

Abstract
The Illumina Infinium HumanMethylation450 BeadChip is a new platform for high-throughput DNA methylation analysis. Several methods for normalization and processing of these data have been published recently. Here we present an integrated analysis pipeline offering a choice of the most popular normalization methods while also introducing new methods for calling differentially methylated regions and detecting copy number aberrations.
Availability and implementation: ChAMP is implemented as a Bioconductor package in R. The package and the vignette can be downloaded at bioconductor.org
Contact: tiffany.morris@ucl.ac.uk
1 INTRODUCTION
DNA methylation is the most studied epigenetic modification. Changes in DNA methylation patterns have been implicated in the development of a number of diseases and have been defined as a major hallmark of cancer (Feinberg, 2007). Technological developments for the genome-wide detection of DNA methylation have grown rapidly in recent years, and several options exist (Bock, 2012). Although bisulphite conversion combined with next-generation sequencing is the most comprehensive approach, it is currently feasible for only small sample sizes, and application to large-scale studies remains challenging. The Infinium HumanMethylation450 BeadChip (www.illumina.com) offers this rapidly moving field an attractive balance with respect to throughput, coverage and cost. It extends the previous 27k array, providing an assessment for >480 000 CpG loci, covering key features of the human genome, including CpG islands, shores and shelves as well as promoters, gene bodies, intergenic and imprinted regions (Bibikova et al., 2011). Based on Pubmed and GEO submissions, the 450k array has established itself as the platform of choice for epigenome-wide association studies (Rakyan et al., 2011).
The challenge with this new technology is in the analysis. There are several important steps a 450k analysis pipeline should include: normalization, batch effect analysis, single nucleotide polymorphism (SNP) flagging, detection of copy number aberrations (CNAs) and segmentation of methylation variable positions (MVPs) into biologically relevant DMRs. Normalization is especially important, as the 450k platform combines two different assays, Infinium I and Infinium II (Bibikova et al., 2011; Sandoval et al., 2011). A number of normalization methods are now available that deal with this issue in slightly different ways (Marabita et al., 2013). In chronological order of development, they are Peak Based Correction (PBC) (Dedeurwaerder et al., 2011), SQN (Touleimat and Tost, 2012), Subset-quantile within array normalisation (SWAN) (Maksimovic et al., 2012) and Beta-mixture quantile normalization (BMIQ) (Teschendorff et al., 2013).
2 DESCRIPTION
The Chip Analysis Methylation Pipeline (ChAMP) package is a pipeline that integrates currently available 450k analysis methods and also offers its own novel functionality. It is implemented in R and can be run on any platform with an existing R (version >3.0) and Bioconductor installation. ChAMP takes the raw IDAT files as input, using the data import, quality control and normalization options offered by minfi (Hansen and Ayree, 2011). By default, raw data are filtered for probes with a detection P > 0.01 in at least one sample. If raw data are not available, users are able to upload a matrix of M-, beta- or raw intensity values. The user can decide to filter out individual probes or probe sets such as the X and Y chromosomes. An option to filter SNPs based on a user-specified minor allele frequency in one of four populations as defined by the 1000 genomes project (The 1000 Genomes Project Consortium, 2012) is also available. This prevents biases due to genetic variation in downstream statistical analyses aimed at identifying differentially methylated CpGs. The batch effect analysis is performed on raw data and can be more thorough if the user provides additional covariate information available for the particular study (i.e. age, gender, etc). Following preprocessing, subsequent steps include normalization, DMR calling and CNA detection, which are illustrated in Figure 1 and described in more detail later.
Fig. 1.
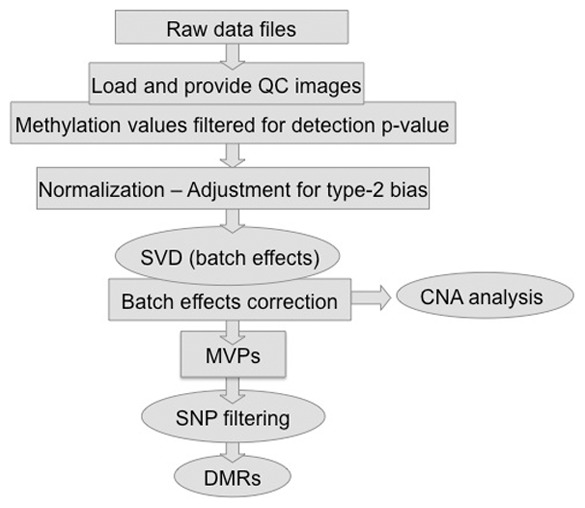
ChAMP includes pre-processing and published methods for adjustment of type 2 bias (squares) and novel methods (circles) for batch effect assessment, DMR correction and CNA analysis
2.1 Adjustment for type2 bias
After running basic quality control metrics, it is recommended to perform intra-array normalization to adjust the data for bias introduced by the Infinium type 2 probe design. ChAMP offers a choice of four methods that have recently been developed specifically for 450k data. As default, ChAMP implements BMIQ (Teschendorff et al., 2013), which was identified by Marabita et al. (2013), as an effective method. The user can also select SWAN (Maksimovic et al., 2012), PBC (Dedeurwaerder et al., 2011) or no normalization.
