mM | 10 mM | |
| NaCl | 98.8±2.5 | 100.3±1.4 |
| KCl | 99.0±1.3 | 100.2±2.2 |
| MgCl2 | 88.3±0.9 | 57.0±4.7 |
| MnCl2 | 87.1±2.2 | 126.0±6.6 |
| CaCl2 | 94.6±3.6 | 85.9±1.2 |
| ZnCl2 | 98.3±1.3 | 78.3±1.5 |
| CoCl2 | 82.9±3.3 | 85.0±6.1 |
| CuCl2 | 70.1±6.4 | 105.3±3.6 |
| HgCl2 | 22.7±1.5 | NDa |
| SDS | ND | ND |
| EDTA | 102.6±5.4 | 86.3±4.8 |
| β-Mercaptoethanol | 101.5±6.3 | 86.5±7.1 |
| DTT | 96.8±3.5 | 80.14±4.2 |
| Control | 100±2.4 | 100±1.9 |
Not determined.
The substrate specificity of BglPm was tested using 2.0 mM of p-nitrophenyl (pNP) and o-nitrophenyl (ONP)-glycosides with α and β configurations. The results showed that BglPm was only active against glucose moiety of pNP-β-D-glucopyranoside and oNP-β-D-glucopyranoside. It showed maximum activity towards pNP-β-D-glucopyranoside and 15% relative activity towards oNP-β-D-glucopyranoside.
3.4. Determination of kinetic parameters
The kinetic parameters of Vmax and Km of BglPm were determined by plotting the substrate concentration vs the initial velocity of each reaction and subjecting the data to no linear regression analysis. The Km, Vmax, kcat and kcat/Km values for 4 substrates were presented in Table 2. The catalytic efficiencies (kcat/Km) for pNPGlc, Rb1, Gyp XVII and Rd decreased in this order: pNPGlc (2.56±0.46 mM−1 S−1) > Rd (1.78±0.46 mM−1 S−1) > Rb1 (1.23±0.25 mM−1 S−1) > Gyp XVII (1.17±0.08 mM−1 S−1). BglPm has higher catalytic efficiency for Rd rather than Rb1 and Gyp XVII, which is attributed that the affinity for the outer glucose moiety at C20 of Rb1 may decrease. The catalytic efficiencies for ginsenosides is lower than that of β-glycosidase from Sulfolobus acidocaldarius [24] for ginsenosides Rd (4.8 mM−1 min−1) and Rb1 (4.8 mM−1 min−1).
Table 2. Kinetic parameters of recombinant BglPm on pNPGlc, Rb1, gypenoside XVII and Rd.
| Substrate | Km (mM) | Vmax (µmol min−1 mg−1) | kcat (s−1) | kcat/Km (mM−1s−1) |
| pNPGlc | 3.24±0.39 | 10.22±0.62 | 8.13±0.50 | 2.56±0.46 |
| Rb1 | 0.384±0.064 | 0.369±0.014 | 0.458±0.018 | 1.23±0.25 |
| Gypenoside XVII | 0.415±0.024 | 0.391±0.006 | 0.487±0.007 | 1.17±0.08 |
| Rd | 0.352±0.053 | 0.484±0.053 | 0.601±0.066 | 1.78±0.46 |
3.5. Ginsenoside-transformation characteristics of BglPm
For the verification of the bioconversion pathways of the seven PPD type ginsenosides (Rb1, Rb2, Rb3, Rc, Rd, Gyp XVII and Rg3) by BglPm, the TLC analyses were performed at regular intervals. As shown Fig. 4, it is clear that the BglPm could transform seven ginsenosides (Rb1, Rb2, Rb3, Rc, Rd, Gyp XVII and Rg3) based on the Rf values. The transformed ginsenosides were also determined using their retention times in the HPLC (data not shown). The proposed biotransformation pathways by BglPm for the PPD ginsenosides are as follows: Rb1 → Gyp XVII → F2; Rd → F2; Rg3(S) → Rh2(S); Rb2 → C-O; Rb3 → C-Mx1; Rc → C-Mc1 via the stepwise hydrolysis of the outer glucose moieties at the C3 position of aglycon (Fig. 5). BglPm hydrolyzes outer glucoses of C3 and C20 position of ginsenosides, similar to the β-glucosidase from Sphingomonas sp. 2F2 [25], but BglPm cannot hydrolyze inner glucose moiety of ginsenosides at C3 and C20.
