

Abstract
The alphaproteobacterial family Beijerinckiaceae contains generalists that grow on a wide range of substrates, and specialists that grow only on methane and methanol. We investigated the evolution of this family by comparing the genomes of the generalist organotroph Beijerinckia indica, the facultative methanotroph Methylocella silvestris and the obligate methanotroph Methylocapsa acidiphila. Highly resolved phylogenetic construction based on universally conserved genes demonstrated that the Beijerinckiaceae forms a monophyletic cluster with the Methylocystaceae, the only other family of alphaproteobacterial methanotrophs. Phylogenetic analyses also demonstrated a vertical inheritance pattern of methanotrophy and methylotrophy genes within these families. Conversely, many lateral gene transfer (LGT) events were detected for genes encoding carbohydrate transport and metabolism, energy production and conversion, and transcriptional regulation in the genome of B. indica, suggesting that it has recently acquired these genes. A key difference between the generalist B. indica and its specialist methanotrophic relatives was an abundance of transporter elements, particularly periplasmic-binding proteins and major facilitator transporters. The most parsimonious scenario for the evolution of methanotrophy in the Alphaproteobacteria is that it occurred only once, when a methylotroph acquired methane monooxygenases (MMOs) via LGT. This was supported by a compositional analysis suggesting that all MMOs in Alphaproteobacteria methanotrophs are foreign in origin. Some members of the Beijerinckiaceae subsequently lost methanotrophic functions and regained the ability to grow on multicarbon energy substrates. We conclude that B. indica is a recidivist multitroph, the only known example of a bacterium having completely abandoned an evolved lifestyle of specialized methanotrophy.
Keywords: methanotrophy, methane monooxygenase, methylotrophy, specialist, generalist
Introduction
Methane is a major greenhouse gas and an alternative fuel. In the environment, methanotrophic microorganisms couple the oxidation of methane to the reduction of sulfate, nitrite or O2 (Stein et al., 2012). The aerobic methanotrophs employ a methane monooxygenase enzyme (MMO), of either a soluble, cytoplasmic type (sMMO), or a particulate, membrane-bound type (pMMO) to oxidize methane to methanol, which is then catabolised for energy production (Stein et al., 2012). Most known aerobic methanotrophs grow only on methane, methanol, formate and some methylated amines. They cannot grow on substrates containing carbon–carbon (C−C) bonds, and are therefore termed ‘obligate methanotrophs' (Wood et al., 2004). The most dramatic exception is the facultative methanotroph Methylocella silvestris, which grows on acetate, ethanol, pyruvate, succinate, malate and propane in addition to methane and methanol (Dedysh et al., 2005; Chen et al., 2010). More limited facultative methanotrophy has also been observed in Methylocapsa aurea and several Methylocystis strains, which utilize acetate (Belova et al., 2010; Dunfield et al., 2010), and Methylocystis sp. strain SB2, which utilizes ethanol (Im and Semrau, 2011). Although not obligate methanotrophs, these are still highly specialized bacteria with few potential growth substrates. We adopt the term ‘specialist methanotrophs' to stress the lack of catabolic substrate diversity in all aerobic methanotrophs.
The reasons behind the specialist nature of methanotrophy are not well understood. Mechanistically, it may be mediated by enzyme lesions-missing elements of key metabolic pathways. An example is the absence of functional alpha-ketoglutarate dehydrogenase in some Gammaproteobacteria methanotrophs, leading to an incomplete TCA cycle (Shishkina and Trotsenko, 1982; Wood et al., 2004); and the lack of gluconeogenic enzymes in some Alphaproteobacteria methanotrophs, leading to an inability to grow on pyruvate or acetate (Shishkina and Trotsenko, 1982). However, no metabolic lesions are universal. The genome of Methylococcus capsulatus contains genes for alpha-ketoglutarate dehydrogenase and systems necessary for sugar metabolism, even though this species cannot grow on sugars or organic acids (Ward et al., 2004; Kelly et al., 2005). It has therefore been hypothesized that obligate methanotrophy, and other obligate metabolisms like ammonia oxidation, may be primarily caused by a limitation of membrane transport systems rather than the absence of key enzymes (Chain et al., 2003; Ward et al., 2004). Experiments on whole-cell suspensions and cell extracts suggest that some obligate methanotrophs can assimilate small exogenous organic acids and alcohols, but not sugars, indicating only limited substrate import into the cytoplasm (Eccleston and Kelly, 1973; Shishkina and Trotsenko, 1982). It should be stressed that these discussions focus on the mechanisms of obligate methanotrophy, not its adaptive logic, which is harder to unravel. Restricting the import and metabolism of alternative substrates must confer an adaptive advantage by improving the efficiency of methane metabolism.
Aerobic methanotrophs are found only in the Gammaproteobacteria and Alphaproteobacteria classes of Proteobacteria, the phylum Verrucomicrobia and the candidate phylum NC10 (Stein et al., 2012). The Gammaproteobacteria methanotrophs (Methylococcaceae) (Stein et al., 2011), Verrucomicrobia methanotrophs ('Methylacidiphilaceae') (Op den Camp et al., 2009), and the two cultured methanotrophs in the NC10 phylum (Ettwig et al., 2010; Zhu et al., 2012) each appear to form monophyletic groups. Alphaproteobacteria methanotrophs belong to two families: the Methylocystaceae and Beijerinckiaceae. The presence of so few phyletic groups of methanotrophs suggests that the phenotype is complex and cannot easily evolve via horizontal transfer of a few genes, a potential example of the ‘complexity hypothesis' (Jain et al., 1999). In support of this, the phylogeny of pmoCAB genes encoding pMMO correspond closely to 16S rRNA gene phylogeny, indicating that lateral transfer of these genes is rare (Kolb et al., 2003; Op den en Camp et al., 2009; Tavormina et al., 2011). Nor, apparently, can species easily reverse their evolution into specialist methanotrophs, or we should observe more non-methanotrophic neighbors of methanotrophic species, rather than unified clades of specialist methanotrophs. All methanotroph clades noted above are composed only of specialist methanotrophs, with the exception of the Beijerinckiaceae, which includes obligate methanotrophs, facultative methanotrophs and versatile chemoorganotrophs that are non-methanotrophic but sometimes methylotrophic (Supplementary Table 1). Although these Beijerinckiaceae species are metabolically diverse, they are evolutionarily close, with a maximum of 3.8% difference among their 16S rRNA gene sequences (Supplementary Table 2). Other methanotrophs show >7% 16S rRNA gene sequence divergence to the closest known non-methanotrophic neighbor.
The Beijerinckiaceae therefore presents a unique opportunity to address methanotroph evolution and specialization. Gene transfer or loss may be more evident in these species than in more deeply-branching methanotrophs. In collaboration with the Joint Genome Institute we have sequenced the genomes of the facultative methanotroph M. silvestris, the obligate methanotroph Methylocapsa acidiphila and the non-methanotrophic chemoorganotroph Beijerinckia indica (Table 1). All were isolated from acidic soil habitats, and share phenotypic traits such as acidophily, exopolysaccharide production and the ability to fix nitrogen (Dedysh et al., 2002; Dunfield et al., 2003; Kennedy, 2005). We hypothesize that much of the genetic variability among them is driven by adaptation to different growth substrates. We compared their genomes in order to: (i) reconstruct the evolutionary history of methanotrophy in the Beijerinckiaceae and (ii) provide insight into the tradeoffs required for a specialist methanotrophic lifestyle compared with a generalist chemoorganotrophic lifestyle.
Table 1. Some physiological and genomic properties of the three study bacteria.
| Organism | Growth on CH4 | Multicarbon substrates used | G+C content (%) | Genome size (Mb) | Genome status | Plasmids | ORFs |
|---|---|---|---|---|---|---|---|
| Methylocella silvestris | + (sMMO) | Acetate, ethanol, propane, pyruvate, succinate, malate | 63.1 | 4.3 | F | 0 | 3971 |
| Methylocapsa acidiphila | + (pMMO) | None | 61.9 | 4.1 | P | 1a | 3762 |
| Beijerinckia indica | — | Many sugars, alcohols, organic acids | 57.0 | 4.4 | F | 2 | 3850 |
Abbreviations: F, finished; P, permanent draft; pMMO, particulate methane monooxygenases; sMMO, soluble methane monooxygenase.
Predicted.
