

Abstract
The accumulation of independent mutations over time in two populations often leads to reproductive isolation. Reproductive isolation between diverging populations may be reinforced by barriers that occur either pre- or postzygotically. Hybrid sterility is the most common form of postzygotic isolation in plants. Four postzygotic sterility loci, comprising three hybrid sterility systems (Sa, s5, DPL), have been recently identified in Oryza sativa. These loci explain, in part, the limited hybridization that occurs between the domesticated cultivated rice varieties, O. sativa spp. japonica and O. sativa spp. indica. In the United States, cultivated fields of japonica rice are often invaded by conspecific weeds that have been shown to be of indica origin. Crop-weed hybrids have been identified in crop fields, but at low frequencies. Here we examined the possible role of these hybrid incompatibility loci in the interaction between cultivated and weedy rice. We identified a novel allele at Sa that seemingly prevents loss of fertility in hybrids. Additionally, we found wide-compatibility type alleles at strikingly high frequencies at the Sa and s5 loci in weed groups, and a general lack of incompatible alleles between crops and weeds at the DPL loci. Our results suggest that weedy individuals, particularly those of the SH and BRH groups, should be able to freely hybridize with the local japonica crop, and that prezygotic factors, such as differences in flowering time, have been more important in limiting weed-crop gene flow in the past. As the selective landscape for weedy rice changes due to increased use of herbicide resistant strains of cultivated rice, the genetic barriers that hinder indica-japonica hybridization cannot be counted on to limit the flow of favorable crop genes into weeds.
Introduction
Population divergence, a critical step in the process of speciation, is often accompanied and reinforced by the evolution of reproductive isolating mechanisms, which can occur pre – or postzygotically. In plants, hybrid sterility is the most common form of postzygotic isolation [1]. Hybrid sterility is thought to evolve according to the Bateson-Dobzhansky-Muller (BDM) theory of speciation, which posits that independent mutations occurring in diverging populations become fixed and then interact negatively in the background of the hybrid [2].
The cultivated rice complex (Oryza sativa L.) affords a rare opportunity to investigate the evolutionary history and underlying genetics of traits influencing postzygotic barriers to hybridization. The sterility observed in crosses of two subspecies of Asian cultivated rice, O. sativa indica and japonica, is one of the most extensively studied of all hybrid incompatibilities in plants [3]–[6]. Indica and japonica cultivars were domesticated ∼10,000 years ago and differ in various morphological characteristics and in their responses to a multitude of biotic and abiotic stresses [5]. Gene exchange between these rice subspecies would be highly beneficial to rice breeding practices, however, full exploitation of hybrid rice is limited by the tendency of hybrids to exhibit some degree of sterility [7], which can vary from 5 to 95% depending on the cross [8]. The identification of sterility-causing loci between these cultivars and subsequent determination of their contributions to sterility has been a topic of much research. Despite the ∼57 hybrid-incompatibility quantitative trait loci (QTL) detected so far in rice [9], only a few have been cloned and subjected to experimental testing. Four such loci, encompassing three hybrid sterility systems, are Sa, s5, and DOPPELGANGER1 (DPL1) and DOPPELGANGER2 (DPL2). Sa and DPL1/2 affect pollen viability and s5 sterility results in embryo-sac abortion [10]–[12].
All three of these hybrid sterility systems have been shown to cause semi-sterility in hybrids between indica and japonica cultivars, and could be partial contributors to the low levels of gene flow observed between these two rice subspecies. However, the possible roles of these loci in influencing gene flow between these main rice varieties and other Oryza groups have not been explored. There is potential for gene flow between indica and japonica cultivars and other Asian rice cultivars (e.g. aus, aromatic), the wild ancestor of cultivated rice (O. rufipogon), and the conspecific weed of cultivated rice known as weedy or red rice (O. sativa). Red rice is a troublesome weed that invades cultivated rice fields worldwide and displays competitive traits such as dormancy, high shattering, and rapid growth [13]. Weedy rice infestations can lead to a reduction of rice yields and considerable financial losses [14]. Gene flow between cultivated and weedy rice can have very negative agricultural consequences, such as the potential for crop traits to escape into weedy rice populations and unfavorable weedy traits contaminating seed stocks.
While weedy red rice is a worldwide problem of rice agriculture [13], the evolutionary origins of weedy rice and its relationship with the local rice crop differs throughout the world [15]–[17], affecting expectations of the potential for gene flow. In the US, local cultivated rice belongs to the tropical japonica variety of the japonica subspecies, while weedy rice is related to the indica-aus lineage. Two main genetically differentiated populations of red rice are known to co-occur in rice fields in the US: the most common straw hull (SH) group, which is characterized by straw colored grains, and the black hull awned (BHA) types, which typically have black colored grains with awns [16]. Studies of polymorphism have shown that the SH and BHA weedy groups are most closely related to, and likely descendant from, the indica and aus cultivated varieties, respectively [16], [18]. Indica and aus are closely related crop varieties, typically grown in lowland tropical regions of Asia, and distinct from the tropical japonica cultivars widely cultivated in the US. Further population structure is observed in the BHA group, which can be partitioned into two subpopulations (BHA1 and BHA2). Additionally, hybridization between SH and BHA groups has given rise to a group of weeds known as the BRH (brown hulled) group that occurs at lower frequency [16].
Although weedy rice is classified as the same species, is interfertile with, and co-occurs in fields of cultivated rice, weedy rice in the US shows limited hybridization with the local japonica crop [19]. Some hybrids between the local US japonica crop and SH or BHA weeds have been identified [16], as well as some evidence for past introgression [20] however there is little genetic evidence for extensive crop-weed hybridization. This may be due to self-pollination tendencies of cultivated and weedy rice [21] or differences in flowering time between crops and weeds in the field [13] (e.g. prezygotic mating barriers). However, since indica and japonica cultivars have been shown to experience limited hybrid compatibility due to various deleterious genetic interactions, it is possible that similar hybrid barriers limit the amount of outcrossing between weedy rice and the local japonica cultivar.
In this study we examine the allelic diversity of the characterized rice hybrid sterility loci (Sa, s5, and DPLs) in US weedy rice populations. We find very few barriers to intercrossing between weedy and cultivated rice at these loci, including the near fixation of a rare allele at the Sa locus, which seemingly confers wide-compatibility to some populations of weedy rice. Despite current low frequencies of hybrids in US rice fields, our results suggest that no postzygotic barriers should prevent widespread gene flow between weedy and cultivated rice.
Materials and Methods
Plant Material
Diverse Oryza seeds obtained from the United States Department of Agriculture (USDA), the International Rice Research Institute (IRRI), collections contributed by Dr. David Gealy of the Dale Bumpers National Rice Research Center, and Susan McCouch of Cornell University (Table S1) were grown at the University of Massachusetts Amherst. Our panel consisted of 107 individuals from multiple Oryza species, including weedy rice (51), O. rufipogon (25), O. nivara (2) and various O. sativa cultivars including aus (7), indica (10), and japonica (10); this latter group contained both US and Asian cultivars. Other AA genome Oryza species, O. meridionalis (1) and O. glaberrima (1), were included as outgroups (Table S1). The weedy groups used in our panel were previously defined by Reagon et al. [16] based on 48 sequence tagged site (STS) markers, and consisted of the main weedy groups in the US that have putative indica and aus ancestry (SH, BHA1 and BHA2), the BRH group believed to be a hybrid between SH and BHA weeds, and rarer weedy individuals classified as MX, which are likely early generation hybrids between the main weedy groups and the local japonica crop. Twenty aus and thirty indica individuals obtained from USDA Genetic Stocks – Oryza Collection (GSOR) and IRRI were later added to our survey for further genotyping at the s5 and SaF loci (Table S2).