Figure 4. Thin layer chromatography (TLC) analyses of biotransformation of Rb1, Rd, Gyp XVII, Rg3(S) in (A) and Rb2, Rb3, Rc in (B) by recombinant BglPm.
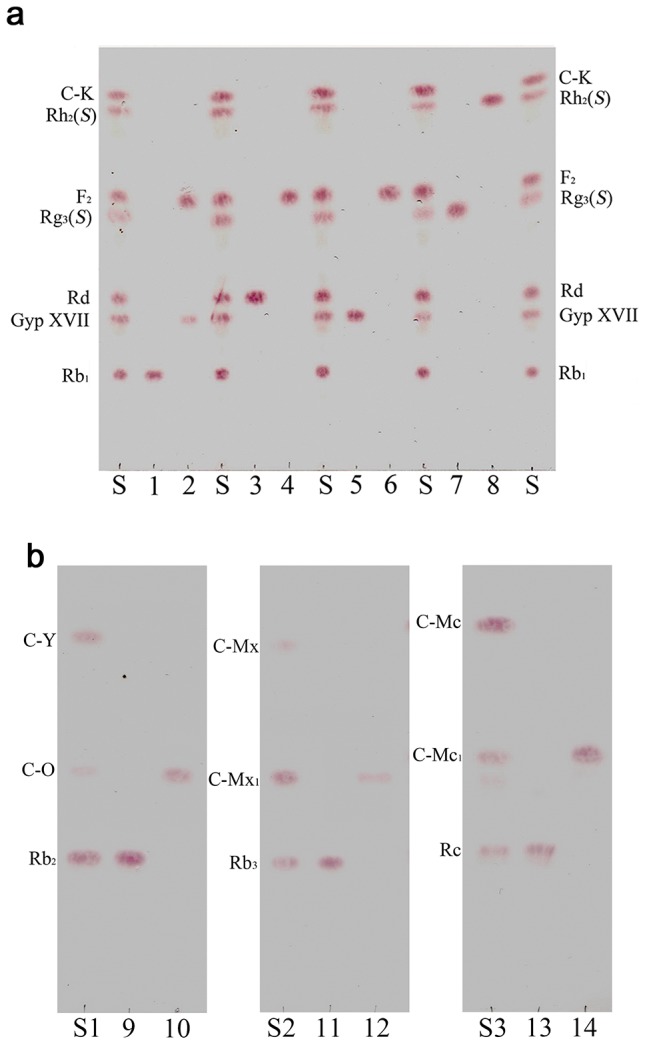
The reaction time was 30: CHCl3-CH3OH-H2O (65∶35∶10, lower phase). (S, S1, S2, S3), ginsenoside standards (PPD type ginsenoside mixtures); Standards: 1, 3, 5, 7, 9, 11, 13; Reaction mixture: 2, 4, 6, 8, 10, 12, 14; 1, Rb1; 2, reaction mixture of Rb1; 3, Rd; 4, reaction mixture of Rd; 5, Gyp XVII; 6, reaction mixture of Gyp XVII; 7, Rg3(S); 8, reaction mixture of Rg3(S); 9, Rb2; 10, reaction mixture of Rb2; 11, Rb3; 12, reaction mixture of Rb3; 13, Rc; 14, reaction mixture of Rc. Abbreviations: C-K, compound K.
Figure 5. Biotransformation pathways of ginsenosides Rb1, Rd, Rb2, Rb3, and Rc by recombinant BglPm and incorporated pathways for F2 production by two glycoside hydrolases.

A few ginsenoside hydrolyzing recombinant enzymes have been characterized to convert major ginsenosides to F2 (Table 3). A β-glucosidase from Sulfolobus solfataricus can hydrolyze Rb1 and Rd into F2, but this enzyme can continuously hydrolyze F2 into C-K [24]. A BglSp from Sphingomonas sp. 2F2 showed F2 production abilities from ginsenosides Rb1, Rb2, Rd, and Rc but the low conversion activity limited its application for F2 production [25] from PPDGM. Until now, the low transformation efficiencies and unconformable conversion pathways of recombinant glycosidase hydrolases limited the applications for the production F2 as gram-scale from PPDGM.
