

Abstract
Metformin treatment has been associated with a decrease in breast cancer risk and improved survival. Metformin induces complex cellular changes, resulting in decreased tumor cell proliferation, reduction of stem cells, and apoptosis. Using a carcinogen-induced rodent model of mammary tumorigenesis, we recently demonstrated that overfeeding in obese animals is associated with a 50% increase in tumor glucose uptake, increased proliferation, and tumor cell reprogramming to an “aggressive” metabolic state. Metformin significantly inhibited these pro-tumorigenic effects. We hypothesized that a dynamic relationship exists between chronic energy excess (glucose by dose) and metformin efficacy/action.
Media glucose concentrations above 5 mmol/L was associated with significant increase in breast cancer cell proliferation, clonogenicity, motility, upregulation/activation of pro-oncogenic signaling, and reduction in apoptosis. These effects were most significant in triple-negative breast cancer (TNBC) cell lines. High-glucose conditions (10 mmol/L or above) significantly abrogated the effects of metformin. Mechanisms of metformin action at normal vs. high glucose overlapped but were not identical; for example, metformin reduced IGF-1R expression in both the HER2+ SK-BR-3 and TNBC MDA-MB-468 cell lines more significantly at 5, as compared with 10 mmol/L glucose. Significant changes in gene profiles related to apoptosis, cellular processes, metabolic processes, and cell proliferation occurred with metformin treatment in cells grown at 5 mmol/L glucose, whereas under high-glucose conditions, metformin did not significantly increase apoptotic/cellular death genes. These data indicate that failure to maintain glucose homeostasis may promote a more aggressive breast cancer phenotype and alter metformin efficacy and mechanisms of action.
Keywords: breast cancer, glucose, metformin, metabolism, therapy
Introduction
Chronic energy excess and physical inactivity are associated with alterations of carbohydrate and fatty acid metabolism, often resulting in obesity, metabolic syndrome, and type 2 diabetes. Both pre- and post-menopausal breast cancers are increased in selected patient subgroups with these disorders.1-11 Obesity and/or type 2 diabetes are also independently associated with a shortened disease-free and overall survival of breast cancer patients. Excessive energy intake relative to total expenditure, as seen in obese and/or overweight individuals, is associated with increased risk of breast cancer. In contrast, physical activity, weight loss, and a lean body habitus are associated with a reduced breast cancer incidence and improved survival.3,12-21 Mechanisms by which obesity and dysregulation of host metabolism may promote breast cancer are reportedly diverse, including upregulation of growth factors, inflammatory cytokines, adipokines, steroid hormones, and circulating nutrients including the hexose sugars, as well as downregulation of immune surveillance and tissue oxygenation.20,22,23 Of these, an increase in insulin and insulin-like growth factors demonstrated very strong associations with breast cancer risk and a shortened breast cancer survival.3,13,14,20,24,25 Women with serum insulin levels in the highest quartile have double the risk of breast cancer recurrence and triple the risk of cancer-associated death, while a 25% reduction in insulin levels was associated with a 5–6% absolute improvement in breast cancer survival.13,20,25-27 This led many to recommend that new breast cancer patients should be screened for alterations in carbohydrate metabolism/high insulin level,3,13,14,20,25,28 and if present, that the anti-diabetic drug metformin (1,1-dimethylbiguanide hydrochloride) should be considered as an additional therapeutic measure. Epidemiological data indicate that metformin has unique anti-cancer activity, reducing risk and improving survival.13,20,29-34 Limited retrospective data also suggests that metformin may improve response to chemotherapy.35,36 The unique anti-cancer benefits of metformin have not been associated with other anti-diabetic therapies, including insulin and sulfonylureas, that reportedly may increase breast cancer risk and diminish survival in patients with the disease.37
In the setting of metabolic syndrome and/or poorly controlled type 2 diabetes, host cells including occult or known breast cancers may experience dramatic shifts in extracellular glucose due to lack of systemic glucose homeostasis, in addition to increased levels of insulin and insulin-like growth factors. Cancer cells are especially adept in transporting extracellular glucose across the cancer cell membrane into the cytoplasm, via upregulation of dedicated transporters known as the “GLUT” proteins.23 High levels of intracellular glucose facilitate cancer and stem cell growth as compared with normal cells, because these cells are particularly dependent on glucose and other energy precursors via shifts in metabolic programing (from anaerobic to aerobic glycolysis, the Warburg Effect).38,39 Aerobic glycolysis is less efficient in producing energy (ATP) from glucose; however, it generates abundant precursor molecules needed for cellular replication.23,38-42 Although cancers frequently undergo metabolic reprogramming and are thus glucose “addicted”, tumor metabolism cannot be assessed on the basis of routine radiologic or pathologic tests without functional studies. Positron emission tomography (PET) imaging using an analog of glucose, FDG, is required to determine if specific tissues and/or tumors have increased glucose consumption (i.e., undergone metabolic reprogramming). PET-FDG studies have shown that breast cancers vary in their ability to take up glucose. In situ and small or well-differentiated neoplasms of lobular or ductal types are the least likely to have undergone metabolic reprogramming, whereas larger, higher-grade invasive ductal carcinomas are the most frequently affected.43 Reprogramming of malignant cells to aerobic glycolysis has been proposed as a broadly applicable target for novel anti-cancer treatment.40,44 One agent with broad activity that appears to significantly reduce this metabolic shift and have anti-cancer activity is the anti-diabetic drug metformin.
We recently reported novel data derived from a carcinogen-induced rat model that merged models of obesity, mammary tumorigenesis, and menopause.45 In that study, premenopausal obesity and menopause-induced overfeeding was associated with greater mammary tumorigenesis and altered mammary tumor glucose and fatty acid metabolism. Nutrient tracer methods were used to study energy utilization patterns in the mammary tumors, as well as benign mammary gland and other tissues. Obesity and overfeeding were associated with a 50% increase in the mammary tumor glucose uptake and a significant nutrient preference for glucose over fat. Normal mammary tissues were generally unresponsive to the energy excess. These findings are characteristic of tumor cell reprogramming to aerobic glycolysis and obesity-associated insulin resistance. In contrast, overfed lean animals showed an increase in glucose uptake by peripheral tissues and benign mammary gland, without an increase in glucose uptake by the mammary tumors. Tumor cells showed no preference for glucose over fat. Thus, lean animals demonstrated normal metabolic response to energy excess. Metformin treatment of both lean and obese tumor-bearing animals was associated with a significant reduction in tumor burden and improved survival, as compared with non-metformin treated animals.45 These provocative findings provide a rationale for the present study in which we sought to interrogate the specific effects of normal vs. supra-physiologic glucose (levels consistent with metabolic syndrome, type 2 diabetes, or higher levels typical of cell culture media) on the biology of human breast cancer cells and determine if “hyperglycemic” conditions would influence metformin efficacy or mechanisms of action.