Sequencing and Genotyping
DNA was extracted from leaf material of all accessions using a CTAB method. Primers were designed using Primer3 [22] to amplify portions of each hybrid sterility locus, taking into account previously described causal polymorphisms at each locus [10]–[12] (Table S3). Indica or japonica type alleles were genotyped for all individuals at each locus using either DNA sequencing, when alleles were differentiated by a single nucleotide polymorphism (SNP), or differential gel migration, when alleles could be visualized by a size difference due to insertion-deletions (indels). DNA sequences were aligned and edited using BioLign Version 2.09.1 (Tom Hall, NC State University). DNA sequences obtained were deposited into GenBank as population datasets under accession numbers KF892880–KF893259.
Genetic Diversity and Phylogenetic Analysis
Summary statistics for each sequenced locus were obtained with DnaSP version 5.0 [23]. Statistics included Watterson’s estimator of nucleotide variation (θW), the average pairwise nucleotide diversity (π) [24] and Tajima’s D (TD) [25]. Summary statistics for each locus were compared against 48 genome-representative STS loci [16] for outlier behavior. Heterozygotes were phased using the haplotype subtraction method [26]. Genealogical relationships among sequenced haplotypes at each locus were determined with Neighbor-Joining analyses using a Kimura-2-parameter model in MEGA5 [27]. For all loci, the Nipponbare sequence, the temperate japonica accession with a sequenced genome, was included as haplotype 1. Weedy accessions were examined for novel alleles, clade membership expectations based on known ancestry, and the likelihood of introgression with US cultivars based on genotypes at each locus. For simplicity, in the remainder of the manuscript the term “haplotype” is used to refer to the DNA sequence content of a given allele, while “allele-type” refers to the functional classification of an allele as indica-type, japonica-type, or (in some cases) wide compatibility-type (Table S4 and Table S5).
Crosses and Quantifying Pollen Viability
Crosses were performed between Oryza accessions to compare pollen production among individuals with different Sa genotypes. Parents were planted in January 2012 and were grown in a walk-in Conviron PGW36 growth chamber at the University of Massachusetts Amherst under 11 hour days at 25°C. Panicles newly emerged from the boot, but not dehisced were chosen as the female. The top of each floret was cut off and the anthers removed with forceps, leaving the stigma intact; 20–30 florets were cut per panicle. A panicle on the verge of dehiscence was chosen to be the male parent. Both male and female panicles were placed in a single glycine bag and secured with a paperclip. Bags were collected after one month to check for hybrid seed. F1 seeds were heat-treated overnight at 34°C and then either plated on a petri dish or planted in soil. Heterozygosity of the F1 was confirmed via PCR.
Both homozygous parents of various Sa genotypes, as well as hybrid offspring were examined for pollen quality. For each sample, pollen grains of six anthers, from six different florets were suspended in 100 µl of Lugol’s Solution (LS), and then serially diluted in 90 µl of LS. Pollen viability was quantified by obtaining a nonviable-to-viable ratio of pollen observed under a Leica MZ 16FA microscope using LS, a potassium iodide stain that reacts with starch in viable cells dying them black; non-viable cells do not stain and appear clear. Three ratios were calculated using ImageJ (http://rsb.info.nih.gov/ij) within a fixed area from three fields of view, and averaged for percent viable pollen.
The quantity of pollen produced was measured using a 0.5 mm deep Nageotte Bright Line Hemacytometer, resulting in a pollen grain/µl concentration. Three measurements were obtained per individual and averaged to calculate mean pollen quantity.
Results
The Genealogy of s5
S5 encodes an aspartic protease on chromosome six that is expressed in ovule tissue. Three allele types have been described: an indica-type (s5-i), a japonica-type (s5-j) and a wide compatibility allele (s5-n) [10]. SNPs C282A and C877T differentiate s5-i and s5-j alleles, while a 136 bp deletion in the N-terminus of s5-n renders it non-functional and confers wide-compatibility [10]. The dimerization of s5-i and s5-j causes embryo sac abortion and has been reported to reduce spikelet fertility by 46% [10].
We sequenced a total of 1897 bp for s5, beginning 949 bp downstream of the start codon in the second exon to 1016 bp after the stop codon, in samples of weedy, wild and cultivated rice. Since sequencing of outgroups failed repeatedly for this locus, we included the published sequence of an O. barthii (Gen Bank Accession # JF298922). Thirty-five SNPs and five indels were identified within this region encompassing thirty-four haplotypes (Figure 1A; Table S4, Table S5A). We classified alleles as either indica- or japonica-type based on the SNPs reported by Chen et. al [10]. Indica-type alleles were further grouped based on whether they contained the wide-compatibility deletion (Table S4).
Figure 1. Neighbor joining trees of hybrid-incompatibility loci sequenced haplotypes.

Numbers on branches correspond to bootstrap percentages from 500 replicates. Bootstrap values below 50 are not shown. Tip labels correspond to haplotype numbers as in Table S4, and to Oryza groups in which the haplotype was found. Numbers in parentheses correspond to the number of alleles found for each haplotype. Green bars designate haplotypes reported as typically from the japonica group, and blue bars designate haplotypes typical of the indica group. A. Haplotype tree for s5. B. Haplotype tree for DPL2. C. Haplotype tree for SaM. D. Haplotype tree for SaF.
Previous analysis of the s5 locus in wild groups found the majority of wild accessions to carry indica-type alleles [28]. Likewise, 97% of wild alleles in our study (29/30) were indica-type (Table 1). Du et al. [28] also found indica and japonica-type alleles fixed within their respective groups, although the aus group was not explicitly characterized. We found that all indica and aus individuals in our panel carried indica-type alleles, while only 62.5% (5/8) of japonica individuals carried the expected japonica allele types (Table 1). The wide-compatibility deletion was detected in 50% of cultivars possessing indica-type alleles, including one grown in the US (sus02), and in 14% of wild indica-type alleles (Figure 1A, Table 1, and Table S4). Interestingly, no indica individuals surveyed possessed the wide-compatibility deletion, but all aus individuals did (Figure 1A, Table 1).
Table 1. Frequency (in percentage) of allele types found at each population and each hybrid incompatibility locus in our core set of accessions.
| Oryza group | Total | |||||||||||
| Locus | Allele Type | indica | aus | japonica | SH | BHA1 | BHA2 | MX | BRH | Wild | samples | |
| s5 | s5-i | 100 | 100 | 37 | 100 | 100 | 63 | 100 | 100 | 97 | ||
| wc# | 0 | 100 | 100 | 86 | 100 | 80 | 100 | 100 | 14 | |||
| s5-j | 0 | 0 | 63 | 0 | 0 | 37 | 0 | 0 | 3 | |||
| alleles sampled * | 6 | 3 | 8 | 14 | 14 | 8 | 5 | 4 | 30 | 92 | ||
| DPL1 | DPL1-K− | 67 | 100 | 21 | 0 | 77 | 100 | 0 | 25 | 44 | ||
| DPL1-N+ | 33 | 0 | 79 | 100 | 23 | 0 | 100 | 75 | 56 | |||
| alleles sampled | 6 | 5 | 14 | 11 | 13 | 7 | 5 | 4 | 32 | 97 | ||
| DPL2 | DPL2-K+ | 100 | 100 | 85 | 100 | 100 | 100 | 80 | 100 | 100 | ||
| DPL2-N− | 0 | 0 | 15 | 0 | 0 | 0 | 20 | 0 | 0 | |||
| alleles sampled | 4 | 4 | 13 | 13 | 13 | 9 | 5 | 3 | 28 | 92 | ||
| SaM | SaM+ | 89 | 80 | 10 | 13 | 100 | 89 | 80 | 20 | 66 | ||
| SaM− | 11 | 20 | 90 | 6 | 0 | 11 | 20 | 0 | 18 | |||
| SaM+X | 0 | 0 | 0 | 75 | 0 | 0 | 0 | 60 | 16 | |||
| SaM-X | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 20 | 0 | |||
| alleles sampled | 9 | 5 | 10 | 16 | 15 | 9 | 5 | 5 | 38 | 112 | ||
| SaF | SaF+ | 78 | 86 | 0 | 0 | 100 | 88 | 80 | 0 | 39 | ||
| SaF− | 22 | 14 | 100 | 7 | 0 | 12 | 20 | 0 | 43 | |||
| SaFX | 0 | 0 | 0 | 93 | 0 | 0 | 0 | 100 | 18 | |||
| alleles sampled | 9 | 7 | 8 | 14 | 15 | 8 | 5 | 4 | 44 | 114 | ||
wc = wide compatibility; wide compatibility percentages are out of total indica-type alleles.