Materials and methods
Organisms and genome sequencing
Genomes were obtained from M. silvestris BL2T (=DSM 15510T= NCIMB 13906T, Genome Accession Number CP001280), B. indica subsp. indica (ATCC 9039T Genome Accession Number CP001016) and M. acidiphila B2T (=DSM 13967T=NCIMB 13765T Project Accession Number PRJNA72841). Genomic and physiological properties are summarized in Table 1. M. acidiphila B2 (Dedysh et al., 2002) is typical of obligate methanotrophs. It grows on methane and methanol only, and contains a pMMO enzyme but no sMMO. B. indica (Starkey and De, 1939) grows on diverse multicarbon compounds, but does not oxidize methane or methanol (Kennedy, 2005; Dedysh, Smirnova et al., 2005). M. silvestris (Dunfield et al., 2003) is the facultative methanotroph with the widest known range of energy substrates (Dedysh et al., 2005). It is thus intermediate to the other two species catabolically (Supplementary Table 1).
The genomes of M. silvestris and B. indica are closed and have been reported as genome announcements (Chen et al., 2010, Tamas et al., 2010). The genome of M. acidiphila was generated at the DoE Joint Genome Institute using a combination of Illumina (San Diego, CA, USA; Bennett, 2004) and Roche 454 technologies (Branford, CT, USA; Margulies, 2005). Sequencing consisted of an Illumina GAii shotgun library (81 153 336 reads totaling 6 167.7 Mb), a 454 Titanium standard library (300 274 reads) and two paired-end 454 libraries with average insert sizes of 4 and 12 kb (347 398 reads) totaling 178.4 Mb of 454 data. All aspects of library construction and sequencing can be found at http://www.jgi.doe.gov/. The 454 data were assembled with Newbler, version 2.6. The Newbler consensus sequences were computationally shredded into 2-kb overlapping fake reads (shreds). Illumina sequencing data were assembled with VELVET, version 1.1.05 (Zerbino and Birney, 2008), and the consensus sequences computationally shredded into 1.5-kb shreds. The shreds and the read pairs in the 454 paired-end library were integrated using parallel phrap, version SPS-4.24 (High Performance Software, LLC). Consed (Gordon et al., 1998) was used in the following finishing process. Illumina data were used to correct potential base errors and increase consensus quality using the software Polisher (Alla Lapidus, unpublished). Possible mis-assemblies were corrected using gapResolution (Cliff Han, unpublished), or Dupfinisher (Han and Chain, 2006). The final assembly was based on 157.2 Mb of 454 draft data (38.3 × coverage) and 1,230 Mb of Illumina draft data (300 × coverage). The final assembly contained two scaffolds: a predicted 186-kb circular plasmid and a circular chromosome in five contigs.
Comparative genomics
Except where noted, genome comparisons were made using the IMG-ER platform (Markowitz et al., 2012). Some comparisons used related genomes, particularly Methylosinus trichosporium OB3b (Stein et al., 2010), Methylocystis strain SC2 (Dam et al., 2012), Methylocystis strain ATCC 49242 (Stein et al., 2011) and several strains of the genius Methylobacterium (Marx et al., 2012). A database of 115 genes involved in methane and methanol oxidation (Supplementary Table 3) including formaldehyde oxidation, formaldehyde fixation and alleviation of stress caused by ammonia cooxidation was assembled and the genomes were searched for these genes via BLAST.
To determine unique and overlapping gene sets in the three genomes, the entire ORF set from each was searched against the other two using BLASTx. Overlapping and core gene sets were based on cutoff thresholds of >60% or >40% amino-acid identity. Rare cases of incomplete agreement of reciprocal BLASTs (for example, A finds a homolog in B and C; but B finds a homolog only in A) were due to gene identities near the cutoff threshold, and were coded as universal genes.
Lateral gene transfer
IMG-ER incorporates a BLAST-based approach to detect lateral gene transfer (LGT). All BLAST hits with bit scores ⩾95% of the best hit are considered. If none of these hits come from a bacterium of the same taxonomic order as the query gene (in our case, the Rhizobiales), then the gene is considered a candidate for LGT (Markowitz et al., 2012; https://img.jgi.doe.gov/er/doc/using_index.html). This approach is questionable when few related genomes are available, as the pan-genome of the group is poorly covered. However, as of October 2012 there were 253 genomes of Rhizobiales on IMG, so the approach should be very robust in our case. As the Rhizobiales contains so many sequenced genomes, we also took this analysis down one taxonomic rank to family. High-resolution phylogeny (see Results) showed that Methylocystaceae, Methylobacteriaceae and Beijerinckiaceae are sister families. Therefore, if the top BLAST hit to a bacterium in one of these three families (24 genomes) had a bitscore <95% of the best hit, the gene was considered a candidate for LGT from another family of Rhizobiales.
Prediction of LGT was also made using three compositional methods. IslandPath-DIMOB is based on codon usage. SIGI-HMM is based on dinucleotide sequence composition bias and the presence of mobility genes. These two methods were implemented online using IslandViewer (Langille and Brinkman, 2009). Finally, Alien Hunter uses variable order motifs (2-mers to 8-mers). It can only identify large regions of LGT (2500-nt windows), as only these provide enough data to estimate nucleotide octamer frequencies (Vernikos and Parkhill, 2006).
Phylogenetic reconstructions
To create a highly resolved phylogenetic tree of the Rhizobiales, we constructed a database of 29 universal, vertically inherited proteins identified by Ciccarelli et al. (2006). The concatenation had an average length of 6399 amino acids. Phylogenies were also calculated for derived proteins with key methanotrophy/methylotrophy functions: including concatenated PmoCAB proteins (subunits of pMMO); MmoX (the hydroxylase component of sMMO); a concatenated set of 21 methylotrophy proteins found in most Alphaproteobacteria methylotrophs (Supplementary Table 3); MxaFJI elements of methanol dehydrogenase (MDH); and NifH and NifD components of nitrogenase. Concatenations were aligned using ClustalW (Thompson et al., 1994). Trees were calculated using neighbor-Joining (Kimura distance correction, Poisson distribution of variable sites, 1000 bootstraps) while excluding gaps in the alignment, using SeaView (Gouy et al., 2010). A 16S rRNA gene phylogeny was constructed via neighbor-joining on the ARB-Silva reference database Version 111 (Quast et al., 2013), using ARB (Ludwig et al., 2004).
Results
Genome characteristics
Genome size, G+C content and number of ORFs are similar among the three Beijerinckiaceae species (Table 1). At 60% identity, there are 1305 genes in the core genome, and 1771-2188 genes unique to each genome (Figure 1). The methanotrophs M. silvestris and M. acidiphila share more genes in common (452) than either does with B. indica (125 and 170). Using an identity cutoff of 40% instead of 60% shows the same general pattern, except with more core genes (1860) and fewer unique genes in each genome (1132–1346). In either case, the three genomes show a remarkable level of individuality. These trends were supported by synteny plot analysis calculated in IMG-ER using Mummer (Supplementary Figure 1). Synteny is low across all genome pairs, but some syntenic regions are evident between the two methanotrophs M. acidiphila and M. silvestris, suggesting a greater similarity of these compared with B. indica.
Figure 1.
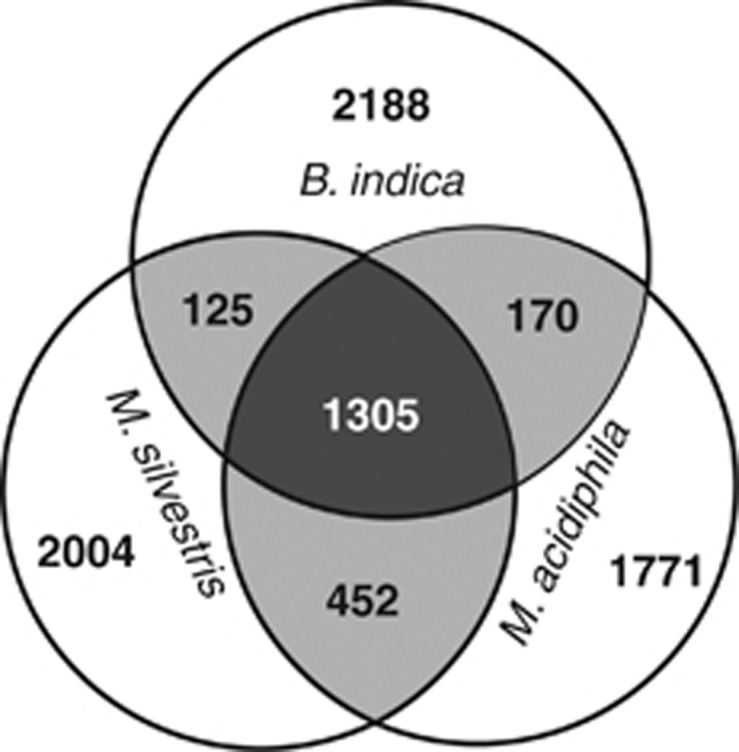
Venn diagram showing genomic overlap of the three study species, based on reciprocal BLAST using an amino-acid identity threshold of 60%. The two methanotrophic species share more genes in common than either does with B. indica, although each genome has a large proportion of unique genes. The diagram was drawn using BioVenn (Hulsen et al., 2008) and manually edited.
