

Abstract
The leading cause of death in the United States is cardiovascular disease. The majority of these cases result from heart failure post-myocardial infarction (MI). We present data providing evidence for use of acetalated dextran (AcDex) microparticles as a delivery vehicle for therapeutics to the heart post-MI. We harnessed the tunable degradation and acid-sensitivity of AcDex in the design of microparticles for intramyocardial injection. The particles released a model protein, myoglobin, and a sensitive growth factor, basic fibroblast growth factor (bFGF), over a wide range of time frames (from days to weeks) based on the percentage of cyclic acetals in the AcDex, which was easily controlled with acetalation reaction time. The release was shown in low pH environments similar to what is found in an infarcted heart. bFGF maintained activity after release from the microparticles. Finally, biocompatibility of the microparticles was assessed.
Keywords: microparticles, tunable, myocardium, acetalated dextran
INTRODUCTION
Cardiovascular disease is the leading cause of death in the United States with the majority of deaths resulting from heart failure post-myocardial infarction (MI).1 Current therapies for MI patients ameliorate symptoms and prolong life, but do not treat the injury at the source. Left-ventricular assist devices (LVADs), which increase risk for stroke and limit patients’ activity, and heart transplantation, for which relatively few donor hearts are available, are currently the only effective treatment interventions for end-stage heart failure, and no therapies prevent the negative LV remodeling process that ensues post-MI.
Recently, injectable biomaterials (hydrogels and microparticles) have been used in the development of experimental therapies.2-5 In contrast to current gold-standard treatment methods, biomaterial-based therapies have the potential to initiate healing at the site of injury. One way of initiating healing is through sustained release of therapeutic agents that promote angiogenesis and positive remodeling. Biomaterials can protect biological cargo from rapid degradation and facilitate delivery over longer time frames than bolus injections.
Several materials have been employed to deliver therapeutic agents to the heart via intramyocardial injection of microparticles. These include delivery of vascular endothelial growth factor (VEGF) in poly(lactic-co-glycolic acid) (PLGA),6 basic fibroblast growth factor (bFGF), VEGF or ginsenoside individually encapsulated in gelatin7-11 and a p38 inhibitor or super oxide dismutase (SOD) in polyketal-based polymers.12, 13 These studies demonstrated encouraging results that support the potential for microparticle-based therapies; however, no single therapeutic achieved restoration of function in the heart post-MI. This is most likely due to the complexity of the healing process in the heart, which will require reducing fibrosis, restoring blood flow, and promoting cellular infiltration. Previous studies using therapeutics to restore blood flow highlight the importance of the time and order of therapeutic agent presentation.14, 15
Angiogenesis, or formation of a fully functioning blood vessel from a preexisting vessel, requires a number of steps, including vessel sprouting, branching and stabilization. Each step occurs in response to a specific set of signals, in a specific order.16 Several groups have already shown in small animal models that the order in which angiogenic factors are presented determines the efficacy of the treatment.14,15 While not yet thoroughly investigated, it is likely that other healing processes needed for full restoration of heart function post-MI will also require therapeutics delivered over specific time frames.
Several promising approaches for tunable delivery of growth factors have been developed, but each is limited to a specific category of payloads or is difficult to manipulate to provide more than two rates of release. The first approach takes advantage of growth factors’ variable heparin binding strength with sulfated materials to create an affinity scaffold to sequentially release VEGF, PDGF-BB, and TGF-β1.14 Here only molecules with heparin affinity or a positive charge can be delivered. Using a similar strategy, polyelectrolyte multilayers were used to control the release of charged growth factors, in this case bFGF (+), layered with heparin (−) and a polymer (+), was released at a rate dependent on the number of layers assembled,17 limiting deliverable cargo to charged molecules. Polyelectrolyte multilayers in the form of microparticles formulated by layer-by-layer (LbL) encapsulation have also been employed to provide sustained release of several payloads including bFGF. However, to date the system has not been specifically designed for delivery to the heart post-MI. For instance, payload release often occurs around neutral pH 18, 19 and in some cases is actually prohibited in mildly acidic conditions19. Some studies have shown permeability of the microcapsules at low pH (<6) and impermeability at high pH (>8) 20 but no studies have shown impermeability at neutral pH (7.4) with permeability in mildly acidic pH (6-7) 21, as desired for this application. Furthermore, in the majority of studies, ~80% of the payload is released in 3-4 days 18, 19, 22, far too short of a time frame for healing post-MI, which requires several weeks. In one study, changing layer thickness modulated release rate, but only by 5% at the time points shown 22. A greater difference in release rate may be achieved by varying the loading method between pre- or post-loading, since this approach caused a larger difference in total protein released at 72 hours, but the long-term release trends were not explored 22. Another approach embedded PDGF-loaded PLGA microspheres in an alginate hydrogel containing VEGF23 to release VEGF followed by PDGF. Creating more than two rates of release with this system would require changing hydrogel density, particle size and/or particle concentration, and no hydrophobic payload could be released from the alginate hydrogel. The growth factors delivered in these studies are likely to be important in some healing pathways post-MI, but other payloads including hydrophobic small molecules, enzymes, and plasmid DNA have also shown promise in aiding healing post-MI. Finally, these approaches are not designed for the mildy acidic environment of an infarct (pH 6.0-7.0).24, 25
Acetalated dextran (AcDex) microparticles allow tunable release, have the potential to deliver charged, uncharged, hydrophilic, and hydrophobic molecules and release the encapsulated therapeutics in response to acidic pH17, 23, 26-33. Tuning release rates from AcDex is straightforward, requiring only variation in the reaction time when acetalating to vary the ratio of cyclic to acyclic acetals; cyclic acetals degrade slower than acyclic acetals. This property represents an advantage of AcDex over materials previously tested in the heart, which have defined release rates in the ischemic environment. Furthermore, payload release from AcDex particles is minimal in neutral pH conditions but is facilitated in acidic conditions. While PLGA degradation can also be catalyzed in acidic environments, the time frame of degradation is long (many weeks) at the pH found in ischemic tissue34. The pH must be low (~2) to see appreciable acceleration35.