Table 3. Major ginsenoside transformation into F2 by the cloned glycoside hydrolases.
| Glycoside hydrolase name | Microorganism | Ginsenoside conversion pathway | Glycoside hydrolase family | Reference |
| BglPm | Paenibacillus mucilaginosus KCTC3870T | Rb1→Gyp XVII→F2, Rd→F2 | 1 | This study |
| β-glycosidase | Sulfolobus solfataricus | Rb1→Rd→F2→C-K, Rb2→Rd→F2→C-K, Rc→C-Mc→C-K | 1 | Noh et al, 2009 |
| BglSp | Sphingomonas sp. 2F2 | Rb1→ Gyp XVII→F2, Rb2→C-O→F2, Rc→C-Mc→F2, Rd→F2 | 1 | Wang et al, 2011 |
| BglF3 | Flavobacterium johnsoniae | Rb1→Rd, Gyp XVII→F2 | 3 | Hong et al, 2012 |
| AbfA | Rhodanobacter ginsenosidimutans Gsoil 3054 | C-Mc1→F2 | 51 | An et al, 2011 |
3.6. Optimization of PPDGM concentration
F2 production efficiency of crude recombinant BglPm were tested with four PPDGM concentration(10 mg/ml, 25 mg/ml, 50 mg/ml, 75 mg/ml) in order to determine the appropriate substrate concentration for the decreasing reactor volume and economical enzyme concentration for reducing production costs. The time course of the ginsenosides (Rb1, Rd, Gyp XVII, and F2) on the process was determined via HPLC analyses and the abundance of ginsenoside F2 was calculated (Fig. 6). In the test condition of a high substrate concentration (50 mg/ml), the ginsenosides mixture of Rb1 and Rd were completely converted ginsenoside F2 within 7 hours. The higher reaction condition (75 mg/ml) did not complete the conversion within 40 hours. Endowing the advantage to the smaller reactor size, the condition of a high concentration substrate (50 mg/ml) was adopted for the next scaled-up biotransformation step. Thus, these three reaction conditions were excluded in the next step.
Figure 6. Effect of the concentration of PPDGM and crude BglPm on the production of ginsenoside F2.

3.7. Scaled-up production of ginsenosides F2
PPDGM was used as substrate for the mass production of ginsenoside F2 with high purity because it is relatively abundant and can be efficiently separated in specialized ways from crude American ginseng extracts [26], [27]. The enzyme reaction occurred using the crude recombinant Abf22-3 followed by BglPm with PPDGM as the substrate with a concentration of 50 mg/ml as the final concentration in 5 L in order to produce F2 (Fig. 5). As shown in Fig. 7, the ginsenoside Rc was completely converted to Rd within six hours after the crude Abf22-3 was applied to the PPDGM. Thus, at this point, the crude lyophilized BglPm was applied to convert both the intact ginsenoside Rb1 and Rd in the PPDGM and transformed Rd via Abf22-3 to F2. The ginsenoside F2 was produced consecutively up to 7 hours after initiation until ginsenosides Rb1 and Rd were exhausted (Fig. 7). The reaction sample of each point were withdrawn and analyzed via HPLC, of which the chromatography images are shown in Fig. 8. It was demonstrated that when the bioconversion rate was nearly complete for ginsenoside Rb1, Rc and Rd which were not detected by the HPLC analysis (Fig. 8-D).
Figure 7. Relative abundance of ginsenosides Rb1, Rc, Rd, Gyp XVII and F2 in scaled-up production reactor in time course.

Figure 8. HPLC results of the transformation of PPDGM via Abf22-3 and followed by GST-BglPm; (a) ginsenoside peaks of PPDGM, (b) reaction mixture after 6 hour treatment with Abf-22-3, (c) reaction mixture after 1 hour treatment with GST-BglPm, (d) reaction mixture after 7 hour treatment with GST-BglPm.