Due to differences in mating system, O. rufipogon genotypes are considered diploid; all other samples are considered as haploid, except in cases of rare heterozygotes.
As expected based on their putative ancestry, all weedy BHA1, SH, and BRH individuals examined possessed indica-type alleles (Table 1, Table S4). All MX weeds also possessed indica-type alleles, despite their mixed weed x japonica ancestry. The only weedy group not fixed for indica-type alleles was BHA2, in which 37% (3/8) of the individuals carried japonica-type alleles (Table 1 and Table S4).
Despite the wide occurrence of indica-type alleles, not all sequenced weedy haplotypes were identical to those detected in cultivars. Nine novel haplotypes were detected in the weedy populations, (Haplotypes 2–8, 14 and 15) (Figure 1A). These haplotypes are not shared with wild or cultivated individuals in our panel, and, except for haplotype 2 [28], have not been previously reported in the literature. Remarkably, not a single SH weed sequenced carried an identical haplotype to any of the indica, its putative progenitor group, in our panel (Figure 1A, Table S4).
Regardless of population, most weeds (87%) contained the wide compatibility deletion (s5-n allele; Figure 1A, Table 1). This deletion occurred in only six of our cultivated individuals, all from the aus and japonica cultivar groups, and, unlike their putative indica progenitors, SH weeds often have the deletion. Based on this finding, we further explored the possible origins of s5-n by genotyping an additional 20 aus and 30 indica accessions from south Asia (Table S2). The deletion was very common in aus, detected in 19/20 individuals. The deletion was rare in indica occurring in only six individuals, mostly from Nepal. The widespread presence of the wide-compatibility allele in US weedy rice groups suggests that this locus poses no postzygotic barrier to hybridization with US crops or hybridization between weedy groups.
Genealogy and Allelic Distribution at the DPL loci
DPL1 (chromosome 1) and DPL2 (chromosome 6) are paralogous hybrid incompatibility genes that encode small plant proteins and are highly expressed in mature anthers [12]. Japonica cultivars have been described as containing a functional copy of DPL1 (DPL1-N+), and a non-functional allele of DPL2 (DPL2-N−) due to a SNP at A434G [12]. Indica and aus cultivars have been described as carrying a non-functional allele of DPL1 (DPL1-K−) due to a 517 bp insertion 204 bp downstream of the start codon, and a functional copy of DPL2 (DPL2-K+) [12]. Pollen carrying non-functional alleles at both loci (e.g. DPL1-K− DPL1-K−//DPL2-N− DPL2-N−) is non-viable [12].
Allelic Distribution of DPL1
DPL1 was genotyped for functionality based on the presence/absence of the 517 bp insertion (Mizuta et. al 2010). Individuals without the insertion were categorized as functional (N+), and individuals with the insertion were classified as non-functional (K−) (Table S4). Previously, the presumably ancestral functional DPL1 alleles were found to be more prevalent in wild rice populations [12]. Similarly, we found DPL1-N+ alleles were present at higher frequencies in our O. rufipogon/nivara sample (56%) (Table 1, Table S4). Within cultivated populations, Mizuta et al. [12] found both alleles at equal frequencies in indica; however we found the DPL1-K− allele at higher frequency than the DPL1-N+ allele in indica (4/6), and fixed in all aus individuals surveyed (Table 1). Mizuta et al. [12] described the japonica group as fixed for DPL1-N+ alleles. Unexpectedly, we found 21% of our japonica individuals to carry nonfunctional DPL1-K− alleles; however, among surveyed US japonica cultivars, all but one carried DPL1-N+ alleles (Table 1, Table S4).
All BHA2 weedy individuals possessed the non-functional indica-type allele (DPL1-K−), which is consistent with their aus ancestry. However, three BHA1 individuals, a group that also has aus ancestry, carried DPL1-N+, which we did not find in any aus individual. MX and SH populations only had DPL1-N+ alleles, even though we found this allele at lower frequency within indica, and the BRH group also carried predominantly DPL1-N+ alleles (Table 1, Table S4).
The Genealogy of DPL2
We sequenced 499 bp of DPL2, encompassing from 16 bp into the 1st exon through 19 bp into the second exon of the gene. Four indels and 22 SNPs were found in our sample, encompassing a total of nine haplotypes (Figure 1B; Table S5B). DPL2-K+ has previously been found to be more frequent in wild populations [12]. Accordingly, this allele was fixed within our wild accessions (Table 1; Table S4). In cultivated populations, the non-functional DPL2-N− and functional DPL2-K+ alleles have previously been reported as fixed within the japonica and indica populations, respectively. The DPL2-K+ allele was also fixed in our indica samples, but, strikingly, the DPL2-N− allele was rare in our japonica individuals, with only 15% (2/13) carrying japonica-like alleles (Table 1; Table S4). Moreover, only one of our US japonica individuals carried DPL2-N− (Table 1).
Similar to wild and domesticated groups, the indica-type allele (DPL2-K+) was nearly fixed in weedy groups (98%, 42/43). The only japonica-type allele was carried by one MX weed thought to be a putative hybrid between an SH weed and a japonica cultivar [16] (Table 1; Table S4). The haplotype carried by this weed, haplotype 9, is the only japonica-type haplotype we found in all our sequences, indicating low sequence diversity for DPL2-N− in contrast with the ancestral DPL2-K+ alleles (Figure 1B). The remaining weedy groups fell within three haplotypes, 1, 4, and 8, with weedy individuals grouping with their domesticated progenitors in haplotypes 1 and 4 (Figure 1B). Haplotype 8, a novel haplotype that occurs in two BHA1 individuals, is characterized by a 1 bp deletion in the coding region of DPL2, which could lead to a non-functional gene.
The Distribution of DPL Genotypes
For simplicity, for hybrid incompatibility loci consisting of two genes, we will list genotypes in haploid format in the remainder of this manuscript; only in the case of hybrid progeny/heterozygotes will we write out the complete diploid genotype. Only two genotypes across both DPL loci were detected in our wild populations: DPL1-K−/DPL2-K+ (42%) and DPL1-N+/DPL2-K+ (58%) (Table S4). Typical japonica cultivars are reported to carry DPL1-N+/DPL2- N− [12], but the predominant genotype within our japonica panel was DPL1-N+/DPL2-K+. Only two of our japonica individuals (one from the US) possessed the genotype expected of typical japonicas, suggesting that previous characterizations of DPL loci may have been too narrow (Table S4). Mizuta et al. [12] found that half of the indica population had DPL1-K−/DPL2-K+ genotypes, similar to what we found in our indica panel (Table S4). This genotype was also fixed among our aus cultivars and was found in three japonica individuals within our panel (Table S4).
In weedy populations, the DPL1-N+/DPL2-K+ genotype, with functional alleles at both loci, was fixed in SH, nearly fixed in MX and BRH individuals, and present in three individuals from the BHA1 group (Table S4). The DPL1-K−/DPL2-K+ genotype was fixed in BHA2 and present at high frequency (73%) in the BHA1 group (Table S4). Given the high frequency of the DPL1-N+/DPL2-K+ genotype in US cultivars as well as several of the weed groups, the DPL loci do not seem to present a barrier to gene flow between cultivated and weedy rice in the US. Only crosses between DPL1-K− carrying weeds (primarily in the BHA groups) and the very rare DPL2-N− carrying cultivars (only 1 out of 8 surveyed cultivars possessed this allele type), would be expected to result in the DPL1-K−/DPL2-N− sterility-causing genotype.