AcDex is synthesized by acetalation of the alcohols on the dextran backbone with vinyl ethers in the presence of an acid catalyst. Acetal functionalization of dextran changes the solubility of the polymer from hydrophilic to hydrophobic, allowing AcDex to encapsulate hydrophobic or hydrophilic payloads via a single oil-in-water (o/w) or double water-in-oil-in-water (w/o/w) emulsion, respectively. In contrast, protein-based microparticles such as gelatin can only be used to deliver hydrophilic payloads. Previous work with AcDex has shown tunable release of a number of payloads including ovalbumin, imiquimod, horseradish peroxidase, rapamycin, plasmid DNA, siRNA and campthomecin17, 23, 26-33. These payloads were encapsulated in particles ranging from 100 nm to 10 μm and released at pH 5.0 and 7.4. The size of the particles and target pH in these studies were designed for cellular uptake and lysosomal degradation for immunotherapy, gene delivery and pulmonary drug delivery. To use AcDex for intramuscular delivery in ischemic conditions, particles need to be designed with a diameter of 20-100 μm to avoid cellular uptake and maximize interstitial retention time,36 a payload release longer than 1-2 days to initiate relevant tissue regeneration processes, and a desired degradation rate in more mildly acidic conditions as seen in ischemic tissue (pH 6.0-7.0). Although not yet tested in vivo AcDex is predicted to have mild degradation products, including dextran, which is used in several FDA approved products, and trace amounts of acetone and methanol.32
In this study we develop AcDex microparticles as a delivery vehicle for therapeutics to the heart post-MI. In mildly acidic conditions (pH 6.0-7.0), tunable release between days and weeks is shown with a model protein and a sensitive growth factor, bFGF. Furthermore, bFGF maintained activity after encapsulation and release. The particles are shown to be injectable into the heart where they are retained in the extracellular space and elicit an acute inflammatory response that followed a biocompatible progression through one month.
EXPERIMENTAL SECTION
Materials
Sunsoft surfactant (818SK) was purchased from Taiyo Int. Inc. (Minneapolis, MN). Dextran (from Leuconostoc mesenteroides; average MW 9,000-11,000), myoglobin (from equine skeletal muscle), poly(vinyl alcohol) (PVA; average MW 30,000-70,000), bovine serum albumin (BSA; Fraction V), and heparin sodium salt (from porcine intestinal mucosa) were purchased from Sigma-Aldrich. Alexa Fluor 594-NHS and basic fibroblast growth factor (bFGF) were purchased from Life Technologies. All other reagents and materials were purchased from Sigma, Fisher or Acros and used as received.
Methods
1H NMR spectra were acquired on a Bruker npa600 and processed using MestreNova software. NMR was used to determine the cyclic:acylic acetal ratio of all synthesized AcDex as previously described37. Briefly, AcDex was dissolved in a stock solution of D2O and DCl and then analyzed. The methanol (3.34 ppm) and acetone (2.08 ppm) peaks were integrated and compared under the assumption that degradation of only acyclic acetals would yield a 1:2 ratio of methanol:acetone, and additional acetone peak area results from cyclic acetal degradation32. GPC was conducted by the UCSB Material Research Laboratory using a Waters Alliance HPLC System with a Waters 2414 Differential Refractometer, Viscotek I-MBHMW and I-Series Mixed Bed High Molecular Weight columns. DMF containing 0.01% of LiBr was used as the eluent (flow rate 1 mL/min). Polystyrene standards were used for calibration. The primary emulsion for all microparticles was created using a QSonica Q700 with Cup Horn sonicator. Microparticles were prepared with a membrane emulsifier from Micropore Technologies. Microparticle size was determined using the Beckman Coulter Multisizer 4. Scanning electron microscopy (SEM) was used to visualize intact particles and particle degradation at various time points throughout the protein release studies. Particles suspended in water were dried on a silicon wafer and imaged using an Agilent 8500 FE-SEM. All fluorescence measurements were performed using a fluorometer (FL-1065; Horiba Jobin Yvon).
Synthesis of Acetalated Dextran with High and Low Catalyst
Acetalated dextran was prepared as described previously.37, 38 Briefly, a flame-dried round-bottom flask (50 mL) was purged with N2. Dextran (Mw = 10,000 g/mol, 1.00 g, 0.095 mmol) and anhydrous DMSO (10 ml) were added and stirred until dextran was completely dissolved. 2-Methoxypropene (3.4 mL, 37 mmol) was added via syringe, followed by pyridinium p-toluenesulfonate (15.6 mg, 0.062 mmol). The system was maintained under positive N2 pressure and then sealed. At designated time points (2, 5, 10, 15, 30, 60, 120, 180 1200 and 1500 minutes) an aliquot (1 mL) was removed and quenched with triethylamine (100 μL TEA/1ml reaction solution, 7 mmol). The solution containing the modified dextran was precipitated in Millipore H2O (pH 8; 1:10 reaction solution:water). The product was isolated by centrifugation (5 000 × g; 10 min). The resulting white pellet was washed with Millipore H2O (pH 8) by vortexing, centrifugation and removal of supernatant. The hydrated pellet was frozen and lyophilized overnight to remove residual water. The following day the pellet was resuspended in acetone and centrifuged (5 000 × g; 5 min) to separate out any impurities. The acetone solution was precipitated in Millipore H2O (pH 8), washed, frozen and lyophilized as described above. The resulting product was a white fluffy powder. To access a wider range of cyclic:acyclic ratios, an identical experiment as described above was completed with one-third the catalyst pyridinium p-toluenesulfonate (5.2 mg, 0.021 mmol).