B. indica has the largest number of genes and M. acidiphila the smallest number in COG categories G: carbohydrate transport and metabolism; C: energy production and conversion; and K: transcription (Figure 2; Supplementary Table 4), reflecting the more versatile catabolic potential of B. indica and M. silvestris compared with the obligate methanotroph M. acidiphila.
Figure 2.

The number of genes belonging to six COG categories in the three genomes, along with the proportion of putative laterally transferred genes in each COG calculated by a BLAST procedure at the taxonomic level of Order (from IMG) or Family. An asterisk indicates that the predicted proportion of LGT is significantly greater in B. indica (right bars) or M. silvestris (left bars) compared with M. acidiphila (centre bars). The raw data and statistical comparisons are shown in Supplementary Table 4.
Phylogenetic constructions
A highly resolved phylogenetic tree constructed from 29 universal vertically inherited genes (Ciccarelli et al., 2006) demonstrated that M. acidiphila and M. silvestris share a common ancestor more recent than their link with B. indica (Figure 3). This is not evident from the phylogeny of the less informative 16S rRNA gene (Supplementary Figure 2), but agrees with the greater genome overlap of the two specialist methanotrophs (Figure 1; Supplementary Figure 1). The tree demonstrates with high support that the family Beijerinckiaceae is linked closely with the Methylocystaceae, and then the Methylobacteriaceae. The Methylocystaceae comprises specialist methanotrophs of the genera Methylosinus and Methylocystis. There are no methanotrophs known in Methylobacteriaceae, although it includes many methylotrophs, including Methylobacterium spp.
Figure 3.

Phylogenetic tree based on a concatenation 29 universally conserved genes, showing the relationships of different members of the Rhizobiales. The tree was constructed using neighbor-joining and a Kimura distance correction, based on a concatenation of core proteins of 6101 amino acids total length. The scale bar represents 0.1 change per amino-acid position. Bootstraps values of 1000 constructions are shown at the nodes when greater than 50%. Genome accession numbers are listed in parentheses.
Officially, the families Methylocystaceae and Beijerinckiaceae contain other genera not included in Figure 3 because public-domain genome sequences for them are not available. These are: Albibacter, Hansschlegelia, Methylopila, Pleomorphomonas and Terasakiella in the Methylocystaceae; and Camelimonas, Chelatococcus, Methylorosula and Methylovirgula in the Beijerinckiaceae (http://www.bacterio.cict.fr/). None of these are methanotrophs, and based on 16S rRNA gene phylogeny it is clear that most are phylogenetically misclassified at the family level (Supplementary Figure 2). Terasakiella does not cluster in the Rhizobiales, rather in the Rhodospirillales. Albibacter, Hansschlegelia, Methylopila and Pleomorphomonas form a monophyletic group distinct from Methylocystis and Methylosinus. Camelimonas and Chelatococcus are not monophyletic with other Beijerinckiaceae. The 16S rRNA gene tree supports the monophyly of most Beijerinckiaceae (Methylocapsa, Methylocella, Methyloferula, Methylorosula, Beijerinckia and Methylovirgula) with the methanotrophic Methylocystaceae (Methylocystis and Methylosinus) and with Rhodoblastus spp. (Supplementary Figure 2). The remainder of this study will ignore the misclassified members of the these families.
Tree topologies of concatenated derived PmoCAB proteins of pMMO (Figure 4), derived MmoX proteins of sMMO (Supplementary Figure 3), concatenated derived MxaFJI proteins of MDH (Supplementary Figure 4), and a concatenation of 21 other derived methylotrophy proteins (Figure 5) all matched well to the highly resolved evolutionary tree shown in Figure 3. This indicates that most key methylotrophic and methanotrophic functions were present in a single common ancestor of the Methylocystaceae and Beijerinckiaceae and vertically inherited through these two families. The relationship of Methylocystaceae, Methylobacteriaceae and Beijerinckiaceae in the MxaFIJ tree (Supplementary Figure 4) is somewhat inconsistent with the other trees, suggesting that these genes have a weak phylogenetic signal, or that there has been some LGT among members of these three families, as noted previously (Heyer et al., 2002; Vorobev et al., 2009). However, the mxa genes in the three families still arose from a common ancestor.
Figure 4.

Phylogenetic tree of concatenated derived PmoCAB proteins in methanotrophs. The tree was constructed using neighbor-joining and a Kimura distance correction, based on a concatenation of 891 amino acids total length. The scale bar represents 0.1 change per amino-acid position. Bootstraps values of 1000 constructions are shown at the nodes. Genome accession numbers are listed in parentheses. The pxmABC operon of Methylomicrobium album has been rearranged to match the pmoCAB order of other operons in the concatenation.
Figure 5.

Phylogenetic tree based on a concatenation of 21 methylotrophy genes (see Supplementary Table 3). The tree was constructed using neighbor-joining and a Kimura distance correction, based on a concatenation of 7186 amino acids total length. The scale bar represents 0.1 change per amino-acid position. Bootstraps values of 1000 constructions are shown at the nodes. Genome accession numbers are listed in parentheses.
As noted before (Dedysh et al., 2004) nifH and nifD have evidently been transferred from Methylocystaceae to other bacteria (Supplementary Figure 5). M. silvestris also appears to have nif genes derived from a Methylocystaceae bacterium. Its genome structure supports this phylogenetic conclusion, as genes for Nif cofactor biosynthesis are located adjacent to nitrogenase genes in M. silvestris, similarly to Methylosinus trichosporium, whereas they are elsewhere in the genomes of M. acidiphila and B. indica. However, despite these apparent LGT events, separate phyletic clusters of most Beijerinckiaceae and Methylocystaceae are joined via a common ancestor, suggesting that a primarily vertical inheritance pattern of nifH and nifD genes has been overlaid with some LGT events.
Lateral gene transfer
The IMG BLAST procedure identified a significantly higher ratio of LGT in B. indica than in the methanotrophs M. acidiphila or M. silvestris (Figure 2; Supplementary Table 4), particularly in COG categories for carbohydrate transport and metabolism (G), energy production and conversion (C) and transcription (K); and to a lesser extent in COG categories P, M and T. Over 20% of B. indica's genes in COG G were predicted to be obtained via LGT; this increased to nearly 60% when we relaxed the BLAST procedure to include related families (Figure 2, Supplementary Table 4). Other COGs in B. indica showed a lower rate of LGT (average around 9%) similar to the rate estimated in the methanotroph genomes (Supplementary Table 4).
The unique gene set of B. indica (Figure 1) was cross-referenced with the putative LGT gene set. Unique genes in B. indica that show evidence of LGT from non-Rhizobiales encode predicted levanase and levansucrases (Bind_2020-2021, Bind_1319), xylanases (Bind_2643; Bind_2759), various alcohol and aldehyde dehydrogenases (for example, Bind_2821-2831; Bind_2505-2508; Bind_3639; Bind_2961, Bind_1830, Bind_1835; Bind_1758-1760), sugar dehydrogenases and sugar kinases (Bind_2541; Bind_3085 and Bind_3045), hydrogenase (Bind_0503-0508) and various membrane transporters (see next section). These data are consistent with B. indica having evolved from a specialist methanotroph via the acquisition of catabolic genes. Based on the closest BLAST hits, many putative LGT genes in B. indica are related to genes from Rhodospirillales (particularly Acetobacter spp. and Gluconacetobacter spp.), Sphingomonadales and Burkholderiales (Supplementary Table 5).