Genealogical Relationships at the Sa Locus
The Sa locus comprises two adjacent genes on chromosome 1, SaM and SaF. SaM encodes a small ubiquitin-like modifier E3 ligase-like protein and SaF encodes an F-box protein [11]. Indica cultivars typically possess a SaM+/SaF+ genotype, while japonica cultivars have a derived SaM−/SaF− genotype. A SaM heterozygote and a SaF+ allele are required to cause male semi-sterility (usually about 50%; [11]). SaM+ and SaM− are differentiated by a G-to-T polymorphism in the fifth intron, resulting in a truncated protein [11]. SaF+ and SaF− are differentiated by a C-to-T transition at position 287 that leads to an amino acid change. Mechanistically, during the uninucleate stage of gamete development, direct interaction of SaF+ with SaM− causes selective abortion of SaM− bearing microspores [11].
The Genealogy of SaM
We amplified a 634 bp portion of SaM starting four bp into the 4th exon through 44 bp downstream of 5th exon. Within this portion of the gene, we found seven indels and 20 SNPs, distributed among 20 haplotypes (Table S5C). Alleles were further classified as either indica (SaM+) or japonica (SaM−)-like based on differentiating polymorphisms assigned by [11] (Table S4). Consistent with two previous studies [11], [29], we found 82% of wild alleles in our panel were SaM+ (Table 1;Table S4). The SaM+ allele has also been documented as the most common in indica cultivars [11], [29]. Likewise, 89% of our indica individuals carried the SaM+ allele (Figure 1C, Table 1). The SaM− allele is nearly fixed in all japonica populations reported to date [11], [29]. Consistently, we found only one japonica individual (str02) with a SaM+ allele in our sample. Consistent with its close relationship with indica, we found that four of five aus individuals had the SaM+ allele (Table 1; Table S4).
As expected based on US weed ancestry, most of our weed alleles (90% or 45/50) were SaM+. Only four individuals from several weedy groups (BHA2, SH, and MX) possessed the SaM− allele (Table 1; Table S4). Weeds largely possessed haplotypes identical to those in progenitor groups or other cultivated groups (Figure 1C). Two novel haplotypes were observed in the MX groups (19 and 20) (Figure 1C).
The Genealogy of SaF
We amplified a 1.3 kb portion of SaF starting 674 bp into the first exon through 357 bp into the 3rd exon. Repeated amplification failures were observed mostly in SH (15/16) and BRH (5/5) groups, so initial genotyping was carried out in the remaining Oryza groups (Table S4). Within this region, we found one indel and 24 SNPs and 18 different haplotypes (Figure 1D; Table S5D). Alleles were further classified as either indica (SaF+) or japonica (SaF−)-like based on differentiating polymorphisms assigned by [11] (Table S4).
Previous reports found wild populations to carry both allele types at relatively equal frequencies, which was consistent with our findings among individuals that amplified (Table 1). Twenty-two percent of our indica individuals had a SaF− allele, while previous reports found 10% of indica individuals carrying this japonica-type allele [11]. SaF+ was nearly fixed within our aus panel (Figure 1D, Table 1; Table S4). SaF− was fixed in our japonica samples (Table 1; Table S4), consistent with previous studies [11], [29].
Among samples that amplified, we found that 90% of weedy individuals (26/29) carried SaF+ alleles (Table S4). Consistent with their aus ancestry, SaF+ is fixed in BHA1, and is found in 87.5% of BHA2 individuals (Table 1; Table S4). The majority of MX weeds also carry SaF+ alleles (Table 1; Table S4). The three weedy individuals detected with japonica-type alleles fall into haplotype 1, the most frequent japonica-type haplotype, and also the only japonica-type haplotype we detected in indica cultivars (Figure 1D).
A Novel SaF Allele: SaFX
Early attempts to sequence the SaF gene were complicated by amplification failures for the majority of samples belonging to the SH and BRH weedy groups and a few other Oryza samples. We obtained the whole-genome sequence of a single SH individual (Young and Caicedo, unpublished information), and noted a large deletion spanning the entire SaF gene. To determine the exact genomic boundaries of the SaF deletion, we designed primers on both sides of the inferred deletion breakpoints (Table S3) and amplified and sequenced four SH samples. We found that the deletion spans 8,628 bp (from coordinates 22,371,187–22,379,815 of chromosome 1 in the MSU 6.0 rice genome) and begins 2,902 bp upstream from the start of SaF. This deletion partially knocks out the gene Os01g39660, a putative transposon protein, as well as the first four exons (1,240 bp) of SaM, the gene located immediately downstream of SaF (Figure 2).
Figure 2. Diagrammatic representation of the 8,628SaFX deletion.
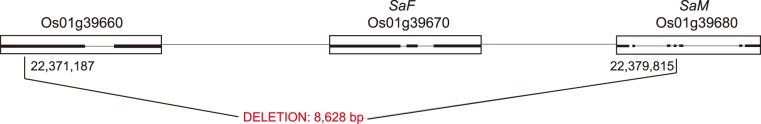
The deletion begins within the gene upstream of SaF, a putative transposon protein, and knocks out the first four exons of SaM.
We found no evidence of this deletion having been reported before, and therefore named the resulting SaF deletion allele SaFX. To genotype for the SaFX allele, a complementary set of primers that amplified specifically only in the presence (SaFdel primers) or absence (SaF primers) of the deletion was used (Table S3). We found that 61% of individuals that failed amplification for SaF (20/33) amplified with our SaFX primers (Table 1; Table S4).
The boundary of the SaFX deletion extends four exons into SaM, but prior to the SaM+/SaM− differentiating SNP in the fifth intron (Figure 2). Taking this into account, we re-assigned SaM alleles in individuals containing SaFX as either SaM+X or SaM-X, to indicate which type of allele it carries (indica (+) or japonica (−)) and that a large portion of the gene is missing (Table 1; Table S4). Only two individuals with SaM-X alleles were found (Table S4), and the deletion breakpoint seemed identical as in the SaM+X alleles.
Given the frequency of this deletion in the SH groups, we expected to find this deletion in both cultivated and wild groups, particularly in indica, which is believed to be ancestral to the SH weed group. While the deletion was found in six wild individuals (Table 1; Table S4), we found no individuals with the deletion among the indica included in our initial panel. We thus expanded our sample set to genotype the same 50 individuals belonging to the aus and indica cultivar groups that were genotyped for s5. The SaFX deletion was detected only in four indica individuals from Nepal (3) and India (1) and one aus cultivar from India, suggesting a low frequency of this allele in wild and cultivated Oryza groups (Table S2).
The Distribution of Sa Genotypes
Wild populations have previously been reported to carry primarily SaM+/SaF+ genotypes [29]. Of the 36 wild samples with genotypes at both loci, 47% carried SaM+/SaF+ and 14% carried SaM−/SaF− (Table 1; Table S4). However, we also detected novel allelic combinations of SaM−/SaF+ (5%), SaM+/SaF− (17%) and SaM+X/SaFX (17%) (Table S4). We believe that SaFX arose in a SaM+ background, since we found it only in combination with SaM+X alleles in wild individuals; however we did find the SaFX/SaM-X combination in two weeds (Table S4). Consistent with previous research [11] the SaM−/SaF− genotype was nearly fixed in our japonica individuals, including the US cultivars surveyed (Table S4). Likewise, the typical indica genotype, SaM+/SaF+, was found to be nearly fixed in our indica and aus panels as well (Table S4).