Large Scale Synthesis of Acetalated Dextran of Different Cyclic to Acylic Ratios
AcDex was prepared on a 10 g scale using the same mole ratios and concentrations as described in the low catalyst procedure above. The 10 g, scaled up reaction, was performed three times, but quenched at different time-points. A low amount of cyclic acetals on dextran (LOW is percent cyclic to acyclic) was obtained by quenching at 30 minutes. A medium amount of cyclic acetals on dextran (MED percent cyclic to acyclic) was obtained by quenching at 60 minutes. A high amount of cyclic acetals on dextran (HIGH percent cyclic to acyclic) was obtained by quenching at 1200 minutes. The cyclic to acyclic ratios on the dextran were determined by 1H NMR as described above.
Myoglobin Labeling and Encapsulation in Microparticles for Release Studies
Myoglobin (6 mg, 339 μM) was dissolved in carbonate-bicarbonate buffer (1mL, pH 8.98). Fluorosceneisocyanate (FITC; 3 mM) in dimethyl sulfoxide (DMSO; 35 uL) was added to the myoglobin solution while stirring. The FITC/myoglobin mixture was stirred for two hours protected from light at 25°C. Free dye was removed by repetitive washing with PBS pH 7.4 through a 3K MWCO Amicon spin filter until no measurable fluorescence was detected in the flow through.
Microparticles were prepared using a w/o/w double emulsion. For the organic phase LOW, MED or HIGH AcDex (630 mg; 10 wt%) was dissolved in dichloromethane (DCM; 6.3 mL) containing Sunsoft surfactant (126 mg; 2 wt%). The primary aqueous phase was composed of Tris buffer (10mM pH 8; 700 μL) with FITC-labeled Myoglobin (1.4 mg; 0.2 wt%) and bovine serum albumin (BSA; 14 mg; 2 wt%).
The organic and primary aqueous phases (9:1 volume ratio) were sonicated (30 seconds; 30% power) and then stirred (2000 rpm, 1 min) three times to generate the primary emulsion. The primary emulsion was injected (rate: 0.5 mL/min) through a 10 μm hydrophobic membrane into a secondary aqueous phase of Tris buffer (10 mM, 150 mL; pH 8) containing 1% poly(vinyl alcohol) (PVA) and stirred (1000 rpm). After the entire primary emulsion was injected, the newly formed particles were stirred in Tris buffer (10 mM, pH 8; 500 mL) with 1% PVA to evaporate the DCM. Following evaporation, the particles were washed three times with Tris Buffer (10 mM, pH 8; 50 mL) and once with Millipore H2O (pH 8; 50 mL) by centrifuging the particles (5 × g, 5 min) and removing the supernatant. Washed particles were frozen and lyophilized before release studies and characterization.
Myoglobin Release from Microparticles
Lyophilized myoglobin-loaded particles were suspended (20 mg/mL) in phosphate buffer saline (PBS; 1 mL) pH 6.0, 6.5 or 7.4 and placed on a rotating tube rack at 37°C. At designated time points, particles were centrifuged (12.1k g, 5 minutes) and supernatant (500 μL) was removed and replaced with fresh PBS of the same pH. The supernatant (400 μL) was mixed with DMSO (800 μL) to dissolve the Sunsoft surfactant and pipetted into a quartz fluorimeter cuvette. The solution was excited at 485 nm and emission at 524 nm was recorded and compared to a previously prepared standard curve for FITC-labeled myoglobin at the given pH. Protein release graphs were created to show percent released over time up to 100%. Complete (100%) release was defined as the amount of protein in solution once particles were fully degraded (ie. there was no pellet upon centrifugation and no particles were visible with SEM).
bFGF Labeling and Encapsulation in Microparticles for Release Studies
Basic fibroblast growth factor (bFGF; 0.1 μM) was dissolved in carbonate buffer (pH 9.0; 500 μL). While stirring on ice, Alexa594-NHS (1 μg) was slowly added to the bFGF solution (100 μL), and then stirred for one hour protected from light. After the conjugation procedure, the labeled bFGF was buffer exchanged into Tris buffer (10 mM) containing 5% trehalose and washed using a 10k Amicon spin filter until no measurable fluorescence was detected in the flow through.
Microparticles were prepared using a w/o/w double emulsion. For the organic phase LOW or HIGH AcDex (475 mg; 10 wt%) was dissolved in DCM (4.75 mL). The primary aqueous phase was composed of Tris buffer (10 mM pH 8; 250 μL) with Alexa594-labeled bFGF (1 μg) and heparin (1 μg). The remainder of the microparticle preparation was identical to that described in the model protein studies above.
bFGF Release from Microparticles
Lyophilized Alexa594-labeled bFGF-loaded particles were suspended (20 mg/mL) in PBS (1 mL) pH 6.0 or 7.4 and placed on a rotating tube rack at 37°C. At designated time points, particles were centrifuged (12.1k g, 5 minutes) and supernatant (800 μL) was removed and pipetted into a quartz fluorimeter cuvette. The solution was excited at 590 nm and emission at 617 nm was recorded and compared to a previously prepared standard curve for Alexa594-labeled bFGF. The supernatant was returned to the particle solution after measurement.
bFGF Encapsulation in Microparticles for the Activity Assay
Microparticles were prepared using a w/o/w double emulsion. Two batches of particles were created, one with Alexa594-labeled bFGF to determine the amount of growth factor to add to cells in the control group of the activity assay, and one with unlabeled bFGF to provide supernatant for the activity assay.
For the organic phase LOW AcDex (285 mg; 10 wt%) was dissolved in DCM (2.85 mL). The primary aqueous phase was composed of Tris buffer (10 mM pH 7.6; 150 μL) with Alexa594-labeled bFGF (600 ng), plus Heparin (1.2 μg) and BSA (12 μg). The remainder of the microparticle preparation was identical to that described in the model protein studies above.