The B. indica and M. acidiphila genomes include predicted plasmids (Table 1), demonstrating one possible mechanism for genomic rearrangements. The M. acidiphila plasmid contains primarily hypothetical genes and DNA modification systems (transposases, helicases, recombinases, polymerases and topoisomerases). There is a single gene from COG G (carbohydrate transport and metabolism). The B. indica plasmid NC_010580 contains DNA modification genes and hypothetical genes, but in addition several catabolic genes from COG G (Supplementary Table 6).
Unlike the BLAST method, the compositional IslandViewer methods did not predict a higher amount of LGT in B. indica, instead predicting similar amounts in each genome (Supplementary Table 7). However, significantly more LGT for genes conferring catabolic diversity (COG categories C and G) were still detected in B. indica (Supplementary Table 8). The M. acidiphila islands predicted by IslandViewer are comprised of typical mobility genes, encoding a type IV secretion pathway, conjugal transfer and DNA modification systems, but no genes from COG categories G or C (Supplementary Table 8). The M. silvestris and B. indica islands contain significantly more genes from these COGs (Supplementary Table 8). The largest island predicted in any genome is in B. indica (Bind_2573-2667) and includes two major facilitator transporters, a xylanase and several dehydrogenases (Supplementary Table 9).
The BLAST results, the plasmid-encoded genes, and the IslandViewer output therefore all suggested that there has been more LGT for catabolic genes in B. indica. The final LGT method used, Alien Hunter, has a large detection window that makes it inaccurate for predicting the origin of individual genes. We therefore used Alien Hunter primarily to support the other methods in identifying large potential islands of LGT in B. indica, which are listed in Supplementary Table 9. This Table does not represent all LGT events, nor do all genes identified represent foreign genes with certainty. However, many of the islands contain genes in COGs C and G and may represent ‘organotrophy islands' similar to the ‘methylotrophy islands‘ found in methylotrophs (Chistoserdova et al., 2003).
A surprising finding of the Alien Hunter analysis was that genes for pMMO in M. acidiphila and for sMMO in M. silvestris were predicted to be foreign (Table 2). These operons are large enough (>2500 bp) to allow accurate detection with this method. Remarkably, every single operon for MMO in the available Methylocystaceae genomes (three in M. trichosporium, three in Methylocystis sp. SC2 and two in Methylocystis sp. ATCC 42492) was also detected as foreign (Table 2). We therefore propose that Alien Hunter did not detect recent transfers of MMO specifically to the Beijerinckiaceae methanotrophs, but instead detected the original LGT events of sMMO and pMMO to the common ancestor of all Alphaproteobacteria methanotrophs. This scenario would explain both the foreign compositional bias of these operons, and their conserved phylogeny within the Alphaproteobacteria (Figure 4; Supplementary Figure 3). Additional detection of MDH genes as foreign in M. acidiphila was unique among the alphaproteobacterial methanotrophs, but agrees with the phylogeny (Supplementary Figure 4) in suggesting some LGT of these genes among Alphaproteobacteria (Heyer et al., 2002; Vorobev et al., 2009).
Table 2. Genes encoding methane monooxygenases in Alphaproteobacteria methanotrophs that were detected as foreign using Alien Hunter compositional analysis.
| Organisms | Enzyme | Genes | Coding loci detected as foreign |
|---|---|---|---|
| Methylocella silvestris | sMMO | mmoXYB | Msil1262_1264 |
| Methylocapsa acidiphila | pMMO | pmoCAB | MetacDRAFT_3746-3748 |
| Methylosinus trichosporium OB3b | sMMO | mmoXYB | MettrDRAFT_2362-2364 |
| Methylosinus trichosporium OB3b | pMMO | pmoCAB | MettrDRAFT_2808-2810 (copy 1) MettrDRAFT_0384-0382 (copy 2) |
| Methylocystis sp. ATCC 49242 | pMMO | pmoCAB | Met49242_2879-2877 (copy 1) Met49242_1455-1453 (copy 2) |
| Methylocystis sp. SC2 | pMMO | pmoCAB | BN69_2826-2828 (copy 1) BN69_3533-3535 (copy 2) |
| Methylocystis sp. SC2 | pMMO2 | pmoCAB2 | BN69_0202-0204 |
Abbreviations: pMMO, particulate methane monooxygenases; sMMO, soluble methane monooxygenase.
The Methylosinus and Methylocystis species possess two nearly identical copies of the pmoCAB1 operon. Methylocystis sp. SC2 also possesses a pmoCAB2 operon, which is divergent from the pmoCAB1 operon and has different kinetic properties (Yimga et al., 2003; Baani and Liesack, 2008). All were detected as compositionally foreign to their respective genomes
Transporters
The number of genes encoding membrane transport functions corresponds to metabolic diversity such that B. indica >M. silvestris >M. acidiphila (Table 3). Significantly more transporter genes are present in B. indica, and a significantly higher percentage of these were predicted to be foreign compared with M. acidiphila (Table 3), particularly genes encoding major facilitator superfamily transporters (uniporter, symporters and antiporters). B. indica has nearly three times more of these transporter-encoding genes than M. acidiphila, and while only four of those identified in M. acidiphila showed signs of LGT, as many as 43 of those in B. indica did. Some transporter-encoding genes have apparently been duplicated in the B. indica genome after LGT acquisition. Three nearly identical copies (>99.3% amino-acid identity) of a carbohydrate-selective porin OprB are present (Bind_2860, Bind_2838 and Bind_3070). The periplasmic-binding protein-encoding genes Bind_3071 and Bind_2837 are 99.0% identical, and also share close identity to three other genes (Bind_2861, Bind_3811 and Bind_2340) that may be genome-specific paralogs (Supplementary Figure 6). To test the hypotheses that these periplasmic-binding proteins may have undergone accelerated evolution and specialization to different substrates after duplication, we performed a nucleotide substitution rate analysis (Ka/Ks) using the Bergen Center for Computational Science online tool (http://services.cbu.uib.no/tools/kaks); (Liberles, 2001). However, no indication of elevated sequence evolution was found.
Table 3. Genome contents of several protein families from the Pfam database involved in membrane transport, along with the number of these predicted to have been acquired via LGT according to BLAST at the level of Order, BLAST at the level of Family, or at least one compositional method (AlienHunter, IslandPath-DIMOB and SIGI-HMM).
| Organism | MFS-1 major facilitator transporter superfamily: PF00083, PF07690, PF05977 | Periplasmic-binding proteins: PF00532, PF00497, PF01094, PF01547 | Carbohydrate-selective porins: PF04966 | Major intrinsic proteins: PF00230 | Total |
|---|---|---|---|---|---|
| Methylocella silvestris | |||||
| Total genes | 25 | 17 | 1 | 1 | 44 |
| LGT Order level BLAST | 2 | 0 | 0 | 0 | 2 |
| LGT Family level BLAST | 14 | 2 | 0 | 1 | 17 |
| LGT Compositional | 4 | 3 | 0 | 0 | 7 |
| Methylocapsa acidiphila | |||||
| Total genes | 14 | 10 | 1 | 1 | 26 |
| LGT Order level BLAST | 0 | 0 | 0 | 0 | 0 |
| LGT Family level BLAST | 4 | 0 | 0 | 0 | 4 |
| LGT Compositional | 2 | 2 | 0 | 0 | 4 |
| Beijerinckia indica | |||||
| Total genes | 43 | 24 | 5 | 3 | 75* |
| LGT Order level BLAST | 18 | 3 | 1 | 2 | 24* |
| LGT Family level BLAST | 26 | 10 | 5 | 2 | 43* |
| LGT Compositional | 9 | 1 | 0 | 0 | 10 |
Abbreviation: LGT, lateral gene transfer.
An asterisk in the ‘Total' column indicates that the number of transporters, or the proportion of predicted LGT, is significantly higher in B. indica than in M. acidiphila, calculated with a Z-ratio test of proportions and a Bonferroni correction for three comparisons (P=0.05). There were no significant differences between M. silvestris and M. acidiphila.
Although not detected as foreign by the LGT methods, several complete operons encoding ABC-type transporters were also found in B. indica but not in the two methanotrophs. These included all elements of a monosaccharide transporter (Bind_2861-2863, together with carbohydrate-selective porin Bind_2860), an amino-acid transporter system (Bind_2455-2457), a multiple sugar transporter (Bind_3071-3074, together with carbohydrate-selective porin Bind_3070) and mannitol–sorbitol transporters in both the genome (Bind_2460-2463) and one plasmid (Bind_3793-3796). The mannitol–sorbitol transporters are both adjacent to mannitol dehydrogenase genes (Bind_2459 and Bind_3797) and share 88–94% identity.