Consistent with aus ancestry, SaM+/SaF+ is fixed and nearly fixed in BHA1 and BHA2, respectively. Given the common occurrence of SaM−/SaF− genotypes in US cultivars, interactions at this locus could limit hybridization with BHA weeds. SaM+X/SaFX was nearly fixed in the BRH and SH groups. However, we were not able to predict how SaFX affects hybrid sterility levels in crop-weed hybrids since this is the first report of this allele. We speculate that SaFX is a non-functional allele since it knocks out the entire SaF gene and four exons of SaM (see below).
The Consequences of SaFX on Pollen Viability
Because the SaFX gene has not been reported in the literature, we attempted to assess the phenotypic consequences of carrying this allele on pollen production. We designed crosses to test the phenotypic consequences of SaFX in various genetic backgrounds (Table 2). We quantified both the quantity and quality of pollen produced in parental individuals and in crosses performed between these parentals.
Table 2. Successful crosses carried out to assess the effects of the SaFX allele.
| Cross | Parental 1genotype | Parental 1accession | Parental 2genotype | Parental 2accession | F1 genotype |
| 1 | SaM+X/SaFX | rr07 | SaM−/SaF− | rr06 | SaM+X/SaM−//SaFX/SaF− |
| 2 | SaM-X/SaFX | rr15 | SaM+/SaF− | or18 | SaM-X/SaM+//SaFX/SaF− |
| 3 | SaM-X/SaFX | rr33 | SaM+/SaF+ | rr03 | SaM-X/SaM+//SaFX/SaF+ |
We assessed five common parental genotypes: SaM+/SaF+, SaM+/SaF−, SaM−/SaF−, SaM+X/SaFX, and SaM-X/SaFX, which were present within our panel. Ideally, our goal was to have each genotype be both the male and female parent, but varied flowering dates prevented us from making all intended crosses (Table 2). The resulting, verified hybrid genotypes we obtained are shown in Table 2.
Pollen Quality
Non-viable pollen ratios obtained for each individual are shown in Table 3. The parental individual with the highest non-viable ratio possessed a SaM-X/SaFX genotype with an average of 44% non-viable pollen (Table 3). However, another individual with the same genotype only had 6.8% non-viable pollen. The homozygous genotype that produced the most viable pollen was SaM+/SaF− with an average of 5.6% non-viability, but variance was high among individuals within the same genotype (Table 3). We performed a t-test comparing the parental individuals with a SaFX allele to those without SaFX and found no effect of SaFX on pollen quality (P = 0.9).
Table 3. Pollen viability in individuals with different Sa genotypes.
| Genotype# | Accession | Average %Non-Viablepollen* | GenotypeAverage(sd)∧ |
| SaM+/SaF+ | rr03 | 8.4 | |
| sin02 | 6.1 | ||
| rr39 | 8.8 | ||
| 7.7 (6.5) | |||
| SaM−/SaF− | rr22 | 12.3 | |
| rr22 | 29.5 | ||
| rr06 | 19 | ||
| rr53 | 6 | ||
| 16.7 (14) | |||
| SaM+/SaF− | or18 | 8 | |
| sin08 | 3.2 | ||
| 5.6 (2.5) | |||
| SaM+X/SaFX | rr07 | 5.4 | |
| rr08 | 6.6 | ||
| rr12 | 15 | ||
| rr11 | 6 | ||
| 8.25 (5.4) | |||
| SaM-X/SaFX | rr33 | 6.8 | |
| rr15 | 44 | ||
| 25.4 (24.4) | |||
| SaM+X/SaM−//SaF−/SaFX | F1 | 13.1 | |
| 13.1 (4.4) | |||
| SaM+/SaM-X//SaF−/SaFX | F1 | 15 | |
| 15 (2) | |||
| SaM+/SaM-X//SaF+/SaFX | F1 | 5.3 | |
| 5.3 (3) |
parental genotypes listed as haploid for simplicity. All parents are homozygous.
Values are averages of three measurements per individual.
Averages and standard deviations calculated from all original raw measurements; standard deviations are in parentheses.
SaFX does not seem to affect pollen quality in the resulting F1 hybrids, though our small sample size limits our statistical power. The typical sterility-causing genotype at Sa, a SaM heterozygote and at least one SaF+ allele, usually leads to a 50% decrease in total pollen viability [11]. Such drastic decreases in pollen viability were not seen in any of our F1 (Table 3). Non-viable ratios among the progeny do not differ significantly from parental values (P = 0.35). Our hypothesis of SaM being non-functional due to the SaFX deletion is supported by the observation of no decrease in pollen quality in our SaM+X/SaM−//SaF+/SaFX hybrid, as a functional SaM heterozygote and SaF+ allele should have resulted in a substantial decrease in total pollen viability.
Pollen Quantity
We also determined if the amount of pollen produced was affected by the presence of a SaFX allele (Table 4). There was considerable variation in the amount of pollen produced among parents regardless of allele type (Table 4). The highest pollen-producing individual had a SaM−/SaF− genotype (97.5 pollen grains/µl) and the lowest was SaM−/SaFX (14.1 pollen grains/µl) (Table 4). We found no statistically significant differences in pollen production between parents with and without the SaFX deletion (t-test, P = 0.54).
Table 4. Pollen quantity in individuals with different Sa genotypes.
| Genotype# | Accession | AveragePollenQuantity(grains/µl)* | GenotypeAverage(sd)∧ |
| SaM+/SaF+ | rr39 | 21.2 | |
| rr03 | 59.6 | ||
| 40.4 (22.2) | |||
| SaM−/SaF− | sus02 | 27.47 | |
| rr56 | 46 | ||
| sus01 | 97.5 | ||
| 57 (37.6) | |||
| SaM+/SaF− | sin08 | 42.4 | |
| 42.4 (11.4) | |||
| SaM+X/SaFX | rr12 | 26.5 | |
| rr07 | 28.5 | ||
| rr11 | 37.5 | ||
| 30.8 (14.6) | |||
| SaM-X/SaFX | rr33 | 32.8 | |
| rr15 | 14.1 | ||
| 23.45 (11.2) | |||
| SaM+/SaM-X//SaF−/SaFX | F1 | 97.2 | |
| 97.2 (20.3) | |||
| SaM−/SaM+X//SaF−/SaFX | F1 | 27.6 | |
| 27.6 (13) | |||
| SaM+/SaM-X//SaF+/SaFX | F1 | 51.7 | |
| 51.7 (1.2) |
parental genotypes listed as haploid for simplicity. All parents are homozygous.
Values are averages of three measurements per individual.
Averages and standard deviations calculated from all original raw measurements; standard deviations are in parentheses.
There was no obvious effect on pollen production in the hybrid progeny carrying the SaFX allele. Pollen production ranged from close to the highest observed in parents (SaM+/SaM-X//SaF−/SaFX), to among the lower values (SaM−/SaM+X//SaF−/SaFX) (Table 4). Hybrid pollen production did not differ significantly from the parents (P = 0.44). As before, the SaM+X/SaM−//SaF+/SaFX hybrid did not display any evidence of reduced pollen production, suggesting that a functional SaM+ protein is not being produced due to the deletion.
Summary Statistics
Genetic diversity statistics were calculated for all members of the main weedy populations, as well as indica, japonica, aus and wild populations at each locus (Table 5). We attempted to look for any common patterns in polymorphism trends across the three hybrid incompatibility loci.