For the organic phase LOW AcDex (475 mg; 10 wt%) was dissolved in DCM (4.75 mL). The primary aqueous phase was composed of Tris buffer (10 mM pH 7.6; 250 μL) with or without bFGF (1 μg), plus Heparin (2 μg) and BSA (20 μg). The remainder of the microparticle preparation was identical to that described in the model protein studies above except that Tris buffer with and without PVA and Millipore H2O were sterile filtered (0.22 μm) before use. All parts of the membrane emulsifier were sprayed with 70% ethanol immediately before use. In addition, the particles were stirred for only four hours to evaporate the DCM.
bFGF Release from Microparticles for the Activity Assay
For the study to determine the amount of growth factor to add to cells in the growth factor activity assay, the same procedure as the growth factor release study was performed at pH 6.0 over five days.
Particles with and without bFGF were suspended (60 mg/mL) in PBS (pH 6.0) and placed at 37°C on a shaker plate. After 24 hours, tubes were centrifuged to pellet particles and supernatant was removed and filtered through a 0.22 μm filter. Removed supernatant was replaced with fresh PBS pH 6.0 and returned to 37°C. After an additional 24 hours, supernatant was removed and filtered.
bFGF Activity Assay
3T3 fibroblasts were seeded at 5,000 cells/well in a 24-well plate in growth media (DMEM, 10% FBS, 0.5% Pen-Strep; 1 mL). After 24 hours at 37°C under 5% CO2, experimental conditions were applied. Growth media (GM; 1 mL; n=4) and low serum (DMEM, 2% FBS, 0.5% Pen-Strep; 1 mL) with fresh bFGF (LS + Fresh; 5 ng bFGF; n=5) were used as positive controls. Low serum (LS; n=5; 1 mL) and low serum with empty particle supernatant (LS + Empty; 250 μL supernatant; n=5) were used as negative controls. Low serum with bFGF supernatant was used as the experimental group (LS + Released; 5 ng bFGF in 250 μL supernatant; n=5). After 24 hours at 37°C under 5% CO2, all media was removed and replaced with the same experimental conditions listed above including the bFGF (10 ng bFGF in 250 μL supernatant) and empty supernatant (250 μL supernatant) from the second day's release and an equivalent amount of fresh bFGF (10 ng bFGF). After 24 hours at 37°C under 5% CO2, cells were trypsinized and counted with a hemocytometer.
Statistical Analysis
Statistical analysis was performed using PRISM. Results are presented as mean ± standard deviation. One-way analysis of variance (ANOVA) was used to evaluate differences in cell number among all groups in the growth factor activity study. Bonferroni's correction was used to detect differences between groups. Significance was defined at p<0.05.
Empty Microparticles for in vivo Biocompatibility
For the organic phase LOW or HIGH AcDex (475 mg; 10 wt%) was dissolved in DCM (4.75 mL). The primary aqueous phase was composed of only Tris buffer (10 mM pH 7.6; 250 μL). The remainder of the microparticle preparation was identical to that described in the model protein studies above except that Tris buffer with and without PVA and Millipore H2O were sterile filtered (0.22 μm) before use. All parts of the membrane emulsifier were sprayed with 70% ethanol immediately before use. In addition, the particles were stirred for only four hours to evaporate the DCM.
Intramyocardial Injections for in vivo Biocompatibility
All experiments were completed in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Diego and the American Association for Accreditation of Laboratory Animal Care.
Intramyocardial injections were performed in female Sprague Dawley rats (225-260 g) as previously described.39 75 μL of microparticles (LOW or HIGH, 20 mg/mL in saline) were injected into the LV free wall with a 27 G needle. Rats were divided into three time points (7, 14 or 28 days) and two groups (LOW and HIGH; n=3 each). Animals were euthanized with an intraperitoneal injection of FatalPlus (Vortech Pharaceuticals; 400 μL) at designated time points to assess histopathology. The heart was resected, rinsed and embedded and fresh frozen in OCT freezing medium. Short axis cross-sections were taken every 350 μm from apex to base throughout each heart and stained with hematoxylin and eosin (H&E). An experienced histopathologist examined the slide for the presence of spindle, small mononuclear, large mononuclear, fibroblast and multinucleate giant cells and provided a score from 0 to 4 (none, minimal, mild, moderate, severe).
RESULTS AND DISCUSSION
Preparation of Acetalated Dextran with Varying Degradation Rates
AcDex is synthesized by reacting dextran with vinyl ethers and an acid catalyst (Scheme 1); specifically, we use methyl vinyl ether and pyridinium p-toluenesulfonate as a catalyst.
Scheme 1.