Results
High glucose promotes cell growth and increases the EC50 of metformin
We have shown that metformin has different biological and molecular anti-cancer effects in breast cancer cells of TNBC as compared with non-TNBC subtypes under standard in vitro culture conditions (with media contained 17 mmol/L glucose [G17]).46-48 In the present study, we interrogated the effects of glucose concentration alone in 13 breast cancer cell lines representing luminal A and B, HER2, and TNBC subtypes. Breast cancer cell growth at 5 mmol/L glucose (G5) (normal physiologic levels) was compared with growth in the absence or presence of glucose (0 [G0], 7 [G7], 10 [G10] or 17 [G17] mmol/L). Cell growth was inhibited in all lines grown in media without glucose; however, TNBC cells showed a greater ability to grow under these conditions (Fig. 1A). As glucose increased from 5 to 7, 10, and 17 mmol/L, significant increases in cell proliferation were demonstrated. Although 17 mmol/L of glucose is incompatible with human life, breast cancer cells show no apparent toxicity from the energy excess.
Figure 1.

Alteration of glucose levels alters cell proliferation in a panel of human breast cancer cell lines. (A) The indicated human breast cancer cell lines were treated with various glucose concentrations (0, 5, 7, 10, 17 mmol/L) for 72 h then assayed for percent proliferation using MTS and normalized to 5 mmol/L glucose. (B–D) Indicated breast cancer cell lines used above were seeded in media containing 5 mmol/L (B), 10 mmol/L (C), or 17 mmol/L (D) glucose and treated with vehicle control, 5 mmol/L or 10 mmol/L metformin for 72 h. The percent cell proliferation was measured by MTS assay and normalized to control treatment. Experiments are representative of triplicate assays. Data are representative of averages of triplicate determinations ± SE.
We next studied the influence of increasing concentrations of glucose with or without metformin at 5 or 10 mmol/L (Fig. 1B–D). In general, TNBC cells showed greater metformin-associated growth inhibition in response to metformin at 5 or 10 mmol/L across the range of glucose concentrations. In the other molecular subtypes of cancer cells, metformin inhibited cell growth more effectively at physiologic (5 mmol/L) glucose. In order to confirm and quantitate these effects, we calculated the EC50s of metformin using nonlinear regression methods for each of the breast cancer cell lines at each glucose concentration. The EC50s of TNBCs for metformin treatment were often less than 50% that of the non-TNBC EC50s (Table 1). Increasing glucose concentrations from 5–17 mmol/L was typically associated with significant increases in the EC50.
Table 1. EC50 for metformin at various glucose levels in human breast cancer cell lines.
| A. Luminal and HER2 breast cancer cell lines | ||||
|---|---|---|---|---|
| Glucose concentration (mmol/L) | ||||
| Cell line | Subtype | 5 mmol/L | 10 mmol/L | 17 mmol/L |
| MCF7 | Luminal A | 19 mmol/L | 21 mmol/L | 26 mmol/L |
| T-47D | Luminal A | 30 mmol/L | 39 mmol/L | 68 mmol/L |
| BT-474 | Luminal B | 16 mmol/L | 25 mmol/L | 31 mmol/L |
| ZR-75–30 | Luminal B | 13 mmol/L | 16 mmol/L | 31 mmol/L |
| MDA-MB-453 | HER2 | 25 mmol/L | 27 mmol/L | 35 mmol/L |
| SK-BR-3 | HER2 | 25 mmol/L | 29 mmol/L | 33 mmol/L |
| B. Triple negative breast cancer cell lines | ||||
|---|---|---|---|---|
| Glucose concentration (mmol/L) | ||||
| Cell line | Subtype | 5 mmol/L | 10 mmol/L | 17 mmol/L |
| MDA-MB-468 | Basal 1 | 3 mmol/L | 5 mmol/L | 5 mmol/L |
| HCC1143 | Basal 1 | 20 mmol/L | 22 mmol/L | 24 mmol/L |
| HCC1937 | Basal 1 | 6 mmol/L | 7 mmol/L | 8 mmol/L |
| HCC70 | Basal 2 | 3 mmol/L | 3 mmol/L | 4 mmol/L |
| HCC1806 | Basal 2 | 2 mmol/L | 2 mmol/L | 3 mmol/L |
| MDA-MB-231 | MSL | 19 mmol/L | 23 mmol/L | 31 mmol/L |
| BT-20 | Unclassified | 2 mmol/L | 3 mmol/L | 3 mmol/L |
High glucose levels promote clonogenicity
We next studied whether glucose levels alone would alter the ability of cells to form anchorage-dependent colonies. We used 4 representative cell lines T-47D (ER+, PR+, HER2-), SK-BR-3 (ER-, PR-, HER2+), MDA-MB-468 (TNBC), and MDA-MB-231(TNBC) supplemented with 5, 10, or 17 mmol/L glucose media, with or without 5 mmol/L metformin, grown for 3 wk in culture (Fig. 2A). Colony formation increased with increasing glucose levels in all cell lines tested. To quantitate these interactions and test them for significance, colonies of at least 50 cells were counted for each treatment group, and the data was plotted graphically (Fig. 2B). Metformin induced a significant reduction in colony formation for each of the 4 cell lines grown under normal and supra-normal glucose concentrations. It induced the greatest reduction of growth in the TNBC cell lines grown with media containing normal glucose levels.
Figure 2.

Reduction of glucose enhances metformin-mediated inhibition of colony formation and motility in TNBC, as compared with Luminal A or HER2 overexpressing cell lines. (A) Luminal A (T-47D), HER2 overexpressing (SK-BR-3), or TNBC (MDA-MB-468, MDA-MB-231) cell lines were treated with 5 mmol/L metformin in media containing various glucose levels (5, 10, 17 mmol/L) as described. (B) Enumeration of colony forming units for each cell line was performed see “Materials and Methods”. Bar graphs show averages of 3 independent experiments ± SE.