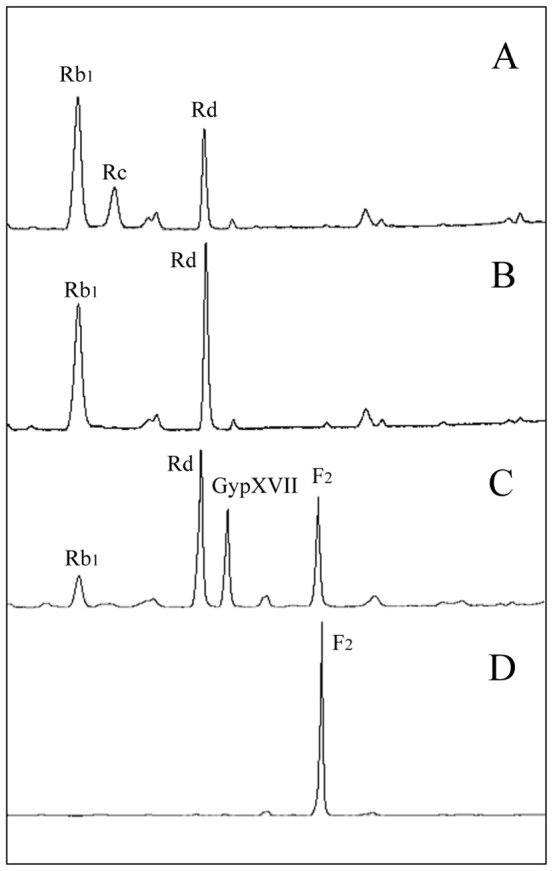
Abbreviations: PPDGM, protopanaxadiol type ginsenoside mixture.
Among the PPDGM, ginsenosides Rb2 and Rb3 occupying approximately 7.6% (mole ratio) were not transformed by Abf22-3, but can be converted by BglPm, and metabolites C-O and C-Mx1 were remained in the solution (Fig. 8-D). The research team behind this paper has searched the ginsenoside hydrolyzing bacteria [28], [29] and constructed several ginsenoside-hydrolyzing recombinant enzymes [15], [21], [25], [30], [31], [32] through the process of surveying the ginsenoside-hydrolyzing enzymes. The finding of efficient BglPm that could efficiently convert Rb1 and Rd to F2 is a key factor in creating F2.
3.8. Purification of biotransformed F2
In order to remove the enzyme, salt, and free sugar from the reaction mixture of the 5 L reaction of PPDGM with Abf22-3 and BglPm, the mixture was centrifuged at 5,000 rpm for 15 min. Most of the ginsenoside F2 was precipitated to form a solid, with a small quantity remaining dissolved in the supernatant (data not shown). After purification step using column chromatography packed with HP20 resin, approximately 24 L of the 95% ethanol eluent was evaporated in vacuo in order to create 152 g of ginsenoside F2. Its chromatographic purity was 80.1% as determined via HPLC (Fig. 8). The impurities were ginsenosides C-O and C-Mx1 derived from the ginsenoside Rb2 and Rb3. As a result, 250 g of PPDGM was used as a substrate for biotransformation and 152 g of ginsenoside F2 with a purity of 80.1% was obtained. The PPDGM was primarily comprised of ginsenosides Rb1, Rc, and Rd in which ginsenosides Re, Rb2, Rb3, Rg3(S), and Rg3(R) were also included. Among these ginsenosides, the total molar amount of Rb1, Rc and Rd that could be biotransformed into F2 using the two recombinant enzymes (Abf22-3 and BglPm) was 162.2 mmol, which corresponds to 173.8 g of 250 g. The 76.2 g of residue was comprised of other types of ginsenosides and unknown impurities. The molar amount of the produced ginsenoside F2 was 155.2 mmol. This indicates that the recovery ratio through the biotransformation process using ginsenosides Rb1, Rc and Rd to F2 reached 95.7% during the entire bioprocess engineering. This is the first report of a cloned enzyme that is capable 100 gram-scale production of F2 through biotransformation.
Conclusion
In this study, we identified and characterized a ginsenoside-transforming glycoside hydrolase (BglPm) from Paenibacillus mucilaginosus. The ginsenoside-transforming pathways and activities of BglPm were proper for mass production of minor ginsenoside F2 using relatively abundant PPDGM consisting of ginsenosides Rb1, Rc, and Rd. Combined usage with Rc-hydrolyzing α-L-arabinofuranosidase, 152 g of ginsenoside F2 with 80.1% chromatography purity was achieved from 250 g of PPDGM using BglPm. This enhanced production method offers an efficient method of preparing the minor ginsenoside F2 in a large scale to meet industrial needs.