Metabolic pathways
We searched each genome against a database of methylotrophy genes. M. acidiphila and M. silvestris each possess an MMO, MDH, a complete serine cycle and complete tetrahydromethanopterin (H4MPT) and tetrahydrofolate (H4F)-linked C1 transfer pathways, but lack the Ethylmalonyl CoA pathway (Supplementary Table 3). As expected from studies of other methylotrophs (Vuilleumier et al., 2009), M. acidiphila and M. silvestris contain large methylotrophy islands (Supplementary Figure 7) for genes involved in H4MPT -linked C1 transfer (Msil_2385-2403 Metac_3701-3721), the serine cycle (Msil_1712-1719, Metac_3401-3408) and MDH (Msil_0471-0482, Metac_0409-0420). Methylotrophy genes are largely absent from B. indica, or where homologs are present they are scattered about the genome (Supplementary Figure 7). The arrangement of the serine cycle islands is nearly identical in the two methanotrophs M. acidiphila and M. silvestris, and these are also highly syntenous with the arrangements in Methylosinus trichosporium and Methylocystis sp. SC2, and more distantly with Methylobacterium extorquens (Supplementary Figure 8). Other Alphaproteobacteria methylotrophs have very different arrangements (for example, Hyphomicrobium sp. MC1; Vuilleumier et al., 2011), or lack this island entirely. The conservation of arrangement mirrors the phylogenetic results, and supports the existense of a single methylotrophic ancestor of the Beijerinckiaceae, Methylocystaceae and Methylobacteriaceae. One difference of the serine cycle island in M. acidiphila and M. silvestris compared with the other methylotrophs shown in Supplementary Figure 8 is the lack of mtdA (methylene-H4MPT/methylene-H4F dehydrogenase) and fch (methenyl-H4F cyclohydrolase), which are involved in conversion of formate to methylene-H4F (Chistoserdova et al., 2003). M. acidiphila does possess these enzymes elsewhere in the genome (Metac_0269-0270); M. silvestris and B. indica do not, but instead possess bifunctional 5,10-methylene-H4F dehydrogenase/methenyl-H4F cyclohydrolases (Msil_1881; Bind_0805).
M. acidiphila contains a single pmoCAB operon (Metac_3743-3748) for pMMO, along with three orphan pmoC genes (Metac_2162, 2258 and 2438). M. silvestris contains an sMMO operon (Msil_1262-1272). M. silvestris and B. indica also possess soluble propane monooxygenases in the same soluble di-iron monooxygenase family as sMMO (Msil_1648-1651; Bind _3394-3397). Although sMMO and pMMO showed compositional evidence of lateral transfer (Table 2), the soluble di-iron propane MOs did not.
A complete TCA cycle is encoded in all three genomes, including genes for the three subunits of 2-oxoglutarate dehydrogenase (Bind_ 3607-3609, Msil_2504-2506 and Metac_3192-3194). Acetate is the most common multicarbon substrate used by facultative methanotrophs. In addition to a complete TCA cycle, metabolism of acetate requires a mechanism to convert acetate to acetyl-CoA, such as acetate kinase plus phosphotransacetylase, or an acetate-CoA ligase. These genes are present (often in multiple homologs) in all three genomes. However, several of the putative acetate kinase-phosphotransacetylase pairs (Msil_2705-2706, Msil_2977-2978, Metac_1244-1245, Bind_0199-0200 and Bind 2453-2454) are linked to poly-3-hydroxyalkanoate synthetase, and may function in carbon storage rather than catabolism. Isocitrate lyase and malate synthase, which are needed for the glyoxylate bypass when growing on 2-C compounds, are present in all three genomes. Interestingly, isocitrate lyase is absent in Methylobacteriaceae (and Methylocystaceae), which instead use the Ethylmalonyl CoA pathway for assimilation of C2 units and for regeneration of glyoxylate in the serine cycle (Erb et al., 2007; Peyraud et al., 2009; Matsen et al., 2013). Genes encoding several key enzymes of the Ethylmalonyl CoA pathway are missing in all the Beijerinckiaceae genomes (Supplementary Table 3).
M. silvestris possesses genes for a respiratory nitrate reductase narGHJI, nitrite reductase nirK, and nitric oxide reductase norB, together with accessory genes and transporters in a single genomic island (Msil_1507-1521). These are absent from M. acidiphila and B. indica. Methanotrophic bacteria often posses nirK and norB genes, possibly for the removal of toxic byproducts of ammonia cooxidation (Stein and Klotz, 2011). The nirK in M. silvestris has apparently been obtained via LGT, as the top BLAST hits are Betaproteobacteria, Deltaproteobacteria, Spirochetes, Gammaproteobacteria and Bacteroidetes, with the first hit to an Alphaproteobacteria having <60% of the bitscore of the top hit. None of the three genomes contain hydroxylamine oxidoreductase (haoA). All three genomes contained complete operons for nitrogenase (Msil_3613-3632, Bind_0473-0490 and Metac_2352-2370).
Discussion
Aerobic methanotrophy has evolved rarely in the tree of life, perhaps as few as four times given that only four phyletic groups of methanotrophs have been discovered. The Beijerinckiaceae presents an intriguing opportunity to address methanotroph specialization and evolution, as bacteria with diverse metabolic potentials are found within this family, and a switch between specialist methanotrophy and generalist organotrophy has occurred in recent evolutionary time (Dedysh and Dunfield, 2010). In this study, we attempted to determine whether the ancestral bacterium in the Beijerinckiaceae was a generalist like B. indica or a specialist methanotroph like M. acidiphila or M. silvestris, and what key genomic properties account for the widely different catabolic potentials in this group. Our findings suggest that the ancestor was a methanotroph, as organotrophy genes in B. indica show more evidence of recent LGT than do methylotrophy genes in M. silvestris and M. acidiphila. This conclusion was supported by BLAST-based and compositional analysis of LGT, by examination of genes in plasmids and genomic islands, and by detailed phylogenetic analyses. We propose the following evolutionary scenario as the most parsimonious.
(1) Methanotrophy arose only once in the Alphaproteobacteria. Phylogenetic analyses clearly place both families of Alphaproteobacteria methanotrophs together: the Beijerinckaceae and the Methylocystaceae. Key methylotrophy modules for methane, methanol and formaldehyde oxidation, as well as C1 carbon fixation, have been vertically inherited through these families, indicating that their common ancestor had methylotrophic/methanotrophic ability (Figures 4 and 5, Supplementary Figures 3–5). Methylotrophy is an obvious prerequisite for methanotrophy, as the key evolutionary constraint for the evolution or acquisition of MMOs would be dealing with the downstream products methanol and formaldehyde. Mechanisms are necessary to limit the accumulation of these toxic intermediates while channeling their electrons into energy conservation or carbon fixation. The fundamental importance of these processes in methylotrophs is attested to by the redundancy in the pathways used to handle them, especially formaldehyde (Chistoserdova et al., 2009; Vuillemier et al., 2009). The close phylogenetic relationship of Beijerinckiaceae and Methylocystaceae to the largely methylotrophic Methylobacteriaceae family in our gene trees supports the idea of a methylotrophic ancestor. The defining event would be when an ancestral methylotroph acquired pMMO and sMMO from a previously evolved methanotroph, likely a member of the Gammaproteobacteria (Figure 4; Supplementary Figure 3). The foreign nature of the sMMO- and pMMO-encoding genes in the Alphaproteobacteria is still detectable via compositional analysis (Table 2). Although the transfer of MMO to a methylotroph seems trivial, it has evidently occurred rarely, perhaps only once in each of the Verrucomicrobia, NC10, Alphaproteobacteria and Gammaproteobacteria (Stein et al., 2012). The transfer of methanotrophic ability may therefore require more than MMO.