High glucose inhibits metformin-induced apoptosis and cell cycle arrest
We have previously shown that metformin induces partial S-phase arrest and apoptosis in TNBC, and partial G1 arrest without apoptosis in non-TNBC cell lines, grown in adherent culture with media containing 17 mmol/L glucose.46,48 In this report, we show the influence of glucose concentration on the ability of metformin to induce apoptosis and cell cycle progression. Little or no apoptosis was detected when each of the 5 cell lines examined in Figure 3 were grown with supra-normal glucose (17 mmol/L). When metformin was added under high glucose conditions, apoptosis was induced in 2 of 3 TNBC cells. In contrast, when these 5 cell lines were grown in media containing 5 mmol/L glucose, apoptosis was detected in 4 of 5 cell lines (all but SK-BR-3). With the addition of metformin, apoptosis dramatically increased in all 5 cell lines. Flow cytometric analysis was used to determine the percent of apoptotic cells (Fig. 3A and B; Fig. S1). Of note, when MDA-MB-468 cells were grown in the absence of glucose (G0 0 mmol/L, see Fig. 3B), metformin induced apoptosis in nearly 50% of cells (consistent with the findings of synthetic lethality, when metformin is administered with glucose withdrawal, recently reported by Menendez et al.49).
Figure 3.

Lowering glucose levels enables metformin to effectively increase cellular apoptosis and decreases levels of cell cycle-regulated proteins. (A) Cell lines MCF7, SK-BR-3, MDA-MB-468, MDA-MB-231, and BT-549 were treated as described. Bar graphs show the percentage of cells in apoptosis (sub G1 fraction) are representative of 3 independent experiments ± SE. (B) Mod Fit analysis of flow cytometry studies performed in (A) shown with the addition of cells treated with 0 mmol/L glucose with or without metformin. Percentage of cells in apoptosis, G1, S, and G2/M are representative of 3 independent experiments. (C) MDA-MB-468 cells were treated as described and western blots performed for E2F1, Cyclins (A, B, D1, and E). Data shown are representative of 3 independent experiments.
Western blots were used to examine the expression of cell cycle-regulatory proteins under similar experimental conditions, with glucose at 5, 7, 10, or 17 mmol/L with or without metformin at 5 mmol/L (Fig. 3C). Metformin treatment was associated with a reduction in E2F1, cyclins A, B, D1, and E expression in MDA-MB-468 cells, which was most pronounced in cells cultured in 5 mmol/L glucose (Fig. 3C). These changes are consistent with the cell cycle data shown in Figure 3B, showing partial S-phase arrest was most significant under low as compared with higher glucose levels. Similar results were observed with MDA-MB-231 cells (data not shown).
Glucose stimulates cell motility and alters metformin inhibition
We investigated the effects of glucose at various concentrations, with or without metformin, on cellular motility as measured by wound-healing assays. As shown in Figure 4A and B (Fig. S2), cell migration of MDA-MB-231 and MDA-MB-468 TNBC cells was significantly higher in cells grown in media with 17 mmol/L, as compared with 5 mmol/L glucose. Metformin significantly inhibited cell migration in each cell line, under both high and low glucose conditions. The extent of inhibition (percent wound closure) was greater at 5 mmol/L glucose than at 17 mmol/L glucose in MDA-MB-231 cells (Fig. 4B; Fig. S2A and C). Immunofluorescent (IF) staining of MDA-MB-231 and MDA-MB-468 was performed on the wound healing adherent culture assays to study the morphology and expression of cytoskeletal proteins moesin (green) and phallodin/F-actin (red). Low power images (10×) of these IF-stained cells is shown in Figure 4A and Figure S2 and 3. High glucose (17 mmol/L) increased the motility of both TNBC cell lines significantly, as compared with control cells cultured with 5 mmol/L glucose-containing media. High-powered microscopic images (100×) of the IF-stained cells are shown in Figure 4C and Figure S2B. Under high glucose conditions, increased cytoplasmic branching and outgrowth, with greater co-localization of the cytoskeletal proteins moesin (MSN) and F-actin within the filopodial cytoplasmic projections was observed (Fig. 4C). Metformin treatment significantly reduced actin filament radial striations, filapodial extensions, and motility, consistent with changes in the expression of moesin and its co-localization with F-actin. These findings are consistent with the significant reduction of motility we observed in both MDA-MB-231 and MDA-MB-468 cell lines treated with metformin.
Figure 4.
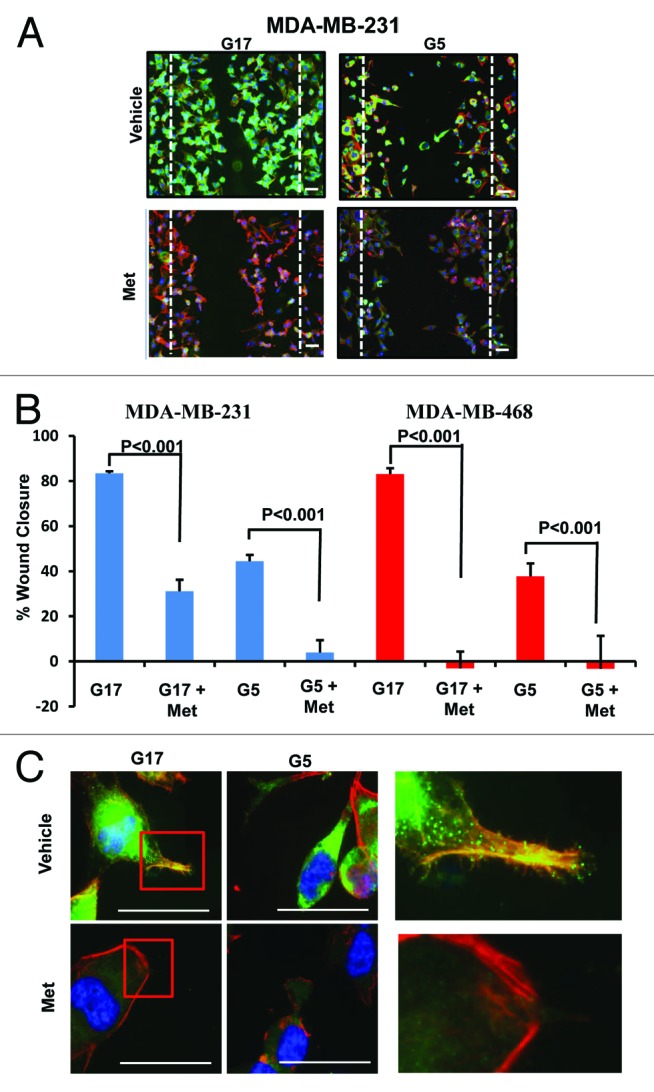
Metformin inhibits cellular motility and wound closure. (A) MDA-MB-231 cell were processed as described. Cover glass was stained by immol/Lunofluorescence (IF) for expression of moesin (green), F-actin (red), and DAPI (blue). Dotted line represents initial scratch at time zero. (B) Percent of wound closure of MDA-MB-231 (left) and MDA-MB-468 (right) was measured relative time 0; bar is representative of 40 μM. Columns are averages of (n = 6) determinants of 3 independent experiments ± SE. (C) MDA-MB-231 cells treated as described above were imaged at 100x on a Nikon microscope. Insets display colocalization of MSN/F-actin and filopodia extension of cells in 17 mmol/L glucose with or without 5 mmol/L metformin, bar is representative of 20 μM. Images are representative of 3 independent experiments.