Funding Statement
This work was supported by the Intelligent Synthetic Biology Center of Global Frontier Project funded by the Ministry of Education, Science and Technology (2011-0031967), Republic of Korea. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cho IH (2012) Effects of Panax ginseng in neurodegenerative diseases. J Ginseng Res 36: 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kang S, Min H (2012) Ginseng, the ‘Immunity Boost’: the effects of Panax ginseng on immune system. J Ginseng Res 36: 354–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park HJ, Kim DH, Park SJ, Kim JM, Ryu JH (2012) Ginseng in traditional herbal prescriptions. J Ginseng Res 36: 225–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Attele AS, Wu JA, Yuan CS (1999) Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol 58: 1685–1693. [DOI] [PubMed] [Google Scholar]
- 5. Christensen LP (2009) Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv Food Nutr Res 55: 1–99. [DOI] [PubMed] [Google Scholar]
- 6. Jia L, Zhao Y, Liang XJ (2009) Current evaluation of the millennium phytomedicine- ginseng (II) : Collected chemical entities, modern pharmacology, and clinical applications emanated from traditional Chinese medicine. Curr Med Chem 16: 2924–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim SK, Park JH (2011) Trends in Ginseng Research in 2010. J Ginseng Res 35: 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim DH (2012) Chemical diversity of Panax ginseng, Panax quinquifolium, and Panax notoginseng . J Ginseng Res 36: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan CS, Wu JA, Osinski J (2002) Ginsenoside variability in American ginseng samples. Am J Clin Nutr 75: 600–601. [DOI] [PubMed] [Google Scholar]
- 10. Choi S, Kim TW, Singh SV (2009) Ginsenoside Rh2-mediated G1 phase cell cycle arrest in human breast cancer cells is caused by p15 Ink4B and p27 Kip1-dependent inhibition of cyclin-dependent kinases. Pharm Res 26: 2280–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JH, Ahn JY, Shin TJ, Choi SH, Lee BH, et al. (2011) Effects of minor ginsenosides, ginsenoside metabolites, and ginsenoside epimers on the growth of Caenorhabditis elegans . J Ginseng Res 35: 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leung KW, Wong AS (2010) Pharmacology of ginsenosides: a literature review. Chin Med 5: 20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park MW, Ha J, Chung SH (2008) 20(S)-ginsenoside Rg3 enhances glucose-stimulated insulin secretion and activates AMPK. Biol Pharm Bull 31: 748–751. [DOI] [PubMed] [Google Scholar]
- 14. Shi Y, Sun C, Zheng B, Gao B, Sun A (2013) Simultaneous determination of ten ginsenosides in American ginseng functional foods and ginseng raw plant materials by liquid chromatography tandem mass spectrometry. Food Anal Method 6: 112–122. [Google Scholar]
- 15. Hong H, Cui CH, Kim JK, Jin FX, Kim SC, et al. (2012) Enzymatic biotransformation of ginsenoside Rb1 and gypenoside XVII into ginsenosides Rd and F2 by recombinant β-glucosidase from Flavobacterium johnsoniae . J Ginseng Res 36: 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu HS, Zhang CZ, Lu MC, Sun F, Fu YY (2007) Purification and characterization of new special ginsenosidase hydrolyzing multi-glycisides of protopanaxadiol ginsenosides, ginsenosidase type I. Chem Pharm Bull 55: 231–235. [DOI] [PubMed] [Google Scholar]
- 17. Mai TT, Moon J, Song Y, Viet PQ, Phuc PV (2012) Ginsenoside F2 induces apoptosis accompanied by protective autophagy in breast cancer stem cells. Cancer Lett 321: 144–153. [DOI] [PubMed] [Google Scholar]