(2) This common ancestor was capable of dinitrogen fixation. Klotz et al. (2008) and Klotz and Stein (2008) suggest that the ability to rapidly oxidize toxic byproducts of ammonia oxidation while conserving electrons was a necessary prerequisite for the evolution of ammonia monooxygenase (AMO). Similarly, the primary prerequisite for a bacterium to evolve or acquire MMO would be mechanisms to deal with methanol and formaldehyde, as discussed above. However, given the promiscuity of ammonia monooxygenase and MMO to each others' substrates, dealing with nitrosative stress might also be important in methanotroph evolution, and dealing with formaldehyde important in nitrifier evolution. Hence, nitrifiers can have pathways to deal with formaldehyde (Klotz et al., 2006) and methanotrophs can have pathways to deal with nitrosative stress (Stein and Klotz, 2011). However, our genomes suggest that dealing with nitrosative stress may have been a weak selective pressure for the secondary acquisition of MMOs by Alphaproteobacteria. The most toxic byproducts of ammonia oxidation are hydroxylamine (NH2OH), nitric oxide (NO) and nitrite (NO2−). Neither M. silvestris nor M. acidiphila have a hydroxylamine oxidoreductase that many other methanotrophs use to detoxify hydroxylamine (Stein and Klotz, 2011), and only M. silvestris has an apparent capacity to reduce NO and NO2−. According to our analyses, these genes in M. silvestris have been recently acquired via LGT, not inherited from the methanotrophic commmon ancestor. However, all three genomes had gene sets for dinitrogen fixation, as do methanotrophs in the Methylocystaceae. These genes are phylogenetically well-conserved (Supplementary Figure 5), so the common ancestor was capable of nitrogen fixation, and adapted to low-nitrogen habitats. Species of Methylocapsa and Methylocella are abundant in oligotrophic peatlands, one of the most nitrogen-limited habitats on Earth (Dedysh, 2011), where they should not require nitrogen detoxification mechanisms. Therefore, systems to handle nitrosative stress were not necessarily a prerequisite for the original alphaproteobacterial methanotroph to obtain MMO, and species have later acquired these functions when dictated by their environment.
(3) The original methanotroph in the Alphaproteobacteria diverged into two families, the Methylocystaceae and Beijerinckiaceae. A duplication of pmoCAB occurred early in the evolution of Methylocystaceae and two enzymes with different kinetic properties evolved (Yimga et al., 2003; Baani and Liesack, 2008). The Beijerinckiaceae includes a group of methanotrophs that are suspected to grow on low, atmospheric levels of methane (upland soil cluster alpha-Figure 4) (Knief et al., 2003; Dunfield, 2007). Evidence suggests that this group may also contain facultative methanotrophs (Dunfield, 2007; Pratscher et al., 2011).
(4) Recidivism occurred in certain lineages of Beijerinckiaceae. B. indica did not stay on the straight and narrow path of methanotrophy, but instead reverted to a generalist lifestyle. M. silvestris followed a similar, but less complete recidivist path. pMMO and/or sMMO genes were lost by these species. The genes acquired to facilitate the recidivism encode primarily transporters and porins (in COG categories G and M), enzymes to catabolise an increased diversity of substrates (COG categories G and C), and regulatory proteins (COG K) to manage this increased metabolic potential. The methods used to detect LGT in this study were all conservative and probably failed to detect most of the foreign genetic material, but they still showed a clear trend toward more LGT of genes conferring catabolic diversity in B. indica >M. silvestris >M. acidiphila (Figure 2). This supports the theory that obligate methanotrophy was the ancestral state of this family, and generalist catabolic functions were acquired later. In support of this scenario, it has been observed that methylotrophy can be lost from Methylobacterium strains during evolution in the laboratory (Lee et al., 2009), and possibly in the field (Ardley et al., 2009). Methylobacterium extorquens displayed rapid, parallel loss of growth on methanol, methylamine and formate during adaptation of replicate populations to succinate in the laboratory (Lee et al., 2009). Although M. extorquens is not so extreme a specialist as an obligate methanotroph, this artificial evolution experiment illustrates the plasticity of these bacteria and their ability to move away from methylotrophy under selection pressure for alternative substrates. A similar, if more complex, process has probably occurred in B. indica.
An alternative explanation for the evolution of methanotrophy in the Alphaproteobacteria is that it occurred twice, once in the Methylocystaceae and once in the Methylocella–Methylocapsa branch of the Beijerinckiaceae. This would require one of two scenarios: (i) that MMO transfer occurred to a common ancestor that retained the capacity to metabolize many multicarbon substrates until specialization later occurred independently in the two families. This seems unlikely given that the combination of methanotrophy and diverse organotrophy, as far as we know, does not exist and may be incompatible (Dedysh et al., 2005; Dedysh and Dunfield, 2010; Semrau et al., 2011). Alternatively: (ii) there was one transfer of MMOs to an ancestor of the Methylocystaceae and then a later transfer from a Methylocystaceae to Methylocella/Methylocapsa. This scenario does not fit well to the phylogenies of pmoCAB, mmoX and other methylotrophy genes, which closely mirror evolutionary trees of conserved core genes. 16S rRNA gene divergence is in fact linearly correlated to mmoX divergence when comparing members of the Methylocystaceae and Beijerinckiaceae (Heyer et al., 2002), demonstrating a clock-like evolution of mmoX genes from a common ancestor.
For unknown reasons methanotrophy entails catabolic specialization to very few substrates (Wood et al., 2004). Until recently it was believed that all methanotrophs were obligate. Now several facultatively methanotrophic species are known (Dedysh et al., 2005; Dunfield et al., 2010; Semrau et al., 2011), and facultative metabolism may be important for the survival of methanotrophic bacteria in some environments (Rahman et al., 2011; Wieczorek et al., 2011). However, even facultative methanotrophs are highly specialized to a few low-molecular-weight growth substrates. Specialization is generally considered a one-way street, particularly in the extreme cases of symbionts and pathogens (Anderson and Kurland, 1998; Moran et al., 2008). However, based on the large genome sizes of some methanotrophs, methanotrophic specialization is not accompanied by drastic genome reduction. Lesions in fundamental metabolic cycles are sometimes evident (Wood et al., 2004), but these are neither excessive nor universal, so the path back to a more generalist lifestyle should not be as difficult as from an obligate symbiont/parasite. Nevertheless, the Methylococcaceae or Methylocystaceae do not contain any known generalist organotrophs, nor any facultative methanotrophs with as diverse a substrate range as Methylocella spp., so recidivism of specialist methanotrophs is not common. This evolutionary path is at present only evident in the Beijerinckiaceae.
It has been speculated that a major cause of the substrate-limited lifestyles of specialist bacteria like methanotrophs and nitrifiers is a paucity of membrane transporters (Chain et al., 2003; Ward et al., 2004; Wood et al., 2004). Our analyses strongly support this speculation (Table 3). B. indica encodes many more transporter elements than M. acidiphila and M. silvestris do, particularly major facilitator proteins and periplasmic-binding proteins. A high proportion of these show evidence of having been acquired via LGT, many from distant taxa such as Rhodospirillales, Sphingomonadales, Myxococcales and Burkholderiales (Supplementary Table 5). Some have been duplicated in the genome after acquisition and are present in separate operons. Other hypotheses about the key defining characteristics of specialist methanotrophs were not borne out by our analyses. There was no obvious lesion in the TCA cycle of the obligate methanotroph M. acidiphila, and it even possessed genes that might theoretically confer the ability to grow on acetate, although it has repeatedly been shown incapable of this in culture (Dedysh et al., 2002; Dedysh et al., 2005; Dunfield et al., 2010).
Our studies have helped to elucidate the metabolic basis of specialization in methanotrophs, as well as the specific evolutionary history of methanotrophy in the Alphaproteobacteria. We propose that methanotrophy arose only once in the Alphaproteobacteria, but that some lineages have reversed their specialization to methane and returned to a catabolically more diverse lifestyle. This recidivism appears to be particularly facilitated by the acquisition of genes encoding membrane transporters. With the increasing ease of obtaining full genome sequences and the large number of specialist methanotrophs that are presently being sequenced by the Joint Genome Institute, we should soon obtain further insights into their functional and evolutionary diversity.