Glucose concentration affects cell signaling and metformin-associated shifts
We have previously shown that metformin has a significant effect on the expression and activation of critical cell signaling proteins.46-48 As shown above, high concentrations of glucose stimulate cell proliferation, clonogenicity, cell cycle progression, and cell motility while suppressing apoptosis, when compared with the rates of these events observed in cells cultured in physiological glucose concentrations. In each of these assays, metformin-associated changes were typically amplified at physiological as compared with supra-physiological glucose. These data provide a rationale to determine whether glucose, with or without metformin, altered the expression and activation of critical cell signaling molecules. As shown in Figure 5, an association between the amount of glucose in the culture media and expression of Akt, mTOR, STAT3, and ERK was not observed. IGF-1R levels were inversely associated with glucose concentration. Increasing glucose was associated with an increase in p-mTOR and p-MAPK. Treatment of these cells with metformin significantly decreased p-Akt but not Akt, p-mTOR but not mTOR, p-STAT3 but not Stat3, p-erbB-2 and erbB2 (in SKBR3 cells), p-EGFR and EGFR, and IGF-1R. These effects were typically observed in cells cultured with 5 mmol/L and 10 mmol/L glucose, but they were less pronounced in cells grown under the highest glucose levels (17 mmol/L) (Fig. 5). There was no effect of either glucose or metformin upon the expression of β-actin (Fig. 5). These data indicate that the concentration of glucose in the media used for adherent cell cultures can affect both basal activation of numerous signaling molecules as well as metformin efficacy.
Figure 5.
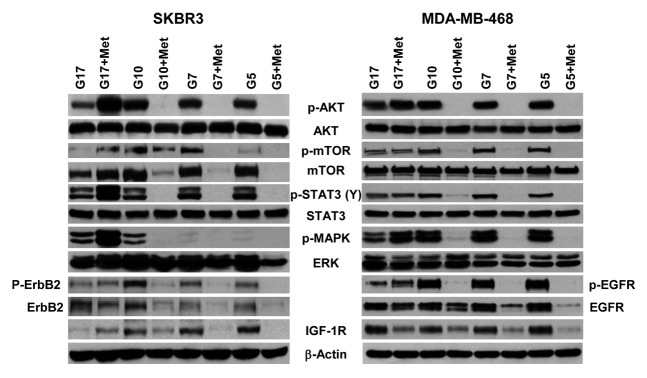
Physiological glucose enables metformin to effectively inhibit procarcinogenic signal transduction in SKBR3 and MDA-MB-468 cells. (A) SK-BR-3 (left) and MDA-MB-468 (right) cells treated with varying concentrations of glucose (5, 7, 10, or 17 mmol/L) in the absence or presence of 5 mmol/L metformin. Western blot membranes were probed for various signaling molecules as described in the “Materials and Methods”. Blots are representative of 3 independent experiments.
Metformin-associated changes in gene expression are altered by glucose concentrations
To extend the results shown above, we used gene expression profiling to characterize changes in gene expression induced by metformin treatment in cells cultured at either physiological glucose (5 mmol/L) or supra-physiological (17 mmol/L) glucose. The networks of genes whose expression were altered were identified using Affymetrix microarrays, followed by the gene ontology (GO) function of the Partek programing suite for analysis. As shown in Figure 6A, metformin caused major shifts in the expression of genes associated with cell metabolism and cell proliferation when the cells were grown in 17 mmol/L glucose. In contrast, when cells were grown at physiological concentrations of glucose (5 mmol/L), metformin induced changes in gene expression in 4 major functional areas, including cell killing, metabolic processes, cell proliferation, and cellular processes. While we cannot draw conclusions based on the percentage of total genes differentially expressed in each category under conditions of normal vs. high glucose concentrations, it is striking to note that the differential expression in genes associated with cell killing following metformin treatment were only observed in cells cultured in 5 mmol/L glucose and not in cells cultured in 17 mmol/L glucose. These results are consistent with data shown in Figure 3 above. In Figure 6B, we show selected genes and the fold change induced by metformin in the MDA-MB-468 cells under conditions of normal (5 mmol/L) or supra-physiologic glucose (17 mmol/L). We then validated the array findings using quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) with probes for fatty acid synthase (FASN), lanosterol synthase (2,3-oxidosqualene-lanosterol cyclase (LSS), insulin-induced gene 1 (INSIG1), moesin (MSN), and growth factor receptor binding-2 (GRB2). The moesin data are consistent with IF-staining patterns demonstrated in Figure 4 and Figures S2 and S3. When the expression of these genes was examined as a function of glucose concentration alone in MDA-MB-468 cells (5 mmol/L vs. 17 mmol/L) there was no significant change in the amount of FASN RNA; however, significant changes in the amount of RNA for LSS, INSIG1, MSN and GRB2 were observed (Fig. 6C). Metformin treatment caused a greater fold change in the amount of RNA for FASN, LSS, and GRB2 in cultures containing 5 mmol/L glucose; however, this was not the case for INSIG1, where metformin was observed to cause a greater increase when cells were cultured in 17 mmol/L glucose (Fig. 6B). In a similar study using MDA-MB-231 (Fig. S4), metformin treatment had significant effects upon the expression of FASN and LSS. In aggregate, these data show that glucose concentration alone, and interactions between glucose by concentration and metformin are variably observed but are significant, particularly in TNBC cells in vitro.
Figure 6.

Microarray analysis of MDA-MB-468 cells cultured in media with normal (5 mmol/L) or high (17 mmol/L) glucose in the presence or absence of metformin. (A) MDA-MB-468 cells were treated as described and analyzed on the Affymetrix Human Gene 1.0 ST Array platform. Gene groups differentially expressed from gene ontology (GO) functions for biological processes as a result of above treatment are shown as a pie chart and representative of biological triplicates. (B) Selected genes differentially regulated by metformin in 5 or 17 mmol/L glucose as determined by Affymetrix gene array. Columns are representative of fold change relative to controls, and are averages of biological triplicate determinants ± SE. (C) Quantitative RT-PCR was performed on MDA-MB-468 treated as indicated above normalized to β-actin control. Columns are representative of triplicate determinants ± SE.