2.2 Batch effects
To assess the magnitude of batch effects in relation to biological variation, singular value decomposition is applied to the data matrix to obtain the most significant components of variation (Teschendorff et al., 2011). A heatmap rendering the strength of association between the principal components and technical/biological factors allows the user to easily visualize whether batch effects are present. If present, there is an option within ChAMP to use ComBat to correct for these effects (Johnson et al., 2007).
2.3 MVP and DMR calling
For MVP calling, ChAMP uses the Bioconductor package Limma (Smyth, 2005) to compare two groups. The MVP calling can be performed on M- or beta- values. Zhuang et al. (2012) recommend that M-values be used for small sample size studies (<10 samples per phenotype). As DNA methylation is highly correlated for up to 1000 bases (Li et al., 2010), unidirectional MVPs can be grouped into biologically more relevant DMRs as implemented by (Jaffe et al., 2012). ChAMP incorporates a novel DMR hunting algorithm ‘probe lasso’ that considers annotated genomic features and their corresponding local probe densities and methylation according to (Li et al., 2010). Probe lasso (Butcher unpublished) varies the requirements for nearest neighbour probe spacing in a given region based on the genomic feature to which the probe is mapped. The appropriate-sized lasso is then centred on each significant CpG probe and retained if the lasso captures an additional minimum user-specified number of significant probes.
2.4 CNA analysis
Finally, ChAMP integrates a method for analyzing 450k intensity values to identify CNAs in a given dataset (Feber et al., 2013). This has the advantage of getting ‘two for one’ analyses of the same sample, which is particularly important in the context of cancer where tumour heterogeneity is a major confounding factor unless the exact same sample is used. The resulting CNA analysis has been compared with SNP data and been shown to yield comparable results (Feber et al., 2013).
3 DISCUSSION
The bottleneck for researchers using the 450k platform as part of systems and disease-oriented projects is the need for an integrated analysis pipeline. We have addressed this need by developing ChAMP and making it publicly available. ChAMP incorporates already published and novel tools and complements existing 450k analysis pipelines such as Illumina Methylation Analyzer (Wang et al., 2012), RnBeads (Assenov et al., 2013) and wateRmelon (Pidsley et al., 2013), providing users a choice for their analyses. The advantage of ChAMP is that it offers three additional methods for the analysis of batch effects, DMR calling and CNA detection over and above the standard functionalities. ChAMP has been tested on studies containing up to 200 samples on a personal machine with 8 GB of memory. For larger epigenome-wide association studies, the pipeline requires more memory, and running it in steps as described in the vignette can break up the time requirements.
Supplementary Material
ACKNOWLEDGEMENT
The authors acknowledge Anna Karpathakis, Charles Breeze and Dirk Paul for their contributions.
Funding: A.E.T. was supported by a Heller Research Fellowship. The Beck laboratory was supported by the Wellcome Trust (084071), Royal Society Wolfson Research Merit Award (WM100023), IMI-JU OncoTrack (115234) and EU-FP7 projects IDEAL (259679), EPIGENESYS (257082) and BLUEPRINT (282510).
Conflict of Interest: none declared.
REFERENCES
- Assenov Y, et al. Comprehension Analysis of DNA Methylation Data with RnBeads. 2013 doi: 10.1038/nmeth.3115. , http://rnbeads.mpi-inf.mpg.de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bock C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- Dedeurwaerder S, et al. Evaluation of the infinium methylation 450k technology. Epigenomics. 2011;3:771–784. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- Feber A, et al. CNA profiling using high density DNA methylation arrays. Genome Biol. 2013 doi: 10.1186/gb-2014-15-2-r30. in process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Hansen K, Ayree M. minfi: Analyze Illumina's 450k methylation arrays. R package version 1.8.3. 2011 [Google Scholar]
- Jaffe A, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 2012;41:200–209. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W, et al. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic J, et al. Swan: subset-quantile within array normalization for illumina infinium humanmethylation450 beadchips. Genome Biol. 2012;13:R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabita F, et al. An evaluation of analysis pipelines for DNA methylation profiling using the illumina humanmethylation450 beadchip platform. Epigenetics. 2013;8:333–346. doi: 10.4161/epi.24008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R, et al. A data-driven approach to preprocessing illumina 450k methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan V, et al. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval J, et al. Validation of a DNA methylation microarray for 450,000 CPG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer, NY: 2005. Limma: linear models for microarray data; pp. 397–420. [Google Scholar]
- Teschendorff A, et al. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011;27:1496–1505. doi: 10.1093/bioinformatics/btr171. [DOI] [PubMed] [Google Scholar]
- Teschendorff A, et al. A beta-mixture quantile normalization method for correcting probe design bias in illumina infinium 450k DNA methylation data. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touleimat N, Tost J. Complete pipeline for infinium((r)) human methylation 450k beadchip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- Wang D, et al. IMA: an R package for high-throughput analysis of illumina’s 450k infinium methylation data. Bioinformatics. 2012;28:729–730. doi: 10.1093/bioinformatics/bts013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, et al. A comparison of feature selection and classification methods in DNA methylation studies using the illumina infinium platform. BMC Bioinformatics. 2012;13:59. doi: 10.1186/1471-2105-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.