Table 5. Summary statistics for sequenced loci.
| Oryza group | ||||||||
| Locus | Statistic | BHA1 | BHA2 | SH | indica | Aus | japonica | wild |
| s5 | π | 0.0013 | 0.004 | 0.0007 | 0.0005 | 0.0004 | 0.003 | 0.0021 |
| θ | 0.002 | 0.003 | 0.0011 | 0.0005 | 0.0004 | 0.0022 | 0.0035 | |
| TD | −1.396 | 1.846 | −1.269 | −0.05 | n/a# | 0.826 | −1.385 | |
| DPL2 | π | 0.0003 | 0.0011 | 0 | 0.0011 | 0 | 0.0031 | 0.0033 |
| θ | 0.0007 | 0.0008 | 0 | 0.0012 | 0 | 0.0031 | 0.0104 | |
| TD | −1.149 | 0.986 | n/a | −0.612 | n/a | 0.022 | −2.374 * | |
| SaM | π | 0 | 0.0014 | 0.0019 | 0.0035 | 0.002 | 0.0021 | 0.0072 |
| θ | 0 | 0.002 | 0.003 | 0.0044 | 0.0023 | 0.0028 | 0.0074 | |
| TD | n/a | −1.609 | −1.183 | −0.901 | −1.048 | −1.035 | −0.096 | |
| SaF | π | 0 | 0.0024 | 0 | 0.0012 | 0.0008 | 0 | 0.003 |
| θ | 0 | 0.0004 | 0 | 0.0014 | 0.0018 | 0 | 0.0042 | |
| TD | n/a | −1.055 | n/a | −0.689 | −1.358 | n/a | −1.037 | |
| STS ∧ | π | 0.0007 | 0.0005 | 0.0006 | 0.0016 | 0.0012 | 0.0011 | 0.0044 |
| θ | 0.0008 | 0.0006 | 0.0004 | 0.0017 | 0.0011 | 0.0014 | 0.0056 | |
| TD | −0.177 | 0.042 | −0.441 | −0.026 | 0.092 | −0.773 | −0.729 | |
TD values only calculated when more then four sequences available.
Bolded values indicate significant TD.
based on averages from Reagon et. al 2010.
A previous study based on STS loci among Oryza and weedy groups and found wild populations to harbor the most genetic diversity, followed by intermediate levels in the cultivars and low levels in US weedy groups, due to a genetic bottleneck upon US colonization [16]. In our study, wild populations were the most diverse, as expected, except at s5 and DPL2, where BHA2 and japonica have the highest levels of nucleotide diversity (π), respectively (Table 5). Levels of diversity for hybrid incompatibility genes in weed groups varied among loci, but were sometimes an order of magnitude larger than the genome-wide averages. Particularly noticeable were the high levels of diversity observed in the BHA groups at s5. At this locus, all three weed groups surpassed the levels of diversity seen in their putative cultivated ancestors (Table 5); moreover, all weed groups possessed novel haplotypes not seen in cultivated or weed groups.
When polymorphism was present, TD values for many weed groups across all hybrid incompatibility loci tended to be more negative than genomic averages, indicating a tendency towards excess of rare mutations. At the s5 locus, SH and BHA1 TD values are consistent with the occurrence of novel alleles. Also, noticeably, BHA2 at s5 is associated with a very positive TD, which is consistent with the moderate frequencies of indica and japonica-type alleles in this group (Figure 1, Table 5).
Discussion
The Origin and Evolution of Hybrid Sterility Loci in US Weedy Groups
The demographic history of US weed groups based on random [16] and “weediness” candidate loci [20], [30] has been previously described, providing a framework for our expectations at hybrid sterility loci. Evidence to date suggests that the most common US weed groups, SH and BHA, which co-occur in crop fields, are directly descended from the indica and aus cultivated groups respectively. Additional low-frequency weedy groups are products of weed-weed hybridization (BRH) or hybridization with the local japonica crop (MX). US weeds tend to harbor very low levels of genetic diversity compared to wild and cultivated Oryza groups, due to a genetic bottleneck upon US colonization. The expectations based on genome-wide surveys, however, were not always borne out at hybrid incompatibility loci.
Levels of diversity among our weedy groups varied across hybrid incompatibility loci, but were sometimes an order of magnitude higher than the genome-wide averages (Table 5). Particularly striking were the high levels of variation observed at s5 (Table 5). At this locus, all weed groups had greater levels of diversity than those seen in their putative cultivated ancestors and possessed novel haplotypes. It should be noted that we sequenced more noncoding sequence at this locus than the other hybrid incompatibility and STS loci. However, this cannot account for greater diversity within weed groups compared to their cultivated ancestors. A possible explanation is that having the wide-compatibility deletion, which makes the s5 gene non-functional, has removed selection at this locus, allowing for higher levels of polymorphism in weedy groups. In the case of the BHA2 group, the high levels of diversity and positive TD could be due to hybridization, given the presence of several japonica-type alleles (Table S4, Table 1). No overall evolutionary trend was observed among hybrid sterility loci in the weedy groups, suggesting that each locus has been subjected to independent evolutionary forces.
As expected, the allele types found within weedy group were also largely found within their ancestral cultivated populations, albeit at varying frequencies (Table 1). However, the occurrence of occasional allele types within the main weedy populations that are not found in their putative ancestors, suggest possible hybridization. For example, several members of BHA1 carry DPL1-N+ and DPL2-K+ alleles, a genotype not observed in any of their putative aus ancestors. This could indicate hybridization with the local japonica crop, or with SH, BRH or MX individuals. As mentioned above, some BHA2 individuals carry s5-j alleles, suggesting possible hybridization with the local japonica crop.
For weed groups of known hybrid origin, contributions of each parental group vary at each locus. BRH, a weedy hybrid of SH and BHA, carries alleles common in both parental groups; however, at DPL2 and Sa, the genotype found in the SH population is more common. The MX individuals, comprising hybrids of japonica cultivars and either SH or BHA weeds, carry alleles found in all three parental groups at s5, DPL2, and Sa. Curiously, however, SaFX is nearly fixed in SH but is not found in any MX individuals.
Origins and Implications of the SaFX Allele
This is the first report of the SaFX allele, a deletion knocking out the SaF gene as well as portions of the SaM gene. We believe SaFX arose before domestication, as it was found in three individuals in our wild rice samples originating from Laos, Cambodia and Papua New Guinea (Table S4). However, it has remained at low frequency in both wild and cultivated populations, where it also seems to be geographically restricted. The indica and aus individuals we detected with the deletion are from Nepal and India (Table S4). The low frequency of this allele in wild and cultivated rice groups suggests lack of selection for the SaFX allele in these populations.
The low frequency of SaFX in wild and cultivated groups is in contrast to its near-fixation in the SH and BRH weedy groups. While further studies need to be done to evaluate how SaFX behaves between inter-subspecific crosses, our analyses suggest that SaFX has no significant effects on pollen quality or quantity. This deletion may counteract the sterility-causing interaction between a SaM heterozygote and SaF+ allele, making SaFX comparable to the wide-compatibility allele at the s5 locus in enabling gene flow between indica and japonica populations. The prominence of SaFX in some weedy groups may be a consequence of founder effects – perhaps SH weeds descend from indica cultivars from Nepal. This view is supported by the high frequency of s5-n alleles in the SH group, which was also only found in indica cultivars from Nepal (Table S2). However, it is also possible that selection may have favored the SaFX allele in weeds, either as a way to decrease hybridization barriers with the local crop or as a way to circumvent other possible fitness effects of the incompatibility interaction.
SaFX may create a new version of a tri-allelic system involved in the evolution of speciation genes. Another wide-compatibility allele has been reported at Sa, characterized by two polymorphisms, a 6 bp insertion in SaM and a SNP in SaF [29]. The occurrence of wide-compatibility alleles in hybrid sterility systems is not uncommon, as these alleles have also been reported at other hybrid sterility loci [31]–[33].
All known wide-compatibility alleles act to restore fertility. Relaxation of sterility barriers can be favored if hybrids with wide-compatibility alleles have higher or equal fitness to their parents. Additionally, as is seen in the killer-protective system at s5, some sterility genes do not function directly in pollen or seed production and deleterious interactions between alleles at these loci, while promoting sterility, can cause other problems within the organism unrelated to gamete development (endoplasmic reticulum stress in the case of s5 [34]. However, intra-cellular complications caused by incompatible interactions at Sa, besides hybrid semi-sterility, have not been reported, though the fitness effects of the SaF+− SaM heterozygote interaction have not been fully investigated. Interestingly, in our study, only the hybrid sterility gene(s) that function directly in gamete development (the DPLs), which are implicated in pollen development; [12] did not display a putative wide-compatibility allele.