Discussion
There has been a resurgence of interest in studying host and cancer cell metabolism in order to identify novel approaches to prevent or treat cancers that arise in many organ systems. While glucose deprivation and starvation generally reduce cancer risk and slow down aging, chronic energy excess, obesity, and type 2 diabetes typically have the opposite effect (see refs. 23 and 29). Breast cancer cell lines grown in adherent cultures using tissue culture media containing physiological glucose (5 mmol/L), nutrients and other growth factors typically grow well, whereas removal of glucose from the media is often incompatible with growth and may induce cell death (Fig. 1A). In actuality, most tissue culture media contains supra-normal glucose (~17 mmol/L) as well as excessive amounts of L-glutamine, which serve as sources to generate energy. These additives complicate direct comparisons of metabolic data derived from in vitro and in vivo studies.
Translational concerns have also been expressed for in vitro studies of metformin use as an anti-cancer agent. Using adherent cultures of human cancer-derived cell lines, mmol/L concentrations of metformin are often required to induce anti-cancer affects, whereas in humans the therapeutic dose is 15–17 µmol/L.50-52 Our data suggests that there are 2 major reasons why such high doses of metformin are required in vitro. First, many experiments use very short-term exposure to metformin (12–48 h). We have shown that metformin treatment of cells for weeks or months, as is required for clonogenicity assays, anti-cancer activity can be detected using doses in the µmol/L range. Second, we have shown that supra-physiologic glucose can significantly increase the EC50 of metformin in short-term adherent and non-adherent cell culture assays. Our data indicates that the so called metformin dosing “paradox”27 may not be translationally significant. We therefore anticipate that ongoing clinical trials of metformin use in breast cancer patients will demonstrate significant anti-cancer activity and improved patient outcomes, as has been suggested by early data (see refs. 36 and 53).
There is increasing consensus that metformin has potent anti-cancer activity, particularly against breast cancer (summarized in detail elsewhere, see refs. 27, 29, 31, and 36). Investigators have linked regulation of genome stability surveillance, cell cycle, and energy metabolism through a central process (the ATM DNA damage response) that may be pivotal to cancer prevention via the recognition and repair of DNA damage and control cell cycle progression.54 Menendez and colleagues have suggested that through perturbation of this defensive mechanism, metformin may upregulate our anti-cancer defense. They theorize that metformin acts as a “tissue sweeper”, eliminating damaged pre-malignant cells before they undergo cell transformation and gain stem-like properties.31,54 Others have focused on the ability of metformin to block immune cell-mediated tumor inflammation that drives cellular transformation and stem-like cell expansion, through inhibition of the NF-κB, IL-6/Stat3 feedback loop.55 Regulation of breast cancer stem cell ontogeny may occur via transcriptional regulation of the epithelial mesenchymal transition (EMT) and may also be related to differences in metformin efficacy in adherent vs. non-adherent cultures, which may also have implications for in vitro study design.56 Finally, Hirsch and colleagues have demonstrated that because metformin has selective abilities to target stem cells, it may selectively kill those cells not damaged or destroyed by standard chemotherapy and thus block tumor growth and prolong remission.57
The ability of metformin to improve disease-free and overall survival of a diverse set of breast cancer patients is currently being tested by randomized clinical trials. There are many unanswered questions, including will metformin benefit patients with and without metabolic dysregulation, should it be combined with other agents (and which ones) or be given sequentially, are there tumor markers (or tests like PET-FDG) to predict which cancers will be the most responsive? Data from retrospective analyses indicates that metformin may be especially effective in patients with metabolic dysregulation. Diabetic patients treated with metformin have shown the highest rates of complete response with neoadjuvant chemotherapy, as compared with diabetic patients on other agents or non-diabetics.35 Bayraktar et al. has reported that type 2 diabetic patients with TNBC, receiving adjuvant metformin, had a significantly lower incidence of tumor metastases as compared with non-metformin-treated diabetic and non-diabetic patients.30 The interactions between glucose dose/dependency and metformin efficacy shown in this report may have significant implications for the clinical use of metformin in breast cancer patients, particularly if they have concomitant metabolic dysregulation. In these patients, maintenance of glucose homeostasis through energy restriction, exercise, and other modalities may further enhance metformin sensitivity and patient outcomes. In a recent review, Gillies and colleagues examined the underlying mechanisms that increase glucose consumption in cancers. They hypothesize that metabolic reprogramming, which uses glucose and generates energy, building blocks, and acids provides a selective advantage for cancer cells, because it facilitates localized invasion through tumor stroma.58 Our findings suggest that metformin has anti-cancer activity against all molecular subtypes of breast cancer, although TNBC appear the most sensitive (lowest EC50 values) across a wide range of glucose concentrations, which may reflect their unique genotype and aggressive phenotype, upregulation of aerobic glycolysis, or a larger subpopulation of highly sensitive stem-like cells.
Menendez and colleagues have reported that metformin is “synthetically lethal” with glucose withdrawal in breast cancer cells. They note that glucose deprivation is a “distinctive feature of the tumor microsystem” and postulate that, in combination, hypoglycemia and metformin will induce apoptosis.49 Our data suggests that under these conditions, the absence of glucose is the major driver of cell death. Furthermore, we have shown that under physiological glucose conditions, metformin can induce significant apoptosis, reduce proliferation, and alter tumor aggression and cell signaling in various breast cancer cell subtypes. Although the concept of synthetic lethality in the absence of glucose is an interesting concept, we are unable to find experimental evidence that this occurs in vivo. Data from a carcinogen-induced rodent model of mammary tumorigenesis has shown that mammary tumors in lean overfed rats take up glucose at approximately the same rate as surrounding mammary and peripheral tissues. In obese overfed animals (with upregulation of insulin, as one might observe in patients with metabolic syndrome) glucose uptake by the tumor cells (and hence microenvironmental availability) was increased by 50%.45 In light of these findings and the demonstrated increase in the EC50s of metformin under high glucose conditions, we hypothesize that altering metformin dose in patients with functional evidence of the Warburg effect (by a positive FDG-PET scan) may increase its anti-cancer activity, particularly in patients with non-TNBC.
In summary, we have shown that energy excess in the form of high glucose promotes cell proliferation, clonogenicity, motility, and upregulation/activation of pro-oncogenic signaling, with a reduction of apoptosis. Supra-physiologic glucose reduces the effectiveness of metformin in various molecular subtypes of cancer cells, but may be overcome by using higher doses of metformin. Alternatively, maintenance of glucose homeostasis may improve metformin efficacy in some patients. These data confirm that metformin induces a broad spectrum of biological and molecular effects against breast cancer cells and may reduce cellular response to critical drivers of tumorigenesis, including insulin, IL-6, and EGF via downregulation of IGF-1R, p-Stat-3, and EGFR.