- 18. Shin JY, Lee JM, Shin HS, Park SY, Yang JE, et al. (2012) Anti-cancer effect of ginsenoside F2 against glioblastoma multiforme in xenograft model in SD rats. J Ginseng Res 36: 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park CS, Yoo MH, Noh KH, Oh DK (2010) Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl Microbiol Biotechnol 87: 9–19. [DOI] [PubMed] [Google Scholar]
- 20. Ye L, Zhou CQ, Zhou W, Zhou P, Chen DF, et al. (2010) Biotransformation of ginsenoside Rb1 to ginsenoside Rd by highly substrate-tolerant Paecilomyces bainier 229-7. Bioresour Technol 28: 857–863. [DOI] [PubMed] [Google Scholar]
- 21. Kim JK, Cui CH, Liu Q, Yoon MH, Kim SC (2013) Mass production of the ginsenoside Rg3(S) through the combinative use of two glycoside hydrolases. Food Chem 141: 1369–1377. [DOI] [PubMed] [Google Scholar]
- 22. Liu QM, Jung HM, Cui CH, Sung BH, Kim JK (2013) Bioconversion of ginsenoside Rc into Rd by a novel α-L-arabinofuranosidase, Abf22-3 from Leuconostoc sp. 22-3: cloning, expression, and enzyme characterization. Anton Leeuw Int JG 103: 747–754. [DOI] [PubMed] [Google Scholar]
- 23. Cleland WW (1979) Statistical analysis of enzyme kinetic data. Methods Enzymol 63: 103–138. [DOI] [PubMed] [Google Scholar]
- 24. Noh KH, Son JW, Kim HJ, Oh DK (2009) Ginsenoside compound K production from ginseng root extract by a thermostable β-glycosidase from Sulfolobus solfataricus . Biosci Biotech Biochem 73: 316–321. [DOI] [PubMed] [Google Scholar]
- 25. Wang L, Liu QM, Sung BH, An DS, Lee HG, et al. (2011) Bioconversion of ginsenosides Rb1, Rb2, Rc and Rd by novel β-glucosidase hydrolyzing outer 3-O glycoside from Sphingomonas sp. 2F2: cloning, expression, and enzyme characterization. J Biotechnol 156: 125–133. [DOI] [PubMed] [Google Scholar]
- 26. Wan JB, Zhang QW, Ye WC, Wang YT (2008) Quantification and separation of protopanaxatriol and protopanaxadiol type saponins from Panax notoginseng with macroporous resins. Sep Purif Technol 60: 198–205. [Google Scholar]
- 27. Zhao Y, Chen B, Yao SZ (2007) Separation of 20(S)-protopanaxdiol type ginsenosides and 20(S)-protopanaxtriol type ginsenosides with the help of macroporous resin adsorption and microwave assisted desorption. Sepa Puri Technol 52: 533–538. [Google Scholar]
- 28. An DS, Wang L, Kim MS, Bae HM, Lee ST, et al. (2011) Solirubrobacter ginsenosidimutans sp. nov., isolated from soil of a ginseng field. Int J Syst Evol Microbiol 61: 2606–2609. [DOI] [PubMed] [Google Scholar]
- 29. Wang L, An DS, Kim SG, Jin FX, Kim SC, et al. (2012) Ramlibacter ginsenosidimutans sp. nov., with ginsenoside-converting activity. J Microbiol Biotech 22: 311–315. [DOI] [PubMed] [Google Scholar]
- 30. An DS, Cui CH, Lee HG, Wang L, Kim SC, et al. (2010) Identification and characterization of a novel Terrabacter ginsenosidimutans sp. nov. beta-glucosidase that transforms ginsenoside Rb1 into the rare gypenosides XVII and LXXV. Appl Environ Microbiol 76: 5827–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cui CH, Kim SC, Im WT (2013) Characterization of the ginsenoside-transforming recombinant β-glucosidase from Actinosynnema mirum and bioconversion of major ginsenosides into minor ginsenosides. Appl Microbiol Biotechnol 97: 649–659. [DOI] [PubMed] [Google Scholar]
- 32. Kim JK, Cui CH, Yoon MH, Kim SC, Im WT (2012) Bioconversion of major ginsenosides Rg1 to minor ginsenoside F1 using novel recombinant ginsenoside hydrolyzing glycosidase cloned from Sanguibacter keddieii and enzyme characterization. J Biotechnol 161: 294–301. [DOI] [PubMed] [Google Scholar]