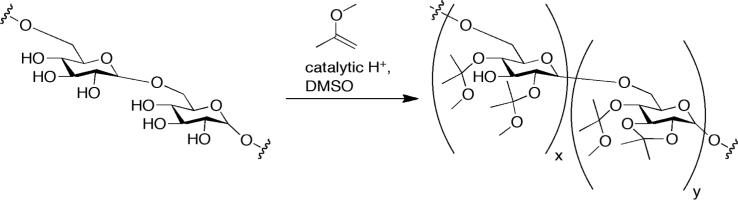
Synthesis of acetalated dextran (AcDex)
Previously it was demonstrated that the length of reaction time controls the amount of cyclic and acyclic acetals on the dextran backbone with a longer the reaction time yielding a higher amount of cyclic acetals.32 Acetalation transforms water-soluble dextran into a hydrophobic polymer, and upon exposure to acid the acetals degrade causing the dextran to become water soluble. The cyclic acetals hydrolyze at a slower rate than the acyclic acetals, allowing for tunable solublization of the AcDex polymer based on the ratio of cyclic:acyclic acetals. The acetal content and ratio of cyclic:acyclic were determined by 1H NMR after acid degradation of the purified polymer. The methanol (3.34 ppm) and acetone (2.08 ppm) peaks in D2O/DCl were used to calculate the cyclic acetal percentage.32 We used three different procedures to synthesize AcDex. First, we investigated the kinetics of acetalation with 0.062 M catalyst and 1 g of dextran (normal catalyst).32 This resulted in a rapid increase of cyclic acetals, with 50% of the polymer containing cyclic acetals after 5 minutes and 66% cyclic acetals (the theoretical maximum) after one hour (Figure S1). Second, in order to access a wider range of cyclic:acyclic ratios we reduced the amount of catalyst by a third (low catalyst). Here, after 10 minutes only 20% cyclic acetals were present, and after 60 minutes 40% cyclic acetals were present (Figure S1). Third, we scaled up the low catalyst procedure to 10 g of dextran to produce a uniform batch for all subsequent studies. We aimed to generate three batches of AcDex with a low, medium and high percent cyclic acetals, respectively. The 1H NMR spectra are shown in Figure S2. This allowed us to investigate a range of payload release profiles once AcDex was formulated into particles. Scaling up the reaction changed the kinetics of acetalation slightly, yielding a narrower range of cyclic acetal percentages. Specifically, the percentage of cyclic acetals from the scale-up was 49% (termed LOW hereafter), 54% (MED), and 59% (HIGH) at 30 min, 60 min, and 1200 min, respectively, compared to 30%, 40% and 60%, respectively, at the smaller scale (Table 1).
Table 1.
Characterization of LOW, MED and HIGH AcDex (10 g batches) used in subsequent studies. Predicted cyclic acetal coverage is based on prior kinetics studies with a 1 g batch.
| Name | Quenching Time (min) | Predicted Cyclic % | Actual Cyclic % | Mw | PDI |
|---|---|---|---|---|---|
| LOW | 30 | 30 | 49 | 7757 | 1.83 |
| MED | 60 | 40 | 54 | 7966 | 1.79 |
| HIGH | 1200 | 60 | 59 | 8199 | 1.82 |
Model Protein Release Studies
Previous work with AcDex fabricated nano- and microparticles with diameters of 100 nm to 10 μm for cellular uptake, whereas for delivery of many therapeutic agents post-MI the particles should be larger (20-100 m)36 to remain in the extracellular space. We aimed to design microparticles that could be used to deliver a payload at a variety of relevant time scales. To demonstrate tunable release over the time-scale relevant to healing post-MI, several days or weeks, we encapsulated a fluorescently labeled model protein and measured its release into the surrounding media over time. Myoglobin was chosen as a model protein because its size (17.7 kDa) matches that of growth factors such as bFGF (18 kDa), which are relevant to post-MI healing. AcDex microparticles containing FITC-labeled myoglobin (FITC-myo) were prepared using a w/o/w double emulsion. The average particle size for the LOW, MED and HIGH particles was 46 ± 14 μm, 58 ± 19 μm and 63 ± 27 μm, respectively. Encapsulation efficiency of myoglobin in the LOW, MED and HIGH microparticles was ~100%, 70% and 47%, respectively. The difference in loading efficiency between groups is likely due to the increase in hydrophobicity of the polymer as the cyclic acetal coverage increases. An SEM image of the LOW, MED and HIGH particles before the release experiment reveals that the particles are spherical with a smooth surface (Figure 1A).
Figure 1.

Tunable release of a model protein from AcDex microparticles. A. Intact LOW, MED and HIGH (left to right) particles containing FITC-myoglobin before model protein release studies. Particles are spherical with a smooth surface. B. Myoglobin release from LOW, MED and HIGH particles at pH 6.0, 6.5 and 7.4. At pH 6.0 complete release of myoglobin was seen at 5, 10 and 60 days for the LOW, MED and HIGH particles, respectively. The same trend was seen at pH 6.5, and at pH 7.4, with progressively slower release; in the latter condition only 50% of encapsulated myoglobin was released after 11, 17 and 63 days, respectively. C. SEM images of the MED particles on day 7 at pH 6.0, 6.5 and 7.4 (left to right). Particle morphology correlates with the observed myoglobin release at this time point. Scale bar: 10 μm.
We examined the release of FITC-myo from the microparticles in mildly acidic conditions, representative of the ischemic infarct (pH 6.0 and pH 6.5) and healthy tissue (pH 7.4). As intended, release of the model myoglobin was dependent on environmental pH (faster at lower pH) and cyclic acetal percentage (faster for particles with lower percentage). At pH 6.0 complete release of myoglobin was seen at 5, 10 and 60 days for the LOW, MED and HIGH particles, respectively. The same trend was seen at pH 6.5, and at pH 7.4, with progressively slower release; in the latter condition only 50% of encapsulated myoglobin was released after 11, 17 and 63 days, respectively (Figure 1B). To corroborate these results, particle degradation was visualized using SEM. The morphology of the MED particles at day 7 is shown in Figure 1C at pH 6.0, 6.5 and 7.4. At pH 6.0 the porous interior of the MED particles is clearly visible and deep cracks have formed throughout the particles. At pH 6.5 some surface cracks appear, but the smooth external surface of the particles is still intact. At pH 7.4 only lines on the surface are visible that suggest where cracks will eventually form. The images correlate well with the observed myoglobin release at this time point: 62%, 34% and 18% for pH 6.0, pH 6.5 and pH 7.4, respectively. These images suggest that the payload only diffused out when the particles degraded, as opposed to passive diffusion through the intact particles.