Materials and Methods
Cell lines and reagents
Human breast cancer-derived cell lines MCF7 and T-47D (luminal A), BT-474 and ZR-75–30 (luminal B), MDA-MB-453 and SK-BR-3 (Her2), MDA-MB-468, HCC70, HCC1806, MDA-MB-231, BT-20, HCC1143, HCC1937, and BT-549 (TNBC) were obtained from the American Type Culture Collection (ATCC). The identity of each cell line was demonstrated to be authentic by analysis of short tandem repeats by the University of Colorado Cancer Center DNA Sequencing and Analysis Shared Resource before the start of this project. All cell lines were confirmed to be free of mycoplasma contamination (using a MycoAlert detection kit, Lonza Walkersville Inc). All cells were maintained in DMEM: nutrient mix F-12 (D-MEM/F-12 1:1; Invitrogen Corp), supplemented with 5–10% fetal bovine serum (FBS; Sigma Chemical Co or Thermo Fisher Scientific) in a 37 °C humidified atmosphere containing 95% air and 5% CO2. Cells to be treated with metformin were starved with glucose-free medium (Invitrogen Corp), then replenished with pure glucose supplemented at 5, 7, 10, or 17 mmol/L glucose (Sigma Chemical Co). Metformin (1,1-dimethylbiguanide hydrochloride: from MP Biomedicals, LLC) was dissolved in sterile water and used at the indicated concentration for each specific study.
Cell proliferation assay
A CellTiter 96 AQ nonradioactive cell proliferation kit (Promega Corp) was used as described48 to determine cell viability. In brief, 5000–10 000 cells were plated in each well of a 96-well plate in DMEM/F-12 medium with 1% FBS and 15 mmol/L HEPES (pH 7.4) for 24 h, and then grown in either DMEM medium containing various glucose concentrations (5, 7, 10, 17 mmol/L glucose) with 0.5% FBS, or the same medium containing various doses of metformin (5 or 10 mmol/L). After 72 h, wells were read at 490 nm with a microplate reader. All conditions were tested in triplicate samples, and the percentage of surviving cells from each group was calculated relative to controls. Control cultures were defined as 100% survival.
Clonogenic assays
Clonogenic assays were performed as described.48 Cells were placed in 6-well plates (in triplicate) at a density of 1000–2000 cells/well, with 2 ml of medium containing 10% FBS. After 24 h, the supernatant was removed and replaced with DMEM media with specific concentrations of glucose (5, 10, or 17 mmol/L) and 0.5–1.0% FBS. Cells were treated with vehicle control or 5 mmol/L metformin for 2–4 wk, and grown in a 37 °C humidified atmosphere containing 95% air and 5% CO2. Colonies were visualized by staining with a 0.5% crystal violet in 25% methanol, followed by 3 washes with tap water to remove excess stain. Colony numbers were counted using a gel doc imager (BioRad Laboratories).
Cell cycle analysis
Cell cycle distribution of treated and untreated cells as previously described.48 Cells were seeded in 60-mm dishes with DMEM medium containing various concentrations of glucose (5, 7, 10, or 17 mmol/L glucose), with or without 5 mmol/L metformin for 24 h. Cells were harvested and fixed in 70% ethanol containing 50 μg/ml propidium iodide and 100 μg/ml RNase I in PBS for 30 min at 37 °C and then stored overnight at 4 °C. Cell cycle distribution was analyzed with the Gallios™ Flow Cytometer (Beckman Coulter Inc) in the University of Colorado Cancer Center Flow Cytometry Shared Resource.
Wound healing/motility assays
MDA-MB-231 or MDA-MB-468 cells were seeded at 3 × 105 cells/well into a 6-well plate and allowed to grow to confluence in DMEM/F12 medium with 10% FBS. Cells were treated with 10 μg/ml mitomycin C (Sigma Chemical Co) for 3 h, scratched with 200 µl pipette tip, and then gently washed 5 times with PBS to remove detached cells. Cells were then starved with DMEM medium containing 0 mmol/L glucose (G0) for 4–6 h then replenished with either 17 mmol/L glucose (G17) or 5 mmol/L glucose (G5) with or without 5 mmol/L (MDA-MB-468) or 10 mmol/L (MDA-MB-231) metformin for 72 h. Phase contrast images were taken at t = 0 h and then again at t = 72 h using an inverted phase microscope. Wound sizes were determined using NIS Elements imaging software (Nikon).
Immunofluorescent imaging
MDA-MB-468 and MDA-MB-231 cells were grown on glass coverslips and treated as described in the wound-healing assay. Medium was removed from each well, and coverslips were washed ×3 in PBS. The cells were fixed for 15 min in ice cold, 4% paraformaldehyde (Electron Microscopy Sciences), pH 7.2–7.3, supplemented with 10% fetal bovine serum (FBS). The cells were permeabilized 3 times for 5 min each, with 0.1% TRITON® X-100 in PBS. The cells were washed 3 times with 0.05% TBS-T then blocked with PBS containing 1% BSA with 4% FBS for 1 h. Cells were then washed 3 times with 0.05% TBS-T then incubated overnight at 4 °C with rabbit anti-moesin (Sigma Chemical Co) in 1× PSB with 4% FBS. The following day, the cells were washed 3 times for 15 min in 0.05% TBS-T before incubation with the secondary antibody Alexa Flour 488 goat-anti-rabbit IgG (Invitrogen Corp) in blocking reagent for 1.5 h at room temperature. Cells were then doubled labeled with phallodin (Alex Flour 568; Invitrogen Corp) per manufacturer’s instructions. Cells were stained with phalloidin for 1 h and then washed 3 times in 0.05% TBS-T. Cells were mounted with Vectashield mounting medium with DAPI (Vector Laboratories)
Western blot analysis
Expression and activation of signaling molecules was quantitated by western blot. SK-BR-3 or MDA-MB-468 cells were grown under varying media conditions as described above. Protein concentrations were determined using the Coomassie Plus protein assay reagent (Pierce Chemical Co). Equal amounts of cell protein lysates were combined with SDS-PAGE loading buffer containing 5% 2-mercaptoethanol, and proteins were resolved by gel electrophoresis using various percent gels, and then transferred to a nitrocellulose membrane (BioRad Laboratories). Antibodies were from the following sources: AKT, anti-phospho Akt (Ser473; p-AKT), mTOR, phospho-mTOR (p-mTOR), Stat3, phospho-Stat3 (Y705; p-Stat3), MAPK (erk2), phospho-MAPK (p44/41 MAP Kinase, Thr202/Tyr204; p-MAPK), erbB-2, phospho-erbB-2 (Tyr 1246; p-erbB2), IGF-1R, Cyclin A2 (BF682), Cyclin B1, and E2F1, were all obtained from Cell Signaling Technology, Inc; Cyclin E (HE12), and Cyclin D1(M-20) were obtained from Santa Cruz Biotechnology; and EGFR (F4), phospho-EGFR (Tyr1068; p-EGFR) and β-actin (AC-75) were obtained from Sigma Chemical Co.