The Potential for Weed-Crop Gene Flow
Because US weedy rice groups descend from closely related indica and aus cultivars, while the local US rice crop is of japonica descent, we expected that typical indica-japonica postzygotic hybrid sterility barriers would occur between these two groups. Surprisingly, examination of three cloned hybrid sterility systems suggests that fewer postzygotic barriers exist between US weeds and the local crop than what is typically observed between indica and japonica cultivated rice subspecies. Given the prominence of the wide compatibility deletion at the s5 locus in all weedy groups, there do not seem to be postzygotic barriers decreasing the possibility of gene flow with the local japonica crop at this locus (Table 1). Functional alleles at both DPL loci predominated in the SH, MX and BRH weed groups, implying that no crosses involving these weed groups can give rise to the sterility causing genotype DPL1-K−/DPL2-N− (Table 1). While BHA groups did have a high incidence of nonfunctional DPL1 alleles, the dearth of nonfunctional alleles at either DPL locus in the local japonica crop also suggests few barriers to gene flow (Table 1). Few barriers for crop-weed gene flow are also apparent at the Sa locus for BRH and SH groups, as SaFX, a seemingly wide-compatibility allele, is nearly fixed. SaFX is absent in both BHA groups, indicating these groups are less likely to able to hybridize without issues of sterility with the local crop. However, the overall lack of postzygotic barriers to gene flow given the ancestry of US weeds is remarkable, and could suggest that japonica-compatible weed types have been favored during weed evolution in the US.
That weed-crop gene flow occasionally occurs in US rice fields is known. As mentioned above, MX weeds are known hybrids of SH or BHA weeds with the local japonica crop [16], and we have also found evidence of more localized genomic introgression of japonica alleles in some members of the BHA1 group [20]. However, the rate of crop-weed hybridization historically in the US has been low, <1% [13], [19]. Most hybrid incompatibility systems described in rice lead to only partial sterility in hybrids, and are expected to decrease the rate of hybridization, not eliminate it, which could account for low rates of gene flow between crops and weeds. Additional possible postzygotic fitness consequences in F1, such as overly late flowering that could compromise seed set [35], have not been sufficiently explored to draw firm conclusions about their overall contribution to reproductive isolation. In general, our study suggests that genetically based postzygotic barriers to hybridization are almost nonexistent, implying that prezygotic barriers [36] are more likely the main barriers to extensive gene flow between weedy and cultivated rice grown in the US. Possible prezygotic barriers to weed-crop outcrossing include the high self fertilizing levels of both weedy and cultivated rice due to short pollen longevity [37], and differences in flowering time among groups. For example, BHA weeds tend to flower later than SH weeds or the japonica crop, although variability in flowering time exists in all groups [20], [38]. Our study also suggests that under the right environmental circumstances or selective pressure, gene flow levels between crops and weeds have the potential to increase.
The use of herbicide resistance (HR) rice cultivars is increasing in the US, and in 2011 60–65% of Southern US rice was reported to be HR [39]. If the hybrid sterility loci used in this study adequately represent how other hybrid sterility loci have evolved within weedy groups, then the capacity of weedy groups to freely cross with these HR cultivars and produce fertile offspring lends itself to the creation of HR red rice, undermining many weed prevention strategies. In this respect, use of HR rice and herbicide applications changes the selective environment for weeds, such that HR hybrid weeds will be selectively favored. Alternatively, gene flow of unfavorable weedy traits, including shattering and dormancy, could be passed into native cultivated fields, which could interfere with uniform harvesting conditions. Likewise, the escape of an HR gene from red rice could also contaminate fields dedicated to non-HR rice [19].
Hybrid sterility is the most common form of postzygotic isolation in plants, and has long been of interest to evolutionary biologists, due to its importance in the speciation process, and to breeders, due to its impact on crop-improvement strategies. The importance of hybrid sterility barriers to crop-weed gene flow has not explicitly been considered, and here we have shown that there is greater potential than expected for crop-weed hybridization in US cultivated rice fields. As planting of HR rice has increased in the southern rice belt over the last 10 years, and reports of herbicide resistant weedy rice begin to surface [40], it is apparent that neither prezygotic nor postzygotic mating barriers are likely to impede weedy rice from acquiring the crop alleles that will enable them to survive in an herbicide-rich environment. Development of effective weed management strategies must take into account the greater than expected capacity for gene flow between weedy rice and its conspecific crop.
Supporting Information
Core Oryza accessions used in the study.
(XLSX)
Additional individuals genotyped for s5 and SaF .
(XLSX)
Primers used for amplification and sequencing of the hybrid incompatibility loci.
(XLSX)
Allele types and haplotypes observed for all individuals in our core set at the hybrid incompatibility loci.
(XLSX)
Haplotype tables for hybrid incompatibility loci.
(XLSX)
Acknowledgments
We are grateful to Lauren Bishop, Sara Weil, and Sherin Perera at UMass Amherst for help with genotyping, and to Caleb Rounds and Peter Hepler at UMass Amherst for all their help with the pollen microscopy. Additional thanks to the Bezanilla lab at UMass Amherst for use of their equipment.
Funding Statement
This project was funded by a grant from the U.S. National Science Foundation Plant Genome Research Program (IOS-1032023) to A.L.C., Kenneth M. Olsen and Yulin Jia (http://www.nsf.gov/awardsearch/showAward?AWD_ID=1032023). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ouyang Y, Liu Y-G, Zhang Q (2010) Hybrid sterility in plant: stories from rice. Curr Opin Plant Biol 13: 186–192 10.1016/j.pbi.2010.01.002 [DOI] [PubMed] [Google Scholar]
- 2. Orr HA (1996) Dobzhansky, Bateson, and the genetics of speciation. Genetics 144: 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harushima Y, Nakagahra M, Yano M, Sasaki T, Kurata N (2002) Diverse variation of reproductive barriers in three intraspecific rice crosses. Genetics 160: 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kubo T, Yamagata Y, Eguchi M, Yoshimura A (2008) A novel epistatic interaction at two loci causing hybrid male sterility in an inter-subspecific cross of rice (Oryza sativa L.). Genes Genet Syst 83: 443–453. [DOI] [PubMed] [Google Scholar]
- 5. Oka H-I (1957) Genic analysis for the sterility of hybrids between distantly related varieties of cultivated rice. J Genet 55: 397–409 10.1007/BF02984059 [DOI] [Google Scholar]
- 6. Yang CY, Chen ZZ, Zhuang CX, Mei MT, Liu YG (2004) Genetic and physical fine-mapping of the Sc locus conferring indica-japonica hybrid sterility in rice (Oryza sativa L.). Chin Sci Bull 49: 1718–1721 10.1360/04wc0197 [DOI] [Google Scholar]
- 7. Reflinur, Chin JH, Jang SM, Kim B, Lee J, et al. (2012) QTLs for hybrid fertility and their association with female and male sterility in rice. Genes Genom 34: 355–365 10.1007/s13258-011-0209-8 [DOI] [Google Scholar]
- 8. Oka H-I (1974) Analysis of Genes Controlling F1 Sterility in Rice by the Use of Isogenic Lines. Genetics 77: 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ouyang YD, Chen JJ, Ding JH, Zhang QF (2009) Advances in the understanding of inter-subspecific hybrid sterility and wide-compatibility in rice. Chin Sci Bull 54: 2332–2341 10.1007/s11434-009-0371-4 [DOI] [Google Scholar]
- 10. Chen J, Ding J, Ouyang Y, Du H, Yang J, et al. (2008) A triallelic system of S5 is a major regulator of the reproductive barrier and compatibility of indica-japonica hybrids in rice. Proc Natl Acad Sci USA 105: 11436–11441 10.1073/pnas.0804761105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Long Y, Zhao L, Niu B, Su J, Wu H, et al. (2008) Hybrid male sterility in rice controlled by interaction between divergent alleles of two adjacent genes. Proc Natl Acad Sci USA 105: 18871–18876 10.1073/pnas.0810108105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mizuta Y, Harushima Y, Kurata N (2010) Rice pollen hybrid incompatibility caused by reciprocal gene loss of duplicated genes. Proc Natl Acad Sci USA 107: 20417–20422 10.1073/pnas.1003124107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delouche JC, Burgos NR, Gealy DR, de San Martin GZ, Labrada R, et al.. (2007) Weedy Rices - Origin, Biology, Ecology, and Control. FAO Plant Production and Protection Paper 188.