Acknowledgments
Sequencing was provided by the Joint Genome Institute of the US Department of Energy via the Community Sequencing Program. The work conducted by the Joint Genome Institute is supported by the Office of Science of the US DoE Under Contract No. DE-AC02-05CH11231. Research into the genomes was supported by Grants from Alberta Innovates-Technology Futures, and the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant Program. AVS was supported by a Grant from the Canada School of Energy and Environment. IT and ZH were supported in part by the Genome Canada/Genome Alberta-sponsored Hydrocarbon Metagenomics Project. We thank Svetlana Dedysh, Stéphane Vuilleumier and four constructive external reviewers for their comments on the manuscript, and acknowledge the support of the OMeGA group.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): Simple prokaryotic genome comparisons. BMC Genomics. 2011;8:12–402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. Reductive evolution of resident genomes. Trends Microbiol. 1998;6:263–268. doi: 10.1016/s0966-842x(98)01312-2. [DOI] [PubMed] [Google Scholar]
- Anthony C. The biochemistry of methylotrophs. Academic Press: London, UK; 1982. [Google Scholar]
- Ardley JK, O'Hara GW, Reeve WG, Yates RJ, Dilworth MJ, Tiwari RP, et al. Root nodule bacteria isolated from South African Lotononis bainesii, L. listii and L. solitudinis are species of Methylobacterium that are unable to utilize methanol. Arch Microbiol. 2009;191:311–318. doi: 10.1007/s00203-009-0456-0. [DOI] [PubMed] [Google Scholar]
- Baani M, Liesack W. Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc Natl Acad Sci USA. 2008;105:10203–10208. doi: 10.1073/pnas.0702643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belova SE, Baani M, Suzina NE, Bodelier PLE, Liesack W, Dedysh SN. Acetate utilization as a survival strategy of peat-inhabiting Methylocystis spp. Environ Microbiol Rep. 2010;3:36–46. doi: 10.1111/j.1758-2229.2010.00180.x. [DOI] [PubMed] [Google Scholar]
- Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- Berestovskaya JJ, Kotsyurbenko OR, Tourova TP, Kolganova TV, Doronina NV, Golyshin PN, et al. Methylorosula polaris gen. nov., sp. nov., an aerobic, facultatively methylotrophic psychrotolerant bacterium from tundra wetland soil. Int J Syst Evol Microbiol. 2012;62:638–646. doi: 10.1099/ijs.0.007005-0. [DOI] [PubMed] [Google Scholar]
- Chain P, Lamerdin J, Larimer F, Regala W, Lao V, Land M, et al. Complete genome sequence of the ammonia-oxidizing bacterium and obligate chemolithoautotroph Nitrosomonas europaea. J Bacteriol. 2003;185:2759–2773. doi: 10.1128/JB.185.9.2759-2773.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Crombie A, Tanvir Rahman Md, Dedysh SN, Liesack W, Stott MB, et al. Complete genome sequence of the aerobic facultative methanotroph Methylocella silvestris BL2. J Bacteriol. 2010;192:3840–3841. doi: 10.1128/JB.00506-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistoserdova L, Chen SW, Lapidus A, Lidstrom ME. Methylotrophy in Methylobacterium extorquens AM1 from a genomic point of view. J Bacteriol. 2003;185:2980–2987. doi: 10.1128/JB.185.10.2980-2987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME. The expanding world of methylotrophic metabolism. Annu Rev Microbiol. 2009;63:477–499. doi: 10.1146/annurev.micro.091208.073600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Dam B, Dam S, Kube M, Reinhardt R, Liesack W. Complete genome sequence of Methylocystis sp. strain SC2, an aerobic methanotroph with high-affinity methane oxidation potential. J Bacteriol. 2012;194:6008–6009. doi: 10.1128/JB.01446-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedysh SN, Liesack W, Khmelenina VN, Suzina NE, Trotsenko YA, Semrau JD, et al. Methylocella palustris gen. nov., sp. nov., a new methane-oxidizing acidophilic bacterium from peat bogs representing a novel sub-type of serine pathway methanotrophs. Int J Syst Evol Microbiol. 2000;50:955–969. doi: 10.1099/00207713-50-3-955. [DOI] [PubMed] [Google Scholar]
- Dedysh SN, Khmelenina VN, Suzina NE, Trotsenko YA, Semrau JD, Liesack W, et al. Methylocapsa acidiphila gen. nov., sp. nov., a novel methane-oxidizing and dinitrogen-fixing acidophilic bacterium from Sphagnum bog. Int J Syst Evol Microbiol. 2002;52:251–261. doi: 10.1099/00207713-52-1-251. [DOI] [PubMed] [Google Scholar]
- Dedysh SN, Ricke P, Liesack W. NifH and nifD phylogenies: an evolutionary basis for understanding nitrogen fixation capabilities of methanotrophic bacteria. Microbiology. 2004;150:1301–1313. doi: 10.1099/mic.0.26585-0. [DOI] [PubMed] [Google Scholar]
- Dedysh SN, Knief C, Dunfield PF. Methylocella species are facultatively methanotrophic. J Bacteriol. 2005;187:4665–4670. doi: 10.1128/JB.187.13.4665-4670.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedysh SN, Smirnova KV, Khmelenina VN, Suzina NE, Liesack W, Trotsenko YA. Methylotrophic autotrophy in Beijerinckia mobilis. J Bacteriol. 2005;187:3884–3888. doi: 10.1128/JB.187.11.3884-3888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedysh SN, Dunfield PF.2010Facultative methanotrophsIn: Timmis KN, (ed)Handbook of Hydrocarbon and Lipid Microbiology Springer: Berlin, Germany; 1967–1976. [Google Scholar]
- Dedysh SN. Cultivating uncultured bacteria from northern wetlands: knowledge gained and remaining gaps. Front Microbiol. 2011;2:184. doi: 10.3389/fmicb.2011.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunfield PF, Khmelenina VN, Suzina NE, Trotsenko YA, Dedysh SN. Methylocella silvestris sp. nov., a novel methane-oxidizing bacterium isolated from an acidic forest cambisol. Int J Syst Evol Microbiol. 2003;53:1231–1239. doi: 10.1099/ijs.0.02481-0. [DOI] [PubMed] [Google Scholar]
- Dunfield PF.2007The soil methane sinkIn: Reay DS, Hewitt CN, Grace J, Smith KA, (eds)Greenhouse Gas Links CAB International: Wallingford, UK [Google Scholar]
- Dunfield PF, Belova SE, Vorob'ev AV, Cornish SL, Dedysh SN. Methylocapsa aurea sp. nov., a facultatively methanotrophic bacterium possessing a particulate methane monooxygenase. Int J Syst Evol Microbiol. 2010;60:2659–2664. doi: 10.1099/ijs.0.020149-0. [DOI] [PubMed] [Google Scholar]
- Eccleston M, Kelly DP. Assimilation and toxicity of some exogenous C-1 compounds, alcohols, sugars and acetate in the methane oxidizing bacterium Methylococcus capsulatus. J Gen Microbiol. 1973;75:211–221. doi: 10.1099/00221287-75-1-211. [DOI] [PubMed] [Google Scholar]
- Erb TJ, Berg IA, Brecht V, Müller M, Fuchs G, Alber BE. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc Natl Acad Sci USA. 2007;104:10631–10636. doi: 10.1073/pnas.0702791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MMM, et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature. 2010;464:543–548. doi: 10.1038/nature08883. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Han C, Chain P.2006Finishing repeat regions automatically with Dupfinisherin Arabnia HR, Valafar H, (eds)Proceedings of the 2006 international conference on bioinformatics & computational biology CSREA Press: Las Vegas, USA; 141–146. [Google Scholar]
- Heyer J, Galchenko VF, Dunfield PF. Molecular phylogeny of type II methane-oxidizing bacteria isolated from various environments. Microbiology. 2002;148:2831–2846. doi: 10.1099/00221287-148-9-2831. [DOI] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im J, Semrau JD. Pollutant degradation by a Methylocystis strain SB2 grown on ethanol: bioremediation via facultative methanotrophy. FEMS Microbiol Lett. 2011;318:137–142. doi: 10.1111/j.1574-6968.2011.02249.x. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: The complexity hypothesis. Proc Natl Acad Sci USA. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, Anthony C, Murrell JC. Insights into the obligate methanotroph Methylococcus capsulatus. Trends Microbiol. 2005;13:195–198. doi: 10.1016/j.tim.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Kennedy C.2005Genus I Beijerinckiain Brenner DJ, Krieg NR, Staley JR, (eds.)Bergeys Manual Systematic Bacteriology2nd edn,Vol. 2, The Proteobacteria Part C The Alpha-, Beta-, Delta- and Epsilonproteobacteria pp423–432.