Real-time quantitative reverse transcriptase-PCR (RT-qPCR)
cDNA was created using the Verso cDNA Synthesis Kit (Cat #AB-1453/A, Thermo Scientific) and 1 μg of total RNA. Predesigned gene-specific primer and probe sets were obtained from SA Biosciences, for the following human genes: lanosterol synthase (2,3-oxidosqualene lanosterol cyclase) (LSS) (PPH06366A) NM_002340.5; insulin induced gene 1 (INSIG1) (PPH06359B) NM_005542.4; fatty acid synthase (FASN) (PPH01012B) NM_004104.4; growth factor receptor-bound protein 2 (GRB2) (PPH00714C) NM_002086.4; moesin (MSN) (PPH13452B) NM_00244.2; and β-actin (ACTB) (PPH00073 G) NM_001101.3. qRT-PCR synthesis was performed using DyNAmo Flash SYBR Green qPCR kit DyNAmo ColorFlash SYBR Green qPCR kit (Thermo Scientific\Applied Biosystems). The assay was performed according to the manufacturer’s protocol on a 7500 Fast Real-time PCR System (Applied Biosystems, Inc). The relative mRNA levels were calculated using the comparative Ct method (ΔΔCt). Briefly, the Ct (cycle threshold) values for the β-actin were subtracted from Ct values of the mRNA of genes listed above to achieve the ΔCt value. The 2−ΔCt was calculated and then divided by a control sample to achieve the relative mRNA levels (ΔΔCt). Reported values are the means and standard errors of 3 biological replicates.
Gene expression microarrays
MDA-MB-468 cells were seeded in DMEM medium containing 0% FBS with either 5 or 17 mmol/L glucose, with or without 10 mmol/L metformin for 24 h. Whole cells were submitted to the UC Denver Microarray Core facility in triplicate for processing. Total RNA was isolated and purified samples were analyzed using the Affymetrix Human Gene 1.0 ST Array microarray containing 33 297 probe set IDs for known genes. RNA quality was RNA quality was assessed and high quality RNA (i.e., RNA integrity number was >7.0) was used in this experiment in accordance with Affymetrix GeneChip© Whole Transcript (WT) sense target labeling assay protocol. rRNA reduction, first round double-strand-cDNA synthesis, ss-cDNA fragment, and labeling were performed in accordance with Affymetrix GeneChip© WT sense target labeling assay manual. Affymetrix Human Exon 1.0 ST microarray were hybridized overnight in accordance to manual guidelines. Signal intensity estimate and P value for each of the processed samples were processed to determine the fold change ratios in each cell line between control and metformin-treated samples. Fold changes over control ≥2.0 or ≤−2.0 were accepted for analysis.
Statistical considerations and calculations of Metformin EC50 by glucose concentration
Statistical analyses of the experimental data were performed using a 2-sided Student t test. Significance was set at P < 0.05. EC50s were calculated using a nonlinear regression using one site competition curve.59 All statistical analysis were performed using Prism5 program (GraphPad Software).
Supplementary Material
Acknowledgments
Support for the UCDenver Micro Array Core Facility is provided in part by the Genomics Shared Resource of Colorado's NIH/NCI Cancer Center Support Grant P30CA046934.
Support for the UCDenver Flow Cytometry Shared Resources Core Facility is provided in part by Colorado's NIH/NCI Cancer Center Support Grant P30CA046934.
Support for the UCDenver Cancer Center DNA Sequence and Analysis Shared Resource Core for cell line authentication is provided in part by Colorado’s NIH/NCI Cancer Center Support Grant P30CA043934
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
Grant support provided by Susan G Komen for the Cure K100575 to RWA, ZF, SME, JR, SMA, and ADT.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26641
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26641
References
- 1.Barone BB, Yeh HC, Snyder CF, Peairs KS, Stein KB, Derr RL, Wolff AC, Brancati FL. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA. 2008;300:2754–64. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 3.Goodwin PJ. Host-related factors in breast cancer: an underappreciated piece of the puzzle? J Clin Oncol. 2008;26:3299–300. doi: 10.1200/JCO.2007.15.4526. [DOI] [PubMed] [Google Scholar]
- 4.Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9:88. doi: 10.1186/1471-2458-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall IJ, Newman B, Millikan RC, Moorman PG. Body size and breast cancer risk in black women and white women: the Carolina Breast Cancer Study. Am J Epidemiol. 2000;151:754–64. doi: 10.1093/oxfordjournals.aje.a010275. [DOI] [PubMed] [Google Scholar]
- 6.Harvie M, Hooper L, Howell AH. Central obesity and breast cancer risk: a systematic review. Obes Rev. 2003;4:157–73. doi: 10.1046/j.1467-789X.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- 7.LeRoith D, Novosyadlyy R, Gallagher EJ, Lann D, Vijayakumar A, Yakar S. Obesity and type 2 diabetes are associated with an increased risk of developing cancer and a worse prognosis; epidemiological and mechanistic evidence. Exp Clin Endocrinol Diabetes. 2008;116(Suppl 1):S4–6. doi: 10.1055/s-2008-1081488. [DOI] [PubMed] [Google Scholar]
- 8.Rose DP, Haffner SM, Baillargeon J. Adiposity, the metabolic syndrome, and breast cancer in African-American and white American women. Endocr Rev. 2007;28:763–77. doi: 10.1210/er.2006-0019. [DOI] [PubMed] [Google Scholar]
- 9.Schapira DV, Kumar NB, Lyman GH, Cox CE. Abdominal obesity and breast cancer risk. Ann Intern Med. 1990;112:182–6. doi: 10.7326/0003-4819-112-3-182. [DOI] [PubMed] [Google Scholar]
- 10.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Stoll BA. Upper abdominal obesity, insulin resistance and breast cancer risk. Int J Obes Relat Metab Disord. 2002;26:747–53. doi: 10.1038/sj.ijo.0801998. [DOI] [PubMed] [Google Scholar]
- 12.Barnett GC, Shah M, Redman K, Easton DF, Ponder BA, Pharoah PD. Risk factors for the incidence of breast cancer: do they affect survival from the disease? J Clin Oncol. 2008;26:3310–6. doi: 10.1200/JCO.2006.10.3168. [DOI] [PubMed] [Google Scholar]
- 13.Goodwin PJ. Insulin in the adjuvant breast cancer setting: a novel therapeutic target for lifestyle and pharmacologic interventions? J Clin Oncol. 2008;26:833–4. doi: 10.1200/JCO.2007.14.7132. [DOI] [PubMed] [Google Scholar]
- 14.Hede K. Doctors seek to prevent breast cancer recurrence by lowering insulin levels. J Natl Cancer Inst. 2008;100:530–2. doi: 10.1093/jnci/djn119. [DOI] [PubMed] [Google Scholar]
- 15.Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE. The impact of diabetes on survival following breast cancer. Breast Cancer Res Treat. 2008;109:389–95. doi: 10.1007/s10549-007-9654-0. [DOI] [PubMed] [Google Scholar]