- 14. Sha XY, Linscombe SD, Groth DE (2007) Field evaluation of imidazolinone-tolerant Clearfield rice (Oryza sativa L.) at nine Louisiana locations. Crop Sci 47: 1177–1185 10.2135/cropsoc2006.09.0592 [DOI] [Google Scholar]
- 15. Bres-Patry C, Lorieux M, Clement G, Bangratz M, Ghesquiere A (2001) Heredity and genetic mapping of domestication-related traits in a temperate japonica weedy rice. Theor Appl Genet 102: 118–126 10.1007/s001220051626 [DOI] [Google Scholar]
- 16.Reagon M, Thurber CS, Gross BL, Olsen KM, Jia Y, et al.. (2010) Genomic patterns of nucleotide diversity in divergent populations of US weedy rice. BMC Evol Biol 10. doi:10.1186/1471-2148-10-180. [DOI] [PMC free article] [PubMed]
- 17. Sun J, Qian Q, Ma D-R, Xu Z-J, Liu D, et al. (2013) Introgression and selection shaping the genome and adaptive loci of weedy rice in northern China. New Phytol 197: 290–299 10.1111/nph.12012 [DOI] [PubMed] [Google Scholar]
- 18. Londo JP, Schaal BA (2007) Origins and population genetics of weedy red rice in the USA. Mol Ecol 16: 4523–4535 10.1111/j.1365-294X.2007.03489.x [DOI] [PubMed] [Google Scholar]
- 19. Shivrain VK, Burgos NR, Gealy DR, Sales MA, Smith KL (2009) Gene flow from weedy red rice (Oryza sativa L.) to cultivated rice and fitness of hybrids. Pest Management Science 65: 1124–1129 10.1002/ps.1802 [DOI] [PubMed] [Google Scholar]
- 20. Reagon M, Thurber CS, Olsen KM, Jia Y, Caicedo AL (2011) The long and the short of it: SD1 polymorphism and the evolution of growth trait divergence in U.S. weedy rice. Mol Ecol 20: 3743–3756 10.1111/j.1365-294X.2011.05216.x [DOI] [PubMed] [Google Scholar]
- 21.Gealy DR, Gressel J (2005) Gene movement between rice (Oryza sativa) and weedy rice (Oryza sativa) - a U.S. temperate rice perspective. Crop Fertility and Volunteerism. CRC Press. 323–354.
- 22. Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132: 365–386. [DOI] [PubMed] [Google Scholar]
- 23. Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 24. Nei M, Li W (1979) Mathematical-Model for Studying Genetic-Variation in Terms of Restriction Endonucleases. Proc Natl Acad Sci USA 76: 5269–5273 10.1073/pnas.76.10.5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clark AG (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 7: 111–122. [DOI] [PubMed] [Google Scholar]
- 27. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Du H, Ouyang Y, Zhang C, Zhang Q (2011) Complex evolution of S5, a major reproductive barrier regulator, in the cultivated rice Oryza sativa and its wild relatives. New Phytol 191: 275–287 10.1111/j.1469-8137.2011.03691.x [DOI] [PubMed] [Google Scholar]
- 29. Wang Y, Zhong ZZ, Zhao ZG, Jiang L, Bian XF, et al. (2010) Fine mapping of a gene causing hybrid pollen sterility between Yunnan weedy rice and cultivated rice (Oryza sativa L.) and phylogenetic analysis of Yunnan weedy rice. Planta 231: 559–570 10.1007/s00425-009-1063-7 [DOI] [PubMed] [Google Scholar]
- 30. Thurber CS, Reagon M, Gross BL, Olsen KM, Jia Y, et al. (2010) Molecular evolution of shattering loci in US weedy rice. Mol Ecol 19: 3271–3284 10.1111/j.1365-294X.2010.04708.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LeiGang S, XiangDong L, Bo L, XingJuan Z, Lan W, et al. (2009) Identifying neutral allele Sb at pollen-sterility loci in cultivated rice with Oryza rufipogon origin. Chin Sci Bull 54: 3813–3821 10.1007/s11434-009-0571-y [DOI] [Google Scholar]
- 32. Wang GW, Cai HY, Xu Y, Yang SH, He YQ (2009) Analysis on the spectra and level of wide-compatibility conferred by three neutral alleles in inter-subspecific hybrids of rice (Oryza sativa L.) using near isogenic lines. Plant Breed 128: 451–457 10.1111/j.1439-0523.2008.01514.x [DOI] [Google Scholar]
- 33. Li JQ, Shahid MQ, Feng JH, Liu XD, Zhao XJ, et al. (2012) Identification of neutral alleles at pollen sterility gene loci of cultivated rice (Oryza sativa L.) from wild rice (O. rufipogon Griff.). Plant Syst Evol 298: 33–42 10.1007/s00606-011-0520-5 [DOI] [Google Scholar]
- 34. Yang J, Zhao X, Cheng K, Du H, Ouyang Y, et al. (2012) A Killer-Protector System Regulates Both Hybrid Sterility and Segregation Distortion in Rice. Science 337: 1336–1340 10.1126/science.1223702 [DOI] [PubMed] [Google Scholar]
- 35. Rajguru SN, Burgos NR, Shivrain VK, McD Stewart J (2005) Mutations in the red rice ALS gene associated with resistance to imazethapyr. Weed Sci 53: 946–946 10.1614/WS-04-111R1.1 [DOI] [Google Scholar]
- 36. Rieseberg LH, Willis JH (2007) Plant Speciation. Science 317: 910–914 10.1126/science.1137729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song ZP, Lu BR, Chen JK (2001) A study of pollen viability and longevity in Oryza rufipogon, O. sativa, and their hybrids. International Rice Research Notes 26: 31–32. [Google Scholar]
- 38. Shivrain VK, Burgos NR, Sales MA, Kuk YI (2010) Polymorphisms in the ALS gene of weedy rice (Oryza sativa L.) accessions with differential tolerance to imazethapyr. Crop Prot 29: 336–341 10.1016/j.cropro.2009.10.002 [DOI] [Google Scholar]
- 39.Salassi ME, Wilson Jr CE, Walker TW (2012) Proceedings of the Thirty-Fourth Rice Technical Working Group Hot Springs Convention Center, Hot Springs, Arkansas. 17–26.
- 40. Burgos NR, Norsworthy JK, Scott RC, Smith KL (2008) Red Rice (Oryza sativa) Status after 5 Years of Imidazolinone-Resistant Rice Technology in Arkansas. Weed Technology 22: 200–208 10.1614/WT-07-075.1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Core Oryza accessions used in the study.
(XLSX)
Additional individuals genotyped for s5 and SaF .
(XLSX)
Primers used for amplification and sequencing of the hybrid incompatibility loci.
(XLSX)
Allele types and haplotypes observed for all individuals in our core set at the hybrid incompatibility loci.
(XLSX)
Haplotype tables for hybrid incompatibility loci.
(XLSX)