- Klotz MG, Arp DJ, Chain PS, El-Sheikh AF, Hauser LJ, Hommes NG, et al. Complete genome sequence of the marine, chemolithoautotrophic, ammonia-oxidizing bacterium Nitrosococcus oceani ATCC 19707. Appl Environ Microbiol. 2006;72:6299–6315. doi: 10.1128/AEM.00463-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz MG, Schmid MC, Strous M, ob den Camp HJM, Jetten MSM, Hooper AB. Evolution of an octahaem cytochrome c protein family that is key to aerobic and anaerobic ammonia oxidation by bacteria. Environ Microbiol. 2008;10:3150–3163. doi: 10.1111/j.1462-2920.2008.01733.x. [DOI] [PubMed] [Google Scholar]
- Klotz MG, Stein LY. Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol Lett. 2008;278:146–156. doi: 10.1111/j.1574-6968.2007.00970.x. [DOI] [PubMed] [Google Scholar]
- Knief C, Lipski A, Dunfield PF. Diversity and activity of methanotrophic bacteria in different upland soils. Appl Environ Microbiol. 2003;69:6703–6714. doi: 10.1128/AEM.69.11.6703-6714.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb S, Knief C, Stubner S, Conrad R. Quantitative detection of methanotrophs in soil by novel pmoA-targeted real-time PCR assays. Appl Environ Microbiol. 2003;69:2423–2429. doi: 10.1128/AEM.69.5.2423-2429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Brinkman FS. Island viewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25:664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Chou HH, Marx CJ. Asymmetric, bimodal trade-offs during adaptation of Methylobacterium to distinct growth substrates. Evolution. 2009;63:2816–2830. doi: 10.1111/j.1558-5646.2009.00757.x. [DOI] [PubMed] [Google Scholar]
- Liberles DA. Evaluation of methods for determination of a reconstructed history of gene sequence evolution. Mol Biol Evol. 2001;18:2040–2047. doi: 10.1093/oxfordjournals.molbev.a003745. [DOI] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucl Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:326–327. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: The Integrated Microbial Genomes database and comparative analysis system. Nucl Acids Res. 2012;40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx CJ, Bringel FO, Chistoserdova L, Moulin L, Farhan Ul Haque M, Fleischman DE, et al. Complete genome sequences of six strains of the genus methylobacterium. J Bacteriol. 2012;194:4746–4748. doi: 10.1128/JB.01009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen JB, Yang S, Stein LY, Beck DAC, Kalyuzhanaya MG. Global molecular analyses of methane metabolism in methanotrophic alphaproteobacterium, Methylosinus trichosporium OB3b. Part I: Transcriptomic study. Front Microbiol. 2013;4:40. doi: 10.3389/fmicb.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Op den en Camp HJM, Islam T, Stott MB, Harhangi HR, Hynes A, Schouten S, et al. Minireview: Environmental, genomic, and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ Microbiol Rep. 2009;1:293–306. doi: 10.1111/j.1758-2229.2009.00022.x. [DOI] [PubMed] [Google Scholar]
- Peyraud R, Kiefer P, Christen P, Massou S, Portais JC, Vorholt JA. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc Natl Acad Sci USA. 2009;106:4846–4851. doi: 10.1073/pnas.0810932106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratscher J, Dumont MG, Conrad R. Assimilation of acetate by the putative atmospheric methane oxidizers belonging to the USCalpha clade. Environ Microbiol. 2011;13:2692–2701. doi: 10.1111/j.1462-2920.2011.02537.x. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MT, Crombie A, Chen Y, Stralis-Pavese N, Bodrossy L, Meir P, et al. Environmental distribution and abundance of the facultative methanotroph Methylocella. ISME J. 2011;5:1061–1066. doi: 10.1038/ismej.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semrau JD, DiSpirito AA, Vuilleumier S. Facultative methanotrophy: False leads, true results, and suggestions for future research. FEMS Microbiol Lett. 2011;323:1–12. doi: 10.1111/j.1574-6968.2011.02315.x. [DOI] [PubMed] [Google Scholar]
- Shishkina VN, Trotsenko YA. Multiple enzymic lesions in obligate methanotrophic bacteria. FEMS Microbiol Lett. 1982;13:237–242. [Google Scholar]
- Starkey RL, De PK. A new species of Azotobacter. Soil Sci. 1939;47:329–343. [Google Scholar]
- Stein LY, Yoon S, Semrau JD, DiSpirito AA, Crombie A, Murrell JC, et al. Genome sequence of the obligate methanotroph Methylosinus trichosporium strain OB3b. J Bacteriol. 2010;192:6497–6498. doi: 10.1128/JB.01144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LY, Bringel FO, DiSpirito AA, Han S, Jetten MSM, Kalyuzhnaya MG, et al. Genome sequence of the methanotrophic alphaproteobacterium Methylocystis sp. Strain Rockwell (ATCC 49242) J Bacteriol. 2011;193:2668–2669. doi: 10.1128/JB.00278-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LY, Roy R, Dunfield PF.2012Aerobic methanotrophy and nitrification: processes and connectionsin Battista J, et al (eds)Encyclopedia of Life Sciences John Wiley and Sons: Chichester. , www.els.net . [Google Scholar]
- Stein LY, Klotz MG. Nitrifying and denitrifying pathways of methanotrophic bacteria. Biochem Soc Trans. 2011;39:1826–1831. doi: 10.1042/BST20110712. [DOI] [PubMed] [Google Scholar]
- Tamas I, Dedysh SN, Liesack W, Stott MB, Alam M, Murrell JC, et al. Complete genome sequence of Beijerinckia indica subsp. indica. J Bacteriol. 2010;192:4532–4533. doi: 10.1128/JB.00656-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavormina PL, Orphan VJ, Kalyuzhnaya MG, Jetten MSM, Klotz MG. A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ Microbiol Rep. 2011;3:91–100. doi: 10.1111/j.1758-2229.2010.00192.x. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;11:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernikos GS, Parkhill J. Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics. 2006;22:2196–2203. doi: 10.1093/bioinformatics/btl369. [DOI] [PubMed] [Google Scholar]
- Vuilleumier S, Chistoserdova L, Lee MC, Bringel FO, Lajus A, Zhou Y, et al. Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS One. 2009;4:e5584. doi: 10.1371/journal.pone.0005584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuilleumier S, Nadalig T, Ul Haque MF, Magdelenat G, Lajus A, Roselli S, et al. Complete genome sequence of the chloromethane-degrading Hyphomicrobium sp. Strain MC1. J Bacteriol. 2011;193:5035–5036. doi: 10.1128/JB.05627-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorob'ev AV, Baani M, Doronina NV, Brady AL, Liesack W, Dunfield PF, et al. Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int J Syst Evol Microbiol. 2011;61:2456–2463. doi: 10.1099/ijs.0.028118-0. [DOI] [PubMed] [Google Scholar]
- Vorob'ev AV, de Boer W, Folman LB, Bodelier PLE, Doronina NV, Suzina NE, et al. Methylovirgula ligni gen. nov., sp. nov., an obligately acidophilic, facultatively methylotrophic bacterium with a highly divergent mxaf gene. Int J Syst Evol Microbiol. 2009;59:2538–2545. doi: 10.1099/ijs.0.010074-0. [DOI] [PubMed] [Google Scholar]
- Ward N, Larsen O, Sakwa J, Bruseth L, Khouri H, Durkin AS, et al. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath) PLoS Biol. 2004;2:E303. doi: 10.1371/journal.pbio.0020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek AS, Drake HL, Kolb S. Organic acids and ethanol inhibit the oxidation of methane by mire methanotrophs. FEMS Microbiol Ecol. 2011;77:28–39. doi: 10.1111/j.1574-6941.2011.01080.x. [DOI] [PubMed] [Google Scholar]
- Wood AP, Aurikko JP, Kelly DP. A challenge for 21st century molecular biology and biochemistry: what are the causes of obligate autotrophy and methanotrophy. FEMS Microbiol Rev. 2004;28:335–352. doi: 10.1016/j.femsre.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Yimga MT, Dunfield PF, Ricke P, Heyer J, Liesack W. Wide distribution of a novel pmoA-like gene copy among type II methanotrophs and its expression in Methylocystis strain SC2. Appl Environ Microbiol. 2003;69:5593–5602. doi: 10.1128/AEM.69.9.5593-5602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, van Dijk G, Fritz C, AJP Smolders, Pol A, Jetten MSM, et al. Anaerobic oxidization of methane in a minerotrophic peatland: enrichment of nitrite-dependent methane-oxidizing bacteria. Appl Environ Microbiol. 2012;78:8657–8665. doi: 10.1128/AEM.02102-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.