Growth Factor Release Studies
To examine whether this system is applicable to growth factor delivery, we encapsulated bFGF. Growth factors are more susceptible to loss of activity upon sonication or exposure to organic solvents than myoglobin, so showing that bFGF can remain active after encapsulation and release from AcDex microparticles would suggest the system's compatibility with any fragile payloads. bFGF has also been employed in post-MI therapies to stimulate new blood vessel formation.7, 9, 10, 40, 41 AcDex microparticles containing Alexa594-labeled bFGF were prepared using a w/o/w double emulsion. For simplicity, in this study the experiment groups were reduced to only the LOW and HIGH microparticles and pH 6.0 and 7.4. Furthermore, while particles in the model protein studies were prepared using Sunsoft as a stabilizing agent, we eliminated the detergent from bFGF-containing formulations to maximize growth factor stability. The average particle size for the LOW and HIGH particles was 60 ± 18 μm and 67 ± 22 μm, respectively. Encapsulation efficiency for bFGF in the LOW microparticles was ~100%. The HIGH microparticles were not fully degraded in this study and thus no encapsulation efficiency was calculated, but it is expected to be similar to that seen with myoglobin, since the LOW values were equivalent. Again, these particles are spherical with smooth surfaces as shown by SEM (Figure 2A).
Figure 2.

Tunable release of a growth factor from AcDex microparticles. A. Intact LOW and HIGH (left to right) particles containing Alexa594-bFGF before bFGF release studies. Particles are spherical with a smooth surface. B. bFGF release from LOW and HIGH particles at pH 6.0 and 7.4. At pH 6.0 complete release was seen after three days from the LOW particles, while only 50% release was seen after 10 days for the HIGH particles. A similar trend was seen at pH 7.4 as 50% of the bFGF was released after one week from the LOW particles, while after one month, only 37% had been released from the HIGH particles. C. SEM images of the LOW particles on day 2 at pH 6.0 and 7.4 (left to right). Particle morphology correlates with the observed bFGF release at this time point. Scale bar: 10 μm.
Release experiments were repeated in the same manner as with the model protein, using the LOW and HIGH AcDex at pH 6.0 and 7.4. At pH 6.0 complete release was seen after three days from the LOW particles, while only 50% release was seen after 10 days for the HIGH particles. A similar trend was seen at pH 7.4 as 50% of the bFGF was released after one week from the LOW particles, while after one month, only 37% had been released from the HIGH particles (Figure 2B). As seen by SEM (Figure 2C), on day 2 at pH 6.0 the particles were mostly degraded, but the remaining intact particles show visible holes and cracks across the surface. The particles at pH 7.4 at the corresponding time point still maintained a smooth surface. While the bFGF was also released at a rate dependent on the cyclic acetal percentage and the environmental pH, release was slightly faster than was seen with the myoglobin. This difference was potentially due to the presence of BSA in the myoglobin-loaded particles, as discussed further in the following section.
Growth Factor Activity Assay
To demonstrate that the encapsulated bFGF was still biologically active, we used a cell-based assay that measures the ability of bFGF to stimulate proliferation of fibroblast cells. LOW AcDex microparticles with and without bFGF were prepared as described above. The average particle size for the bFGF-encapsulating and empty particles was 68 ± 22 μm and 58 ± 20 μm, respectively. Both types of microparticles were suspended in PBS pH 6.0 for two days at 37°C. Each day supernatant was removed and added to cells in low serum media (Released bFGF; Empty Supernatant). As a positive control, bFGF that had not been processed in any way (Fresh bFGF) was added to cells at the same concentration as the released bFGF as determined by the release experiment described in the Methods section. An additional negative control was used consisting of low serum media with no bFGF (No bFGF). After 24 hours, the Fresh bFGF and Released bFGF groups both had significantly more cells per well than either the No bFGF or Empty Supernatant groups. In addition, the Fresh bFGF and Released bFGF groups were not statistically different from one another (Figure 3). These results demonstrate that the bFGF maintained its activity after encapsulation in and release from the microparticles and suggest that any number of formulation-sensitive growth factors could be delivered using this particle system, if encapsulated with an appropriate stabilizing agent.
Figure 3.

bFGF activity after release from AcDex microparticles. Cell number after 24 hours of growth in growth factor activity assay (mean ± SD; *p < 0.05). The LS + fresh bFGF and LS + released bFGF groups both had significantly more cells per well than either control group (LS alone or LS + empty). In addition, the LS + Fresh bFGF and LS + Released bFGF groups were not statistically different from one another. *p<0.05 to both negative control groups.
The tunable release of myoglobin and bFGF suggests that this system could be used to release a variety of payloads. Myoglobin and bFGF were likely in different charge states inside the particles, yet released at comparable rates. At the pHs in these studies, myoglobin (isoelectric point of 7 to 7.5) was likely slightly positively charged, while bFGF (isoelectric point of 9.6) was likely negatively charged. This suggests that electrostatic interactions did not play a role in keeping either protein inside the particles. In addition, AcDex is hydrophobic and both payloads are hydrophilic, so hydrophobic interactions were also not likely playing a role. As a result, the small 17 kDa proteins were likely kept inside the particles simply by physical entrapment until the particles broke down. Therefore, it is expected that the release of a larger protein would be easily controlled as well. All of these considerations suggest that AcDex provides a more versatile delivery vehicle than previously developed systems because compatible payloads are not limited by affinity, size, or charge.
The bFGF release studies were performed without BSA for simplicity, but it was later found that the BSA was needed for optimal bFGF activity, in addition to heparin. The hypothesis mentioned in the previous section about BSA changing release rate was supported by the release experiment completed prior to the activity studies. In this experiment, particles containing labeled bFGF, heparin and BSA (Figure S3) displayed slower release of bFGF than observed in the bFGF release study, which omitted BSA (Figure 2B). This slowed release rate was more similar to that seen with myoglobin (Figure 1B).
Biocompatibility
While AcDex has been predicted to have mild degradation products, it has not been tested in an in vivo system. Therefore, to examine the biocompatibility of AcDex, empty LOW (69 ± 24 μm) and HIGH (74 ± 20 μm) particles were injected into the hearts of healthy rats. At 7, 14 and 28 days following injection, animals were euthanized and the hearts excised and processed for histological analysis. Spherical voids created by the particles were visible for both the LOW and HIGH groups at all three time points in H&E stained slides (Figure 4).
Figure 4.

Images showing AcDex biocompatibility in healthy rat hearts. H&E images from day 7 (A), 14 (B) and 28 (C) of the biocompatibility study. Spherical voids created by the particles (black arrows) are visible for both the LOW (left) and HIGH (right) groups. As particles degrade, the voids, representative of particle morphology, becomes less spherical. Scale bar: 100 μm.
An experienced histopathologist examined the cellular response to the microparticles using semi-quantitative scoring (1 or minimal = cells of that type are rarely present (<1% of the population); 2 or mild = small numbers of the cells are present, generally not in contact with each other or scattered (<10%); 3 or moderate = moderate numbers of cells are present, which are sometimes in contact with each other (10 - <50%); 4 or marked = cells of the specific type are the primary cell present (> 50%)). On day 7 the cell population around the particles consisted of spindle (immature fibroblast) cells as well as small and large mononuclear cells (all mild to moderate). No mature fibroblasts or multinucleate giant cells were present at this time point (Figure 5). On day 14 the cell population shifted to include fewer spindle (immature fibroblast) and mononuclear cells (minimal to mild), with the addition of mature fibroblasts (mild) and a few multinucleate giant cells (none to minimal) (Figure 5). On day 28 the cell population consisted primarily of mature fibroblasts with some remaining mononuclear cells (minimal) and a few multinucleate giant cells (none to minimal) (Figure 5). Overall the response was described as progressing normally from acute inflammation towards a fibrotic resolution. While not fully resolved at day 28, due to the presence of some mononuclear cells, there was no indication of a severe or abnormal response that would suggest incompatibility of the material.
Figure 5.

Cell type scores from AcDex biocompatibility study. On day 7 the cell population around the particles consisted of spindle (immature fibroblast) cells as well as small and large mononuclear cells. No mature fibroblasts or multinucleate giant cells were present at this time point. On day 14 the cell population shifted to include fewer spindle and mononuclear cells, with the addition of fibroblasts and a few multinucleate giant cells. On day 28 the cell population consisted primarily of mature fibroblasts with some remaining mononuclear cells and a few multinucleate giant cells. Score: none (0), minimal (1), mild (2), moderate (3) and severe (4).
CONCLUSIONS
In summary, we have harnessed the tunable degradation rate and acid sensitivity of AcDex in the design of microparticles for intended delivery of therapeutics to the heart post-MI. Microparticles were created within an optimal size range for intramuscular injection and were retained for up to 28 days in the heart. The particles reproducibly released a model protein and formulation-sensitive growth factor at rates varying from within days to over months, tuned by altering the percentage of cyclic acetals. Controlled payload release was shown over a pH range similar to what is likely found in an infarcted heart. A sensitive growth factor maintained activity after encapsulation in and release from the microparticles, suggesting the system's compatibility with fragile payloads. While not tested here, AcDex particles can also encapsulate and release hydrophobic payloads, increasing the system's versatility. Finally, microparticles were easily injected into the heart, where they elicited an acute inflammatory response that followed a biocompatible progression through one month, similar to the response to an injection of saline. All these results support the potential of AcDex microparticles as a delivery vehicle for therapeutics to the heart post-MI.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Eric Jacquinet for his histological evaluation in the biocompatibility study.
Funding Sources
The authors acknowledge the National Science Foundation Graduate Research Fellowship Program, the NIH Director's New Innovator Award 1DP2OD006499-01, and King Abdulaziz City for Science and Technology center grant to the CEN at UCSD for funding this work.
ABBREVIATIONS
- AcDex
acetalated dextran
- bFGF
basic fibroblast growth factor
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information Available
Supporting information includes kinetics of acetalation reaction using low and normal amount of catalyst, 1H NMR spectra for 10 g batches of AcDex used in all studies, and release of bFGF experiment used to determine amount of bFGF to be added to cells in activity assay. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rane AA, Christman KL. Journal of the American College of Cardiology. 2011;58:2615–2629. doi: 10.1016/j.jacc.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Christman T. D. J. a. K. L. 2012:1–14. [Google Scholar]
- 4.Li Z, Guan J. Polymers. 2011;3:740–761. [Google Scholar]
- 5.Tous E, Purcell B, Ifkovits JL, Burdick JA. J Cardiovasc Transl Res. 2011;4:528–542. doi: 10.1007/s12265-011-9291-1. [DOI] [PubMed] [Google Scholar]
- 6.Formiga FR, Pelacho B, Garbayo E, Abizanda G, Gavira JJ, Simon-Yarza T, Mazo M, Tamayo E, Jauquicoa C, Ortiz-de-Solorzano C, Prosper F, Blanco-Prieto MJ. Journal of Controlled Release. 2010;147(1):30–37. doi: 10.1016/j.jconrel.2010.07.097. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Sun L, Huan Y, Zhao H, Deng J. Eur J Cardiothorac Surg. 2006;30:103–7. doi: 10.1016/j.ejcts.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 8.Wei HJ, Yang HH, Chen CH, Lin WW, Chen SC, Lai PH, Chang Y, Sung HW. J Control Release. 2007;120(1-2):27–34. doi: 10.1016/j.jconrel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Iwakura A, Fujita M, Kataoka K, Tambara K, Sakakibara Y, Komeda M, Tabata Y. Heart Vessels. 2003;18:93–9. doi: 10.1007/s10380-002-0686-5. [DOI] [PubMed] [Google Scholar]
- 10.Sakakibara Y, Tambara K, Sakaguchi G, Lu F, Yamamoto M, Nishimura K, Tabata Y, Komeda M. Eur J Cardiothorac Surg. 2003;24:105–11. doi: 10.1016/s1010-7940(03)00159-3. discussion 112. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Suto N, Okubo T, Mikuniya A, Hanada H, Yagihashi S, Fujita M, Okumura K. Jpn Circ J. 2001;65:439–44. doi: 10.1253/jcj.65.439. [DOI] [PubMed] [Google Scholar]
- 12.Seshadri G, Sy JC, Brown M, Dikalov S, Yang SC, Murthy N, Davis ME. Biomaterials. 2010;31:1372–9. doi: 10.1016/j.biomaterials.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sy JC, Seshadri G, Yang SC, Brown M, Oh T, Dikalov S, Murthy N, Davis ME. Nat Mater. 2008;7(11):863–8. doi: 10.1038/nmat2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman I, Cohen S. Biomaterials. 2009;30:2122–31. doi: 10.1016/j.biomaterials.2008.12.057. [DOI] [PubMed] [Google Scholar]
- 15.Tengood JE, Ridenour R, Brodsky R, Russell AJ, Little SR. Tissue Engineering Part A. 2011;17:1181–1189. doi: 10.1089/ten.tea.2010.0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otrock ZK, Mahfouz RAR, Makarem JA, Shamseddine AI. Blood Cells Mol Dis. 2007;39:212–20. doi: 10.1016/j.bcmd.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Macdonald ML, Rodriguez NM, Shah NJ, Hammond PT. Biomacromolecules. 2010;11:2053–9. doi: 10.1021/bm100413w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.She Z, Wang C, Li J, Sukhorukov GB, Antipina MN. Biomacromolecules. 2012;13:2174–80. doi: 10.1021/bm3005879. [DOI] [PubMed] [Google Scholar]
- 19.Itoh Y, Matsusaki M, Kida T, Akashi M. Biomacromolecules. 2008;9:2202–6. doi: 10.1021/bm800321w. [DOI] [PubMed] [Google Scholar]
- 20.Sukhorukov GB, Antipov AA, Voigt A, Donath E, hwald HM. Macromol. Rapid Commun. 2001;22:44–46. [Google Scholar]
- 21.De Geest BG, Sanders NN, Sukhorukov GB, Demeester J, De Smedt SC. Chem. Soc. Rev. 2007;36:636. doi: 10.1039/b600460c. [DOI] [PubMed] [Google Scholar]
- 22.She Z, Antipina MN, Li J, Sukhorukov GB. Biomacromolecules. 2010;11:1241–7. doi: 10.1021/bm901450r. [DOI] [PubMed] [Google Scholar]
- 23.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotechnol. 2001;19:1029–34. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 24.Khabbaz KR, Zankoul F, Warner KG. Ann Thorac Surg. 2001;72:S2227–33. doi: 10.1016/s0003-4975(01)03284-2. discussion S2233-4, S2267-70. [DOI] [PubMed] [Google Scholar]
- 25.Kumbhani DJ, Healey NA, Birjiniuk V, Crittenden MD, Josa M, Treanor PR, Khuri SF. Surgery. 2004;136:190–8. doi: 10.1016/j.surg.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Fréchet JMJ. J Am Chem Soc. 2008;130:10494–5. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachelder EM, Beaudette TT, Broaders KE, Fréchet JMJ, Albrecht MT, Mateczun AJ, Ainslie KM, Pesce JT, Keane-Myers AM. Mol Pharm. 2010;7:826–35. doi: 10.1021/mp900311x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen JA, Beaudette TT, Cohen JL, Broaders KE, Bachelder EM, Fréchet JMJ. Adv. Mater. 2010;22:3593–3597. doi: 10.1002/adma.201000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen JL, Schubert S, Wich PR, Cui L, Cohen JA, Mynar JL, Fréchet JMJ. Bioconjugate Chem. 2011 doi: 10.1021/bc100542r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kauffman KJ, Kanthamneni N, Meenach SA, Pierson BC, Bachelder EM, Ainslie KM. Int J Pharm. 2013;422:356–363. doi: 10.1016/j.ijpharm.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 31.Meenach SA, Kim YJ, Kauffman KJ, Kanthamneni N, Bachelder EM, Ainslie KM. Mol Pharm. 2012;9:290–8. doi: 10.1021/mp2003785. [DOI] [PubMed] [Google Scholar]
- 32.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Fréchet JMJ. Proc Natl Acad Sci USA. 2009;106:5497–502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanthamneni N, Sharma S, Meenach SA, Billet B, Zhao J-C, Bachelder EM, Ainslie KM. Int J Pharm. 2012:1–10. doi: 10.1016/j.ijpharm.2012.04.043. [DOI] [PubMed] [Google Scholar]
- 34.Holy CE, Dang SM, Davies JE, Shoichet MS. Biomaterials. 1999;20:1177–85. doi: 10.1016/s0142-9612(98)00256-7. [DOI] [PubMed] [Google Scholar]
- 35.Zolnik BS, Burgess DJ. Journal of controlled release : official journal of the Controlled Release Society. 2007;122:338–44. doi: 10.1016/j.jconrel.2007.05.034. [DOI] [PubMed] [Google Scholar]
- 36.Esposito E, Cortesi R, Nastruzzi C. Biomaterials. 1996:1–12. doi: 10.1016/0142-9612(95)00325-8. [DOI] [PubMed] [Google Scholar]
- 37.Frechet JMJ, Broaders KE, Cohen JA, Beaudette TT, Bachelder EM. P Natl Acad Sci USA. 2009;106(14):5497–5502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JM. J Am Chem Soc. 2008;130(32):10494–5. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christman KL, Vardanian AJ, Fang Q, Sievers RE, Fok HH, Lee RJ. J Am Coll Cardiol. 2004;44(3):654–60. doi: 10.1016/j.jacc.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 40.Garbern JC, Minami E, Stayton PS, Murry CE. Biomaterials. 2011;32:2407–2416. doi: 10.1016/j.biomaterials.2010.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seif-Naraghi SB, Horn D, Schup-Magoffin PJ, Christman KL. Acta Biomaterialia. 2012;8:3695–3703. doi: 10.1016/j.actbio.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.