- 16.Pasanisi P, Berrino F, De Petris M, Venturelli E, Mastroianni A, Panico S. Metabolic syndrome as a prognostic factor for breast cancer recurrences. Int J Cancer. 2006;119:236–8. doi: 10.1002/ijc.21812. [DOI] [PubMed] [Google Scholar]
- 17.Peairs KS, Barone BB, Snyder CF, Yeh HC, Stein KB, Derr RL, Brancati FL, Wolff AC. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol. 2011;29:40–6. doi: 10.1200/JCO.2009.27.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Protani M, Coory M, Martin JH. Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis. Breast Cancer Res Treat. 2010;123:627–35. doi: 10.1007/s10549-010-0990-0. [DOI] [PubMed] [Google Scholar]
- 19.Wolf I, Sadetzki S, Catane R, Karasik A, Kaufman B. Diabetes mellitus and breast cancer. Lancet Oncol. 2005;6:103–11. doi: 10.1016/S1470-2045(05)01736-5. [DOI] [PubMed] [Google Scholar]
- 20.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol. 2009;27:3271–3. doi: 10.1200/JCO.2009.22.1630. [DOI] [PubMed] [Google Scholar]
- 21.Davis AA, Kaklamani VG. Metabolic syndrome and triple-negative breast cancer: a new paradigm. Int J Breast Cancer. 2012;2012:809291. doi: 10.1155/2012/809291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guastamacchia E, Resta F, Triggiani V, Liso A, Licchelli B, Ghiyasaldin S, Sabbà C, Tafaro E. Evidence for a putative relationship between type 2 diabetes and neoplasia with particular reference to breast cancer: role of hormones, growth factors and specific receptors. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:59–66. doi: 10.2174/1568008043339965. [DOI] [PubMed] [Google Scholar]
- 23.Young CD, Anderson SM. Sugar and fat - that’s where it’s at: metabolic changes in tumors. Breast Cancer Res. 2008;10:202. doi: 10.1186/bcr1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodwin PJ, Ennis M, Bahl M, Fantus IG, Pritchard KI, Trudeau ME, Koo J, Hood N. High insulin levels in newly diagnosed breast cancer patients reflect underlying insulin resistance and are associated with components of the insulin resistance syndrome. Breast Cancer Res Treat. 2009;114:517–25. doi: 10.1007/s10549-008-0019-0. [DOI] [PubMed] [Google Scholar]
- 25.Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE, et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008;68:10238–46. doi: 10.1158/0008-5472.CAN-08-2755. [DOI] [PubMed] [Google Scholar]
- 26.Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, Hartwick W, Hoffman B, Hood N. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002;20:42–51. doi: 10.1200/JCO.20.1.42. [DOI] [PubMed] [Google Scholar]
- 27.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2:778–90. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 28.Josefson D. High insulin levels linked to deaths from breast cancer. BMJ. 2000;320:1496. [PMC free article] [PubMed] [Google Scholar]
- 29.Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY) 2010;2:760–74. doi: 10.18632/aging.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayraktar S, Hernadez-Aya LF, Lei X, Meric-Bernstam F, Litton JK, Hsu L, Hortobagyi GN, Gonzalez-Angulo AM. Effect of metformin on survival outcomes in diabetic patients with triple receptor-negative breast cancer. Cancer. 2012;118:1202–11. doi: 10.1002/cncr.26439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–5. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–73. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 35.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9:1057–64. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 37.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes Care. 2006;29:1990–1. doi: 10.2337/dc06-0997. [DOI] [PubMed] [Google Scholar]
- 38.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 42.Zhivotovsky B, Orrenius S. The Warburg Effect returns to the cancer stage. Semin Cancer Biol. 2009;19:1–3. doi: 10.1016/j.semcancer.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Quon A, Gambhir SS. FDG-PET and beyond: molecular breast cancer imaging. J Clin Oncol. 2005;23:1664–73. doi: 10.1200/JCO.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 44.Hamanaka RB, Chandel NS. Targeting glucose metabolism for cancer therapy. J Exp Med. 2012;209:211–5. doi: 10.1084/jem.20120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giles ED, Wellberg EA, Astling DP, Anderson SM, Thor AD, Jindal S, Tan AC, Schedin PS, Maclean PS. Obesity and overfeeding affecting both tumor and systemic metabolism activates the progesterone receptor to contribute to postmenopausal breast cancer. Cancer Res. 2012;72:6490–501. doi: 10.1158/0008-5472.CAN-12-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–15. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 47.Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, Wahdan-Alaswad R, Thor AD. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–76. doi: 10.4161/cc.11.2.18813. [DOI] [PubMed] [Google Scholar]
- 48.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–40. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 49.Menendez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2012;11:2782–92. doi: 10.4161/cc.20948. [DOI] [PubMed] [Google Scholar]
- 50.Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. 2012;48:R31–43. doi: 10.1530/JME-12-0007. [DOI] [PubMed] [Google Scholar]
- 51.Kourelis TV, Siegel RD. Metformin and cancer: new applications for an old drug. Med Oncol. 2012;29:1314–27. doi: 10.1007/s12032-011-9846-7. [DOI] [PubMed] [Google Scholar]
- 52.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–9. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 53.Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, Escallon J, Leong WL, McCready DR, Reedijk M, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. 2012;135:821–30. doi: 10.1007/s10549-012-2223-1. [DOI] [PubMed] [Google Scholar]
- 54.Menendez JA, Cufí S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L, Vazquez-Martin A. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging (Albany NY) 2011;3:1063–77. doi: 10.18632/aging.100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A. 2013;110:972–7. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9:3807–14. doi: 10.4161/cc.9.18.13131. [DOI] [PubMed] [Google Scholar]
- 57.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 59.Ding WQ, Liu B, Vaught JL, Palmiter RD, Lind SE. Clioquinol and docosahexaenoic acid act synergistically to kill tumor cells. Mol Cancer Ther. 2006;5:1864–72. doi: 10.1158/1535-7163.MCT-06